Particle Formation in a Complex Environment

 ,
, .jpg) , , , ,
, , , ,
Abstract
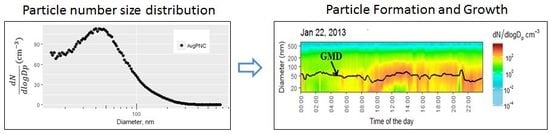
:
1. Introduction
2. Materials and Methods
2.1. Site Description and Weather Conditions
2.2. Instrumentation and Data Analysis
2.3. Classification, Growth Rate and Condensation Sinks of Particle Formation Events
- (a)
- A “strong” particle formation event is when:N > 10,000 cm−3 for at least 1 h anddN/dt > 10,000 cm−3 h−1
- (b)
- A “weak” particle formation event is when:5000 < N < 10,000 cm−3 for at least 1 h and5000 < dN/dt < 10,000 cm−3 h−1
3. Results and Discussion
3.1. General Characteristics of Class I Particle Formation Events
3.2. Factors That Influence Particle Formation and Growth Events
3.2.1. Sulfate (SO)
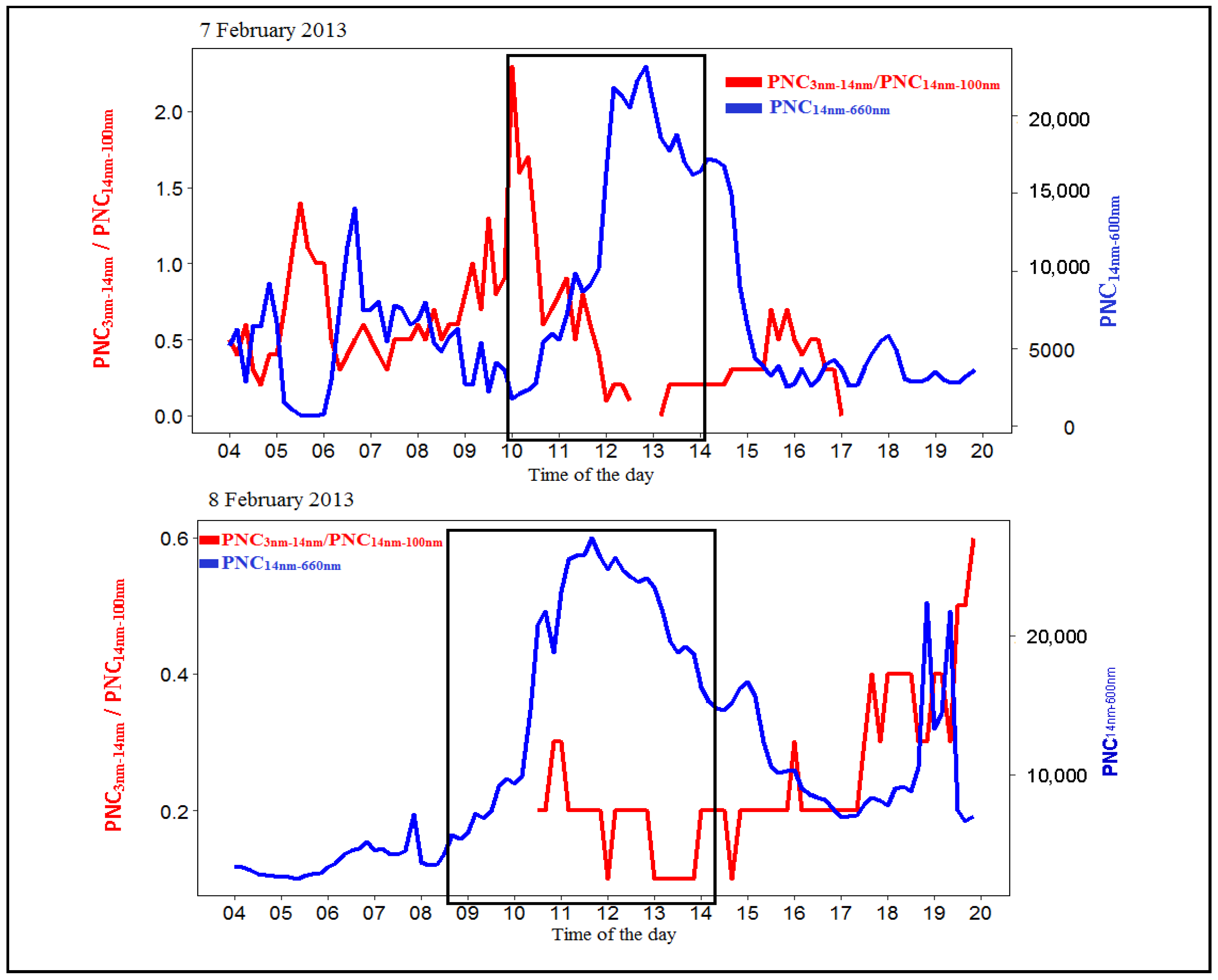
3.2.2. Particle Number Size Distributions and Condensation Sinks
3.2.3. Meteorological Conditions
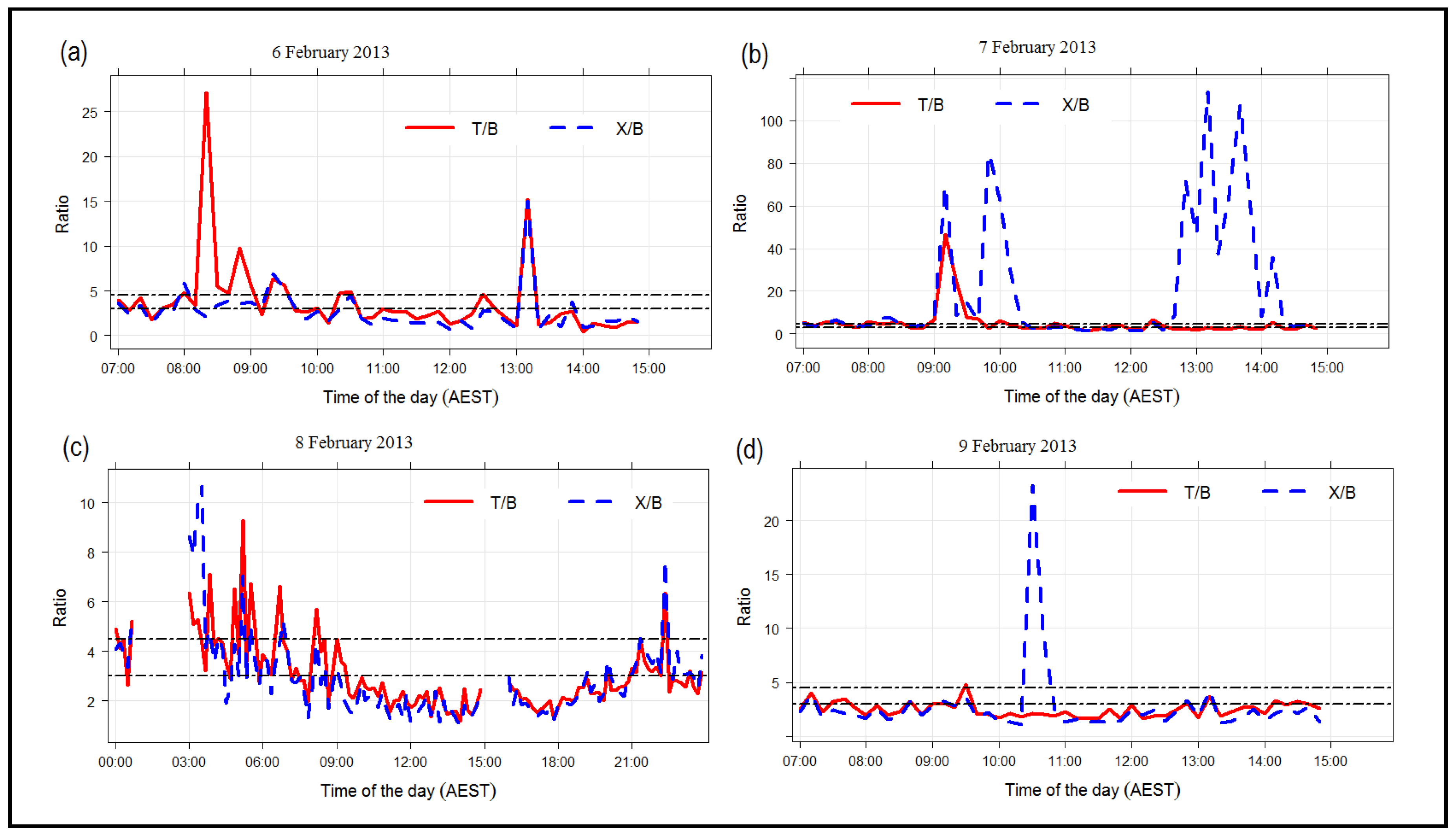
3.2.4. CO, NO, O and Isoprene
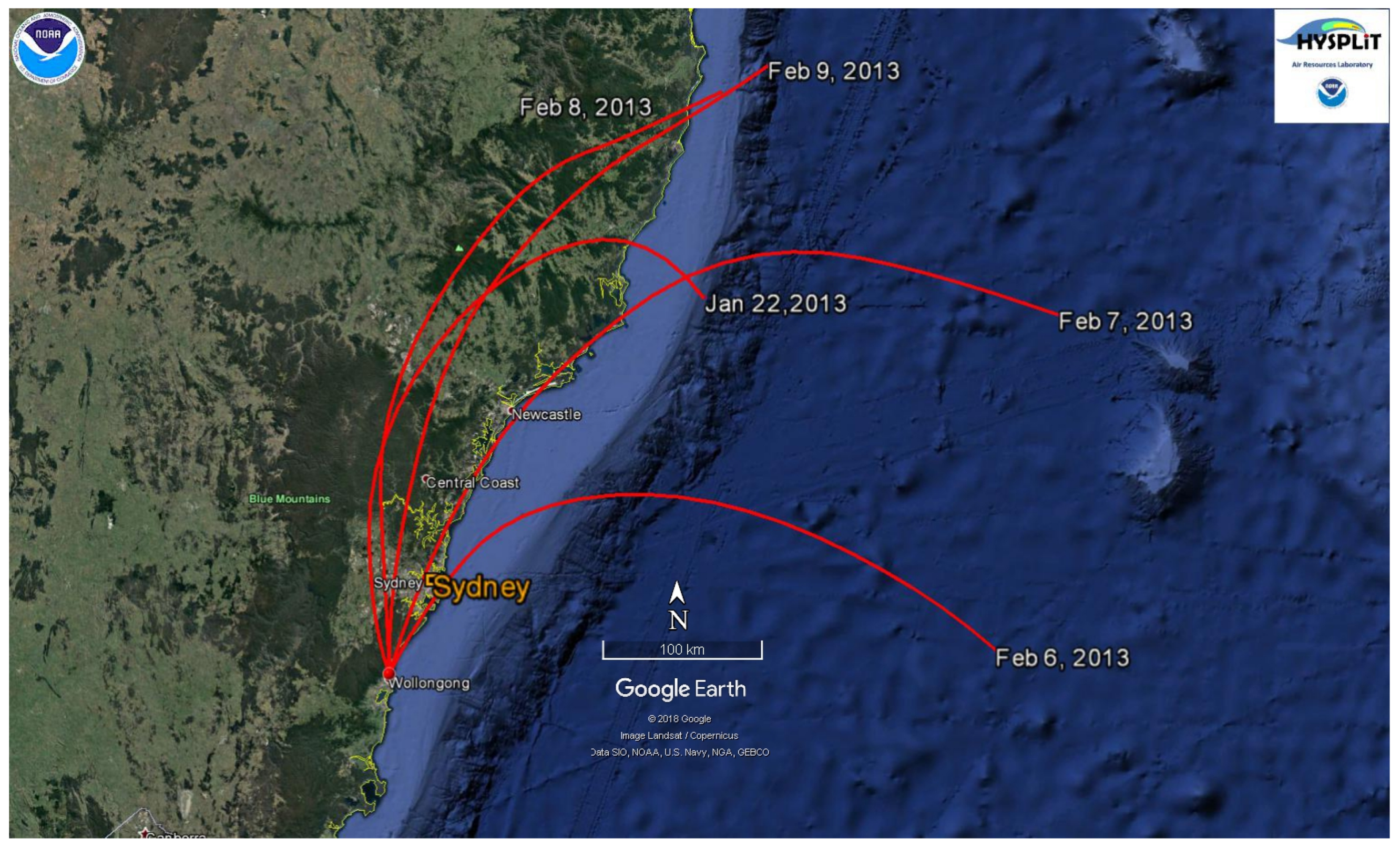
3.2.5. Photochemical Age of Air Masses
3.3. Particle Growth from Smaller Size Particles
3.4. Effect of Particles Produced during the Class I Event Days on Cloud Condensation Nuclei
4. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- U.S. Environmental Protection Agency (USEPA). Health and Environmental Effects of Particulate Matter (PM); Technical Report; U.S. Environmental Protection Agency: Washington, DC, USA, 2018.
- State of Global Air (SOGA). State of Global Air 2018: A Special Report on Global Exposure to Air Pollution and Its Disease Burden; Technical Report; Health Effects Institute: Boston, MA, USA, 2018. [Google Scholar]
- Intergovernmental Panel on Climate Change (IPCC). Summary for Policymakers. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T., Qin, D., Plattner, G.K., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P., Eds.; Cambridge University Press: Cambridge, UK, 2013; Chapter SPM; pp. 1–30. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.K.; Miller, M.R.; Shah, A.S. Air pollution and stroke. J. Stroke 2018, 20, 2–11. [Google Scholar] [CrossRef]
- Parker, J.D.; Kravets, N.; Vaidyanathan, A. Particulate matter air pollution exposure and heart disease mortality risks by race and ethnicity in the United States: 1997 to 2009 National Health Interview Survey with mortality follow-up through 2011. Circulation 2018, 137, 1688–1697. [Google Scholar] [CrossRef]
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Morales Betancourt, R.; Nenes, A. Understanding the contributions of aerosol properties and parameterization discrepancies to droplet number variability in a global climate model. Atmos. Chem. Phys. 2014, 14, 4809–4826. [Google Scholar] [CrossRef] [Green Version]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK, 2013; pp. 571–657. [Google Scholar]
- Seinfeld, J.H.; Bretherton, C.; Carslaw, K.S.; Coe, H.; DeMott, P.J.; Dunlea, E.J.; Feingold, G.; Ghan, S.; Guenther, A.B.; Kahn, R.; et al. Improving our fundamental understanding of the role of aerosol- cloud interactions in the climate system. Proc. Natl. Acad. Sci. USA 2016, 113, 5781–5790. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Dong, Y.; Li, X.; Liu, X.; Dai, J.; Chen, C.; Dong, Z.; Du, C.; Wang, Z. Different Characteristics of New Particle Formation Events at Two Suburban Sites in Northern China. Atmosphere 2017, 8, 258. [Google Scholar] [CrossRef]
- Dall’Osto, M.; Beddows, D.; Asmi, A.; Poulain, L.; Hao, L.; Freney, E.; Allan, J.; Canagaratna, M.; Crippa, M.; Bianchi, F.; et al. Novel insights on new particle formation derived from a pan-european observing system. Sci. Rep. 2018, 8, 1482. [Google Scholar] [CrossRef] [Green Version]
- Kulmala, M.; Vehkamäki, H.; Petäjä, T.; Dal Maso, M.; Lauri, A.; Kerminen, V.M.; Birmili, W.; McMurry, P.H. Formation and growth rates of ultra-fine atmospheric particles: A review of observations. J. Aerosol Sci. 2004, 35, 143–176. [Google Scholar] [CrossRef]
- Kerminen, V.M.; Chen, X.; Vakkari, V.; Petäjä, T.; Kulmala, M.; Bianchi, F. Atmospheric new particle formation and growth: Review of field observations. Environ. Res. Lett. 2018, 13, 103003. [Google Scholar] [CrossRef]
- Sipilä, M.; Berndt, T.; Petäjä, T.; Brus, D.; Vanhanen, J.; Stratmann, F.; Patokoski, J.; Mauldin, R.L.; Hyvärinen, A.P.; Lihavainen, H.; et al. The role of sulphuric acid in atmospheric nucleation. Science 2010, 327, 1243–1246. [Google Scholar] [CrossRef]
- Jokinen, T.; Berndt, T.; Makkonen, R.; Kerminen, V.M.; Junninen, H.; Paasonen, P.; Stratmann, F.; Herrmann, H.; Guenther, A.B.; Worsnop, D.R.; et al. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. USA 2015, 112, 7123–7128. [Google Scholar] [CrossRef] [Green Version]
- Kirkby, J.; Duplissy, J.; Sengupta, K.; Frege, C.; Gordon, H.; Williamson, C.; Heinritzi, M.; Simon, M.; Yan, C.; Almeida, J.; et al. Ion-induced nucleation of pure biogenic particles. Nature 2016, 533, 521. [Google Scholar] [CrossRef] [PubMed]
- Riccobono, F.; Schobesberger, S.; Scott, C.E.; Dommen, J.; Ortega, I.K.; Rondo, L.; Almeida, J.; Amorim, A.; Bianchi, F.; Breitenlechner, M.; et al. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science 2014, 344, 717–721. [Google Scholar] [CrossRef]
- Kirkby, J.; Curtius, J.; Almeida, J.; Dunne, E.; Duplissy, J.; Ehrhart, S.; Franchin, A.; Gagné, S.; Ickes, L.; Kürten, A.; et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 2011, 476, 429. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.; Schobesberger, S.; Kürten, A.; Ortega, I.K.; Kupiainen-Määttä, O.; Praplan, A.P.; Adamov, A.; Amorim, A.; Bianchi, F.; Breitenlechner, M.; et al. Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere. Nature 2013, 502, 359. [Google Scholar] [CrossRef]
- Sorribas, M.; Martín, J.G.; Hay, T.; Mahajan, A.; Cuevas, C.; Reyes, M.A.; Mora, F.P.; Gil-Ojeda, M.; Saiz-Lopez, A. On the concentration and size distribution of sub-micron aerosol in the Galápagos Islands. Atmos. Environ. 2015, 123, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Dada, L.; Paasonen, P.; Nieminen, T.; Buenrostro Mazon, S.; Kontkanen, J.; Peräkylä, O.; Lehtipalo, K.; Hussein, T.; Petäjä, T.; Kerminen, V.M.; et al. Long-term analysis of clear-sky new particle formation events and nonevents in Hyytiälä. Atmos. Chem. Phys. 2017, 17, 6227–6241. [Google Scholar] [CrossRef] [Green Version]
- De España, C.D.; Wonaschütz, A.; Steiner, G.; Rosati, B.; Demattio, A.; Schuh, H.; Hitzenberger, R. Long-term quantitative field study of New Particle Formation (NPF) events as a source of Cloud Condensation Nuclei (CCN) in the urban background of Vienna. Atmos. Environ. 2017, 164, 289–298. [Google Scholar] [CrossRef]
- Mohr, C.; Lopez-Hilfiker, F.D.; Yli-Juuti, T.; Heitto, A.; Lutz, A.; Hallquist, M.; D’Ambro, E.L.; Rissanen, M.P.; Hao, L.; Schobesberger, S.; et al. Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth. Geophys. Res. Lett. 2017, 44, 2958–2966. [Google Scholar] [CrossRef]
- Németh, Z.; Rosati, B.; Zíková, N.; Salma, I.; Bozó, L.; de España, C.D.; Schwarz, J.; Ždímal, V.; Wonaschütz, A. Comparison of atmospheric new particle formation events in three Central European cities. Atmos. Environ. 2018, 178, 191–197. [Google Scholar] [CrossRef]
- Vehkamäki, H.; Maso, M.D.; Hussein, T.; Flanagan, R.; Hyvärinen, A.; Lauros, J.; Merikanto, P.; Mönkkönen, M.; Pihlatie, K.; Salminen, K.; et al. Atmospheric particle formation events at Värriö measurement station in Finnish Lapland 1998-2002. Atmos. Chem. Phys. 2004, 4, 2015–2023. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Luo, G. Simulation of particle size distribution with a global aerosol model: Contribution of nucleation to aerosol and CCN number concentrations. Atmos. Chem. Phys. 2009, 9, 7691–7710. [Google Scholar] [CrossRef]
- Pierce, J.; Adams, P. Uncertainty in global CCN concentrations from uncertain aerosol nucleation and primary emission rates. Atmos. Chem. Phys. 2009, 9, 1339–1356. [Google Scholar] [CrossRef] [Green Version]
- Spracklen, D.V.; Carslaw, K.S.; Kulmala, M.; Kerminen, V.M.; Sihto, S.L.; Riipinen, I.; Merikanto, J.; Mann, G.W.; Chipperfield, M.P.; Wiedensohler, A.; et al. Contribution of particle formation to global cloud condensation nuclei concentrations. Geophys. Res. Lett. 2008, 35. [Google Scholar] [CrossRef] [Green Version]
- Gordon, H.; Kirkby, J.; Baltensperger, U.; Bianchi, F.; Breitenlechner, M.; Curtius, J.; Dias, A.; Dommen, J.; Donahue, N.M.; Dunne, E.M.; et al. Causes and importance of new particle formation in the present-day and preindustrial atmospheres. J. Geophys. Res. Atmos. 2017, 122, 8739–8760. [Google Scholar] [CrossRef] [Green Version]
- Box, E.O. Vegetation analogs and differences in the northern and southern hemispheres: A global comparison. Plant Ecol. 2002, 163, 139–154. [Google Scholar] [CrossRef]
- Mitchell, R.M.; Forgan, B.W.; Campbell, S.K. The climatology of Australian aerosol. Atmos. Chem. Phys. 2017, 17, 5131–5154. [Google Scholar] [CrossRef]
- Rotstayn, L.; Collier, M.; Chrastansky, A.; Jeffrey, S.; Luo, J.J. Projected effects of declining aerosols in RCP4. 5: unmasking global warming? Atmos. Chem. Phys. 2013, 13, 10883–10905. [Google Scholar] [CrossRef]
- Pushpawela, B.; Jayaratne, R.; Morawska, L. Temporal distribution and other characteristics of new particle formation events in an urban environment. Environ. Pollut. 2018, 233, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Salimi, F.; Rahman, M.; Clifford, S.; Ristovski, Z.; Morawska, L. Nocturnal new particle formation events in urban environments. Atmos. Chem. Phys. 2017, 17, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Morawska, L.; Ristovski, Z. Observation of new particle formation in subtropical urban environment. Atmos. Chem. Phys. 2011, 11, 3823–3833. [Google Scholar] [CrossRef] [Green Version]
- Mejia, J.; Wraith, D.; Mengersen, K.; Morawska, L. Trends in size classified particle number concentration in subtropical Brisbane, Australia, based on a 5 year study. Atmos. Environ. 2007, 41, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Suni, T.; Kulmala, M.; Hirsikko, A.; Bergman, T.; Laakso, L.; Aalto, P.; Leuning, R.; Cleugh, H.; Zegelin, S.; Hughes, D.; et al. Formation and characteristics of ions and charged aerosol particles in a native Australian Eucalypt forest. Atmos. Chem. Phys. 2008, 8, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Modini, R.L.; Ristovski, Z.; Johnson, G.R.; He, C.; Surawski, N.; Morawska, L.; Suni, T.; Kulmala, M. New particle formation and growth at a remote, sub-tropical coastal location. Atmos. Chem. Phys. 2009, 9, 7607–7621. [Google Scholar] [CrossRef] [Green Version]
- Cainey, J.M.; Keywood, M.; Grose, M.R.; Krummel, P.; Galbally, I.E.; Johnston, P.; Gillett, R.W.; Meyer, M.; Fraser, P.; Steele, P.; et al. Precursors to Particles P2P at Cape Grim 2006: campaign overview. Environ. Chem. 2007, 4, 143–150. [Google Scholar] [CrossRef]
- Guo, H.; Ding, A.; Morawska, L.; He, C.; Ayoko, G.; Li, Y.s.; Hung, W.t. Size distribution and new particle formation in subtropical eastern Australia. Environ. Chem. 2008, 5, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Dominick, D.; Wilson, S.R.; Paton-Walsh, C.; Humphries, R.; Guérette, E.A.; Keywood, M.; Kubistin, D.; Marwick, B. Characteristics of airborne particle number size distributions in a coastal-urban environment. Atmos. Environ. 2018, 186, 256–265. [Google Scholar] [CrossRef]
- Spracklen, D.; Carslaw, K.; Kulmala, M.; Kerminen, V.M.; Mann, G.; Sihto, S.L. The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales. Atmos. Chem. Phys. 2006, 6, 5631–5648. [Google Scholar] [CrossRef] [Green Version]
- Kulmala, M.; Kontkanen, J.; Junninen, H.; Lehtipalo, K.; Manninen, H.E.; Nieminen, T.; Petäjä, T.; Sipilä, M.; Schobesberger, S.; Rantala, P.; et al. Direct observations of atmospheric aerosol nucleation. Science 2013, 339, 943–946. [Google Scholar] [CrossRef]
- Paton-Walsh, C.; Guérette, É.A.; Kubistin, D.; Humphries, R.; Wilson, S.R.; Dominick, D.; Galbally, I.; Buchholz, R.; Bhujel, M.; Chambers, S.; et al. The MUMBA campaign: Measurements of urban, marine and biogenic air. Earth Syst. Sci. Data 2017, 9, 349–362. [Google Scholar] [CrossRef]
- Guérette, E.A.; Paton-Walsh, C.; Kubistin, D.; Humphries, R.; Bhujel, M.; Buchholz, R.R.; Chambers, S.; Cheng, M.; Davy, P.; Dominick, D.; et al. Measurements of Urban, Marine and Biogenic Air (MUMBA): Characterisation of Trace Gases and Aerosol at the Urban, Marine and Biogenic Interface in Summer in Wollongong, Australia. Earth Syst. Sci. Data 2017. [Google Scholar] [CrossRef]
- Paton-Walsh, C.; Guérette, É.A.; Emmerson, K.; Cope, M.; Kubistin, D.; Humphries, R.; Wilson, S.; Buchholz, R.; Jones, N.; Griffith, D.; et al. Urban air quality in a coastal city: Wollongong during the MUMBA campaign. Atmosphere 2018, 9, 500. [Google Scholar] [CrossRef]
- Draxler, R.R.; Stunder, B.; Rolph, G.; Stein, A.; Taylor, A. HYSPLIT 4 User’s Guide; Version 4 Report; NOAA Air Resources Laboratory: Silver Spring, MD, USA, 2018.
- Draxler, R.R.; Rolph, G. HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model Access; NOAA Air Resources Laboratory: Silver Spring, MD, USA, 2003. Available online: http://www.arl.noaa.gov/ready/hysplit4.html (accessed on 24 June 2018).
- Stein, A.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.; Cohen, M.; Ngan, F. NOAA’s HYSPLIT atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Fleming, Z.L.; Monks, P.S.; Manning, A.J. Untangling the influence of air-mass history in interpreting observed atmospheric composition. Atmos. Res. 2012, 104, 1–39. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Carslaw, D.C.; Ropkins, K. Openair—An R package for air quality data analysis. Environ. Model. Softw. 2012, 27, 52–61. [Google Scholar] [CrossRef]
- Dal Maso, M.; Kulmala, M.; Riipinen, I.; Wagner, R.; Hussein, T.; Aalto, P.P.; Lehtinen, K.E. Formation and growth of fresh atmospheric aerosols: Eight years of aerosol size distribution data from SMEAR II, Hyytiälä), Finland. Boreal Environ. Res. 2005, 10, 323–336. [Google Scholar]
- Zhang, Q.; Stanier, C.O.; Canagaratna, M.R.; Jayne, J.T.; Worsnop, D.R.; Pandis, S.N.; Jimenez, J.L. Insights into the chemistry of new particle formation and growth events in Pittsburgh based on aerosol mass spectrometry. Environ. Sci. Technol. 2004, 38, 4797–4809. [Google Scholar] [CrossRef] [PubMed]
- Sorribas, M.; Adame, J.; Olmo, F.; Vilaplana, J.; Gil-Ojeda, M.; Alados-Arboledas, L. A long-term study of new particle formation in a coastal environment: Meteorology, gas phase and solar radiation implications. Sci. Total Environ. 2015, 511, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Kulmala, M.; Petäjä, T.; Nieminen, T.; Sipilä, M.; Manninen, H.E.; Lehtipalo, K.; Dal Maso, M.; Aalto, P.P.; Junninen, H.; Paasonen, P.; et al. Measurement of the nucleation of atmospheric aerosol particles. Nat. Protoc. 2012, 7, 1651. [Google Scholar] [CrossRef]
- Dal Maso, M.; Kulmala, M.; Lehtinen, K.E.; Mäkelä, J.; Aalto, P.; O’Dowd, C. Condensation and coagulation sinks and formation of nucleation mode particles in coastal and boreal forest boundary layers. J. Geophys. Res. Atmos. 2002, 107, PAR 2-1–PAR 2-10. [Google Scholar] [CrossRef]
- Stanier, C.O.; Khlystov, A.Y.; Pandis, S.N. Nucleation events during the Pittsburgh Air Quality Study: Description and relation to key meteorological, gas phase, and aerosol parameters special issue of aerosol science and technology on findings from the fine particulate matter supersites program. Aerosol Sci.Technol. 2004, 38, 253–264. [Google Scholar] [CrossRef]
- Yue, D.; Hu, M.; Zhang, R.; Wang, Z.; Zheng, J.; Wu, Z.; Wiedensohler, A.; He, L.; Huang, X.; Zhu, T. The roles of sulphuric acid in new particle formation and growth in the mega-city ofBeijing. Atmos. Chem. Phys. 2010, 10, 4953–4960. [Google Scholar] [CrossRef]
- Fuchs, N.; Sutugin, A. High-dispersed aerosols. In Topics in Current Aerosol Research; Hidy, G., Brock, J., Eds.; International Reviews in Aerosol Physics and Chemistry: Pergamon, Turkey, 1971; p. 1. [Google Scholar] [CrossRef]
- Massman, W. A review of the molecular diffusivities of H2O, CO2, CH4, CO, O3, SO2, NH3, N2O, NO, and NO2 in air, O2 and N2 near STP. Atmos. Environ. 1998, 32, 1111–1127. [Google Scholar] [CrossRef]
- Cope, M.; Keywood, M.; Emmerson, K.; Galbally, I.; Boast, K.; Chambers, S.; Cheng, M.; Crumeyrolle, S.; Dunne, E.; Fedele, R.; et al. Sydney Particle Study–Stage II; The Centre for Australian Weather and Climate Research: Aspendale, Australia, 2014.
- Kulmala, M.; Petäjä, T.; Mönkkönen, P.; Koponen, I.; Maso, M.D.; Aalto, P.; Lehtinen, K.; Kerminen, V.M. On the growth of nucleation mode particles: Source rates of condensable vapour in polluted and clean environments. Atmos. Chem. Phys. 2005, 5, 409–416. [Google Scholar] [CrossRef]
- Zhu, Y.; Sabaliauskas, K.; Liu, X.; Meng, H.; Gao, H.; Jeong, C.H.; Evans, G.J.; Yao, X. Comparative analysis of new particle formation events in less and severely polluted urban atmosphere. Atmos. Environ. 2014, 98, 655–664. [Google Scholar] [CrossRef]
- Grythe, H.; Ström, J.; Krejci, R.; Quinn, P.; Stohl, A. A review of sea-spray aerosol source functions using a large global set of sea salt aerosol concentration measurements. Atmos. Chem. Phys. 2014, 14, 1277–1297. [Google Scholar] [CrossRef] [Green Version]
- Millero, F.J.; Feistel, R.; Wright, D.G.; McDougall, T.J. The composition of Standard Seawater and the definition of the Reference-Composition Salinity Scale. Deep Sea Res. Part I Oceanogr. Res. Pap. 2008, 55, 50–72. [Google Scholar] [CrossRef]
- Ayers, G.; Cainey, J.; Gillett, R. Sulfur dioxide and dimethyl sulphide in marine air at Cape Grim, Tasmania. Oceanogr. Lit. Rev. 1998, 1, 49. [Google Scholar]
- CSIRO. Cape Grim Baseline Air Pollution Station Data. 2017. Available online: https://research.csiro.au/acc/capabilities/cape-grim-baseline-air-pollution-station/cgbaps/ (accessed on 12 February 2018).
- Derek, N.; Krummel, P.; Cleland, S. Baseline Atmospheric Program Australia 2009–2010; Technical Report; Australian Government Bureau of Meteorology: Melbourne, Australia, 2014. Available online: http://www.bom.gov.au/inside/cgbaps/baseline.shtml (accessed on 12 February 2018).
- Zhang, J.; Chen, Z.; Lu, Y.; Gui, H.; Liu, J.; Wang, J.; Yu, T.; Cheng, Y. Observations of New Particle Formation, Subsequent Growth and Shrinkage during Summertime in Beijing. Aerosol Air Qual. Res. 2016, 16, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Guérette, E.A. Measurements of Volatile Organic Compound Sources and Ambient Concentrations in South-East Australia. Ph.D. Thesis, School of Chemistry, University of Wollongong, Wollongong, NSW, Australia, 2016. [Google Scholar]
- Hamed, A.; Korhonen, H.; Sihto, S.L.; Joutsensaari, J.; Järvinen, H.; Petäjä, T.; Arnold, F.; Nieminen, T.; Kulmala, M.; Smith, J.N.; et al. The role of relative humidity in continental new particle formation. J. Geophys. Res. 2011, 116. [Google Scholar] [CrossRef] [Green Version]
- Claeys, M.; Graham, B.; Vas, G.; Wang, W.; Vermeylen, R.; Pashynska, V.; Cafmeyer, J.; Guyon, P.; Andreae, M.O.; Artaxo, P.; et al. Formation of secondary organic aerosols through photooxidation of isoprene. Science 2004, 303, 1173–1176. [Google Scholar] [CrossRef]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef] [Green Version]
- Borbon, A.; Fontaine, H.; Veillerot, M.; Locoge, N.; Galloo, J.; Guillermo, R. An investigation into the traffic-related fraction of isoprene at an urban location. Atmos. Environ. 2001, 35, 3749–3760. [Google Scholar] [CrossRef]
- Kansal, A. Sources and reactivity of NMHCs and VOCs in the atmosphere: A review. J. Hazard. Mater. 2009, 166, 17–26. [Google Scholar] [CrossRef]
- Kalabokas, P.; Hatzianestis, J.; Bartzis, J.; Papagiannakopoulos, P. Atmospheric concentrations of saturated and aromatic hydrocarbons around a Greek oil refinery. Atmos. Environ. 2001, 35, 2545–2555. [Google Scholar] [CrossRef]
- Molteni, U.; Bianchi, F.; Klein, F.; Haddad, I.E.; Frege, C.; Rossi, M.J.; Dommen, J.; Baltensperger, U. Formation of highly oxygenated organic molecules from aromatic compounds. Atmos. Chem. Phys. 2018, 18, 1909–1921. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Hanai, Y.; Masunaga, S. Ambient levels of volatile organic compounds in the vicinity of petrochemical industrial area of Yokohama, Japan. Air. Qual. Atmos. Health 2010, 3, 65–75. [Google Scholar] [CrossRef]
- Miller, L.; Xu, X.; Wheeler, A.; Atari, D.O.; Grgicak-Mannion, A.; Luginaah, I. Spatial variability and application of ratios between BTEX in two Canadian cities. Sci. World J. 2011, 11, 2536–2549. [Google Scholar] [CrossRef]
- Khoder, M.I. Ambient levels of volatile organic compounds in the atmosphere of Greater Cairo. Atmos. Environ. 2007, 41, 554–566. [Google Scholar] [CrossRef]
- Miller, L.; Lemke, L.D.; Xu, X.; Molaroni, S.M.; You, H.; Wheeler, A.J.; Booza, J.; Grgicak-Mannion, A.; Krajenta, R.; Graniero, P.; et al. Intra-urban correlation and spatial variability of air toxics across an international airshed in Detroit, Michigan (USA) and Windsor, Ontario (Canada). Atmos. Environ. 2010, 44, 1162–1174. [Google Scholar] [CrossRef]
- Barletta, B.; Meinardi, S.; Rowland, F.S.; Chan, C.Y.; Wang, X.; Zou, S.; Chan, L.Y.; Blake, D.R. Volatile organic compounds in 43 Chinese cities. Atmos. Environ. 2005, 39, 5979–5990. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Chameides, W.; Cardelino, C.; Blake, D.; Streets, D. Source characteristics of volatile organic compounds during high ozone episodes in Hong Kong, Southern China. Atmos. Chem. Phys. 2008, 8, 4983–4996. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, D.; Lohmann, U.; Raga, G.B.; O’Dowd, C.D.; Kulmala, M.; Fuzzi, S.; Reissell, A.; Andreae, M.O. Flood or drought: How do aerosols affect precipitation? Science 2008, 321, 1309–1313. [Google Scholar] [CrossRef]
- Kazil, J.; Stier, P.; Zhang, K.; Quaas, J.; Kinne, S.; O’donnell, D.; Rast, S.; Esch, M.; Ferrachat, S.; Lohmann, U.; et al. Aerosol nucleation and its role for clouds and Earth’s radiative forcing in the aerosol-climate model ECHAM5-HAM. Atmos. Chem. Phys. 2010, 10, 10733–10752. [Google Scholar] [CrossRef]
- Wang, M.; Penner, J. Aerosol indirect forcing in a global model with particle nucleation. Atmos. Chem. Phys. 2009, 9, 239–260. [Google Scholar] [CrossRef] [Green Version]
- Dusek, U.; Frank, G.; Hildebrandt, L.; Curtius, J.; Schneider, J.; Walter, S.; Chand, D.; Drewnick, F.; Hings, S.; Jung, D.; et al. Size matters more than chemistry for cloud-nucleating ability of aerosol particles. Science 2006, 312, 1375–1378. [Google Scholar] [CrossRef]
- Petters, M.; Kreidenweis, S. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons. Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Leng, C.; Zhang, Q.; Tao, J.; Zhang, H.; Zhang, D.; Xu, C.; Li, X.; Kong, L.; Cheng, T.; Zhang, R.; et al. Impacts of new particle formation on aerosol cloud condensation nuclei (CCN) activity in Shanghai: Case study. Atmos. Chem. Phys. 2014, 14, 11353–11365. [Google Scholar] [CrossRef]
- Lihavainen, H.; Kerminen, V.M.; Komppula, M.; Hatakka, J.; Aaltonen, V.; Kulmala, M.; Viisanen, Y. Production of “potential” cloud condensation nuclei associated with atmospheric new-particle formation in northern Finland. J. Geophys. Res. 2003, 108, 4782. [Google Scholar] [CrossRef]
- Kulmala, M.; Pirjola, L.; Mäkelä, J.M. Stable sulphate clusters as a source of new atmospheric particles. Nature 2000, 404, 66. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
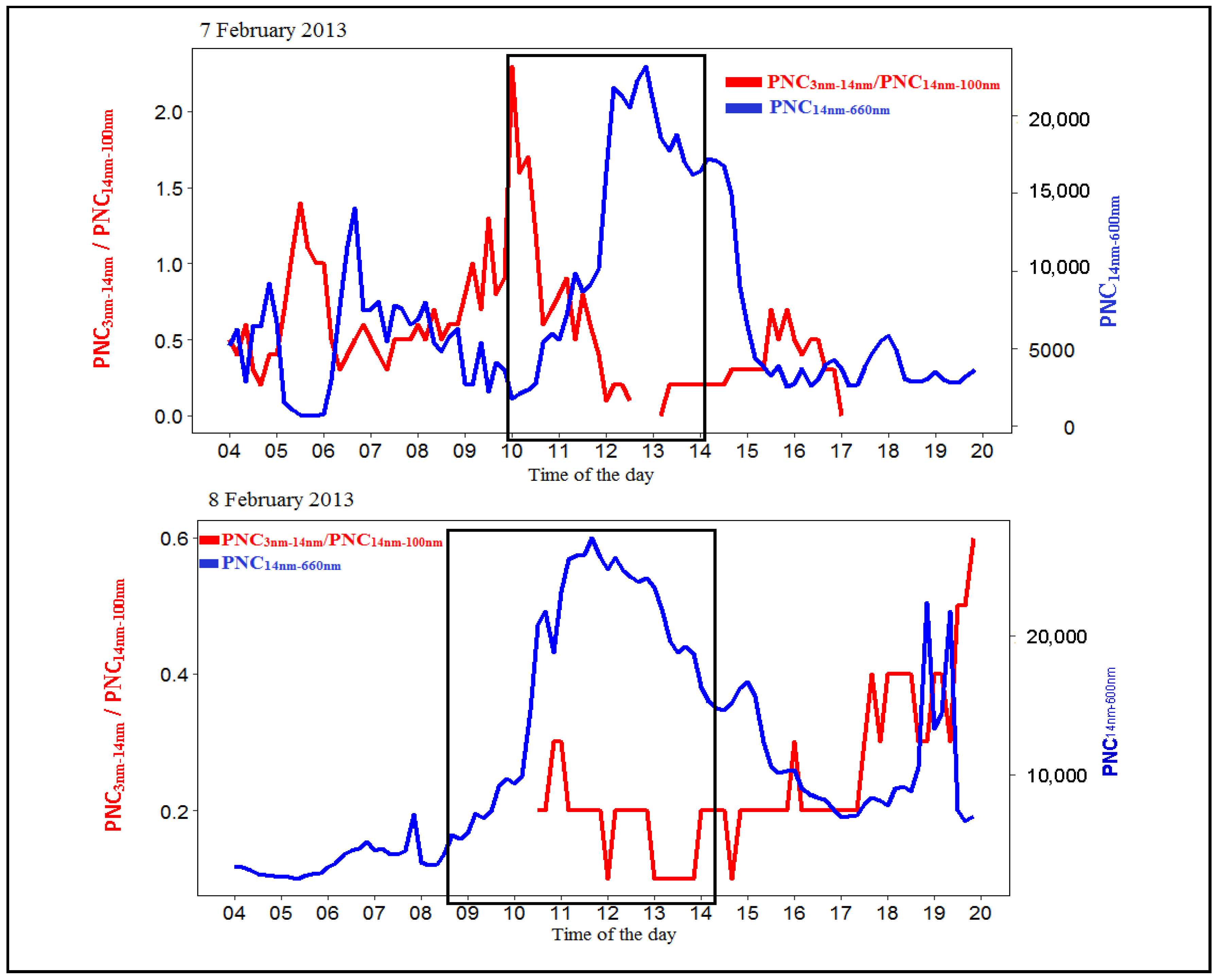{kind=link}
{kind=link}
{kind=link}
| Date | Time (Start) | Time (End) | Duration (hours) | Primary GMD (nm) | Final GMD (nm) | GR (nm/h) | Classification |
|---|---|---|---|---|---|---|---|
| 22 January 2013 | 8:30 | 14:00 | 6.3 | 30.0 | 70.0 | 6.3 | Weak |
| 6 February 2013 | 10:00 | 13:00 | 3.0 | 25.0 | 50.0 | 8.3 | Weak |
| 7 February 2013 | 10:00 | 14:00 | 4.0 | 24.0 | 55.0 | 7.8 | Weak |
| 8 February 2013 | 8:30 | 14:30 | 6.0 | 30.0 | 60.0 | 5.0 | Weak |
| Average | 4.8 | 27.2 | 58.8 | 6.9 |
| Class I | Condensation Sink, CS (s) (10) | Class II | Condensation Sink (CS) (s) (10) |
|---|---|---|---|
| 22 January 2013 | 0.6 ± 0.3 | 17 January 2013 | 0.9 ± 0.2 |
| 6 February 2013 | 0.2 ± 0.03 | 27 January 2013 | 0.5 ± 0.2 |
| 7 February 2013 | 0.3 ± 0.1 | 29 January 2013 | 0.6 ± 0.3 |
| 8 February 2013 | 0.6 ± 0.2 | 9 February 2013 | 1.0 ± 0.2 |
| Condensation Sink (s) (10) | ||||||
|---|---|---|---|---|---|---|
| Minimum | Maximum | Mean | Median | Std | ||
| Class I | AM | 0.11 | 1.49 | 0.59 | 0.54 | 0.03 |
| PM | 0.10 | 3.24 | 1.35 | 1.18 | 0.06 | |
| Class II | AM | 0.09 | 2.62 | 0.89 | 0.83 | 0.06 |
| PM | 0.18 | 3.17 | 0.90 | 0.84 | 0.06 | |
| Non_event | AM | 0.11 | 3.47 | 0.73 | 0.58 | 0.02 |
| PM | 0.13 | 0.13 | 0.35 | 0.22 | 0.03 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominick, D.; Wilson, S.R.; Paton-Walsh, C.; Humphries, R.; Guérette, É.-A.; Keywood, M.; Selleck, P.; Kubistin, D.; Marwick, B. Particle Formation in a Complex Environment. Atmosphere 2019, 10, 275. https://doi.org/10.3390/atmos10050275
Dominick D, Wilson SR, Paton-Walsh C, Humphries R, Guérette É-A, Keywood M, Selleck P, Kubistin D, Marwick B. Particle Formation in a Complex Environment. Atmosphere. 2019; 10(5):275. https://doi.org/10.3390/atmos10050275
Chicago/Turabian StyleDominick, Doreena, Stephen R. Wilson, Clare Paton-Walsh, Ruhi Humphries, Élise-Andrée Guérette, Melita Keywood, Paul Selleck, Dagmar Kubistin, and Ben Marwick. 2019. "Particle Formation in a Complex Environment" Atmosphere 10, no. 5: 275. https://doi.org/10.3390/atmos10050275
APA StyleDominick, D., Wilson, S. R., Paton-Walsh, C., Humphries, R., Guérette, É.-A., Keywood, M., Selleck, P., Kubistin, D., & Marwick, B. (2019). Particle Formation in a Complex Environment. Atmosphere, 10(5), 275. https://doi.org/10.3390/atmos10050275