
Nitrogen Oxides (NOx) in the Arctic Troposphere at Ny-Ålesund (Svalbard Islands): Effects of Anthropogenic Pollution Sources

 , , ,
, , , 


Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
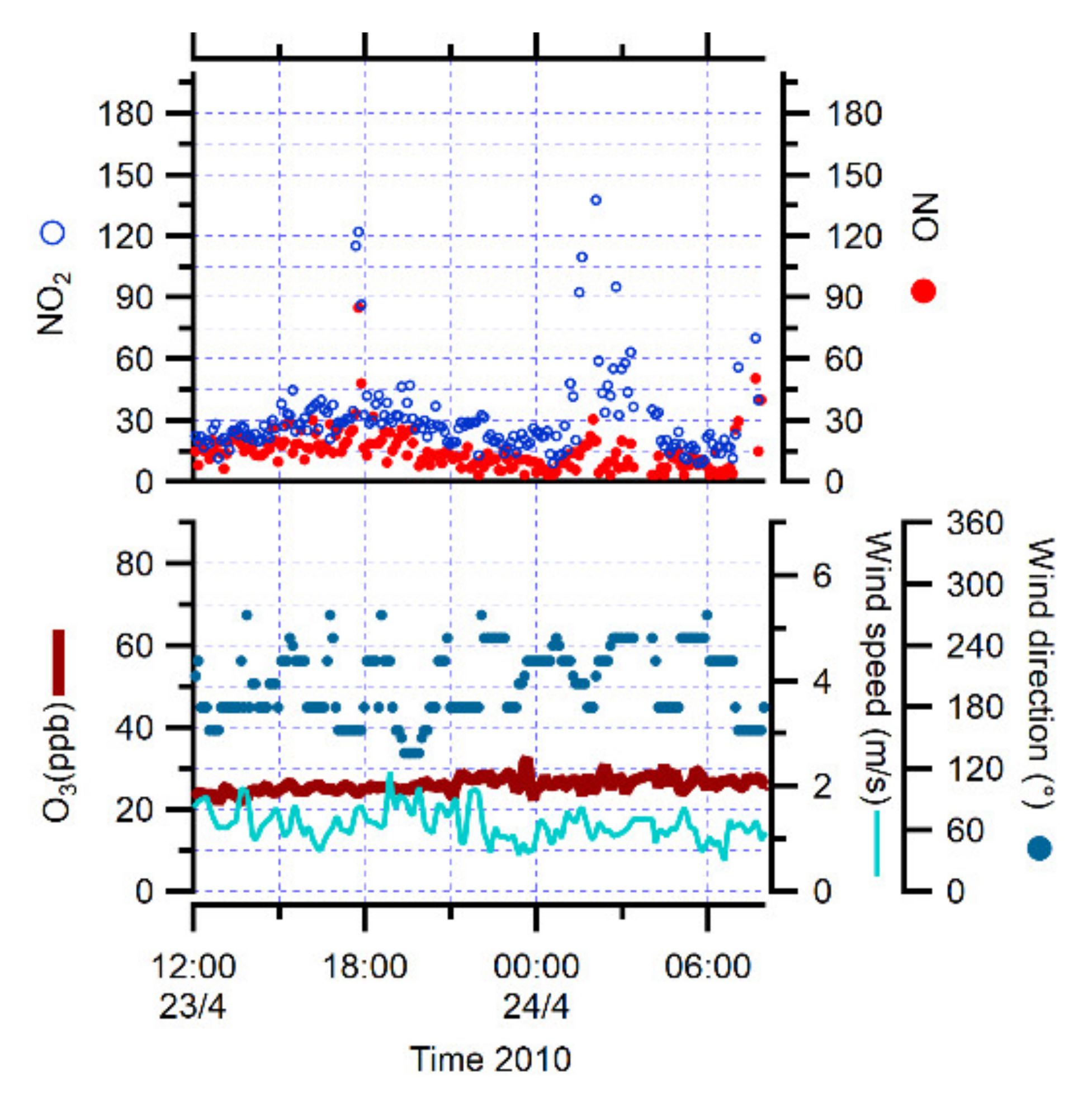
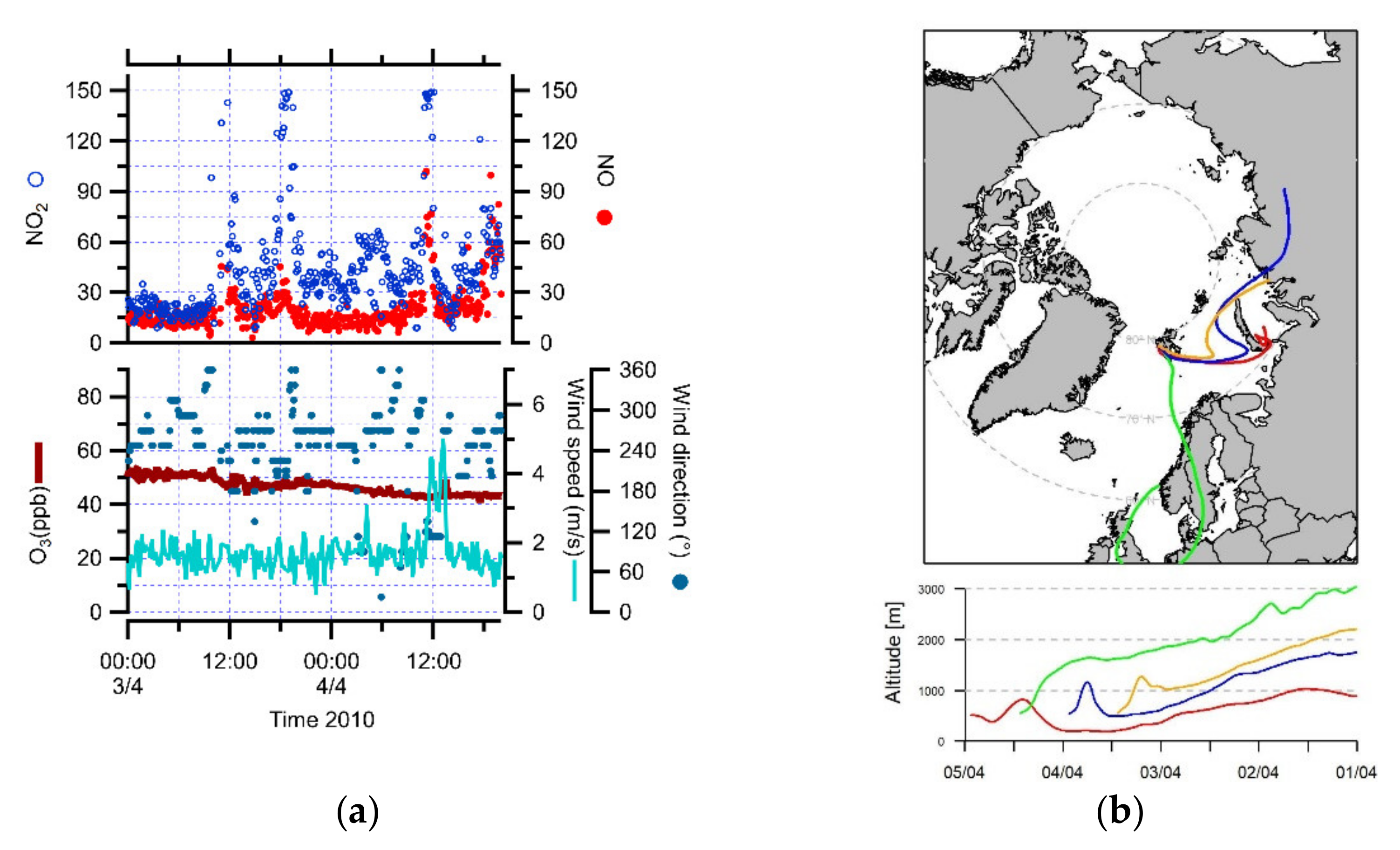
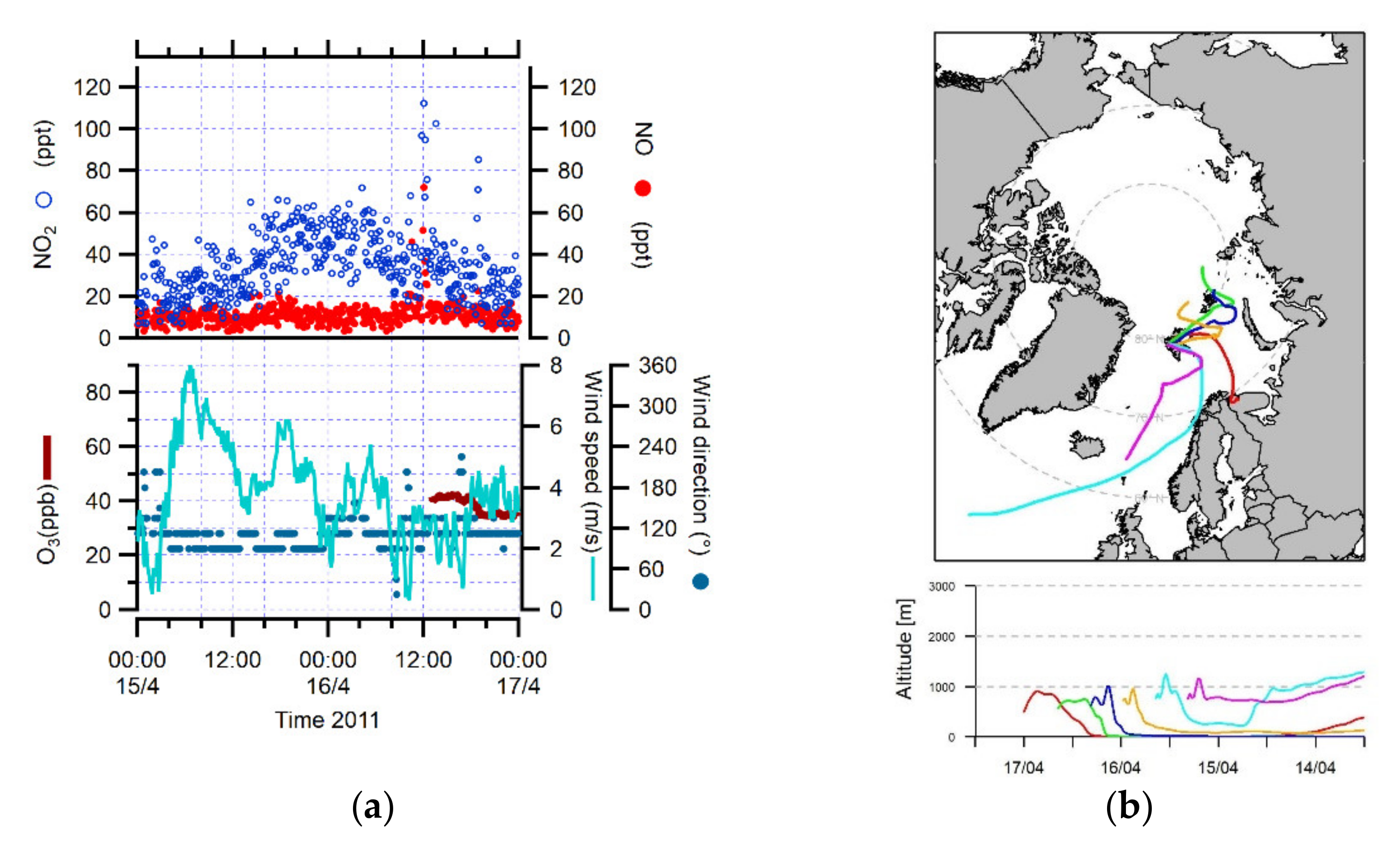
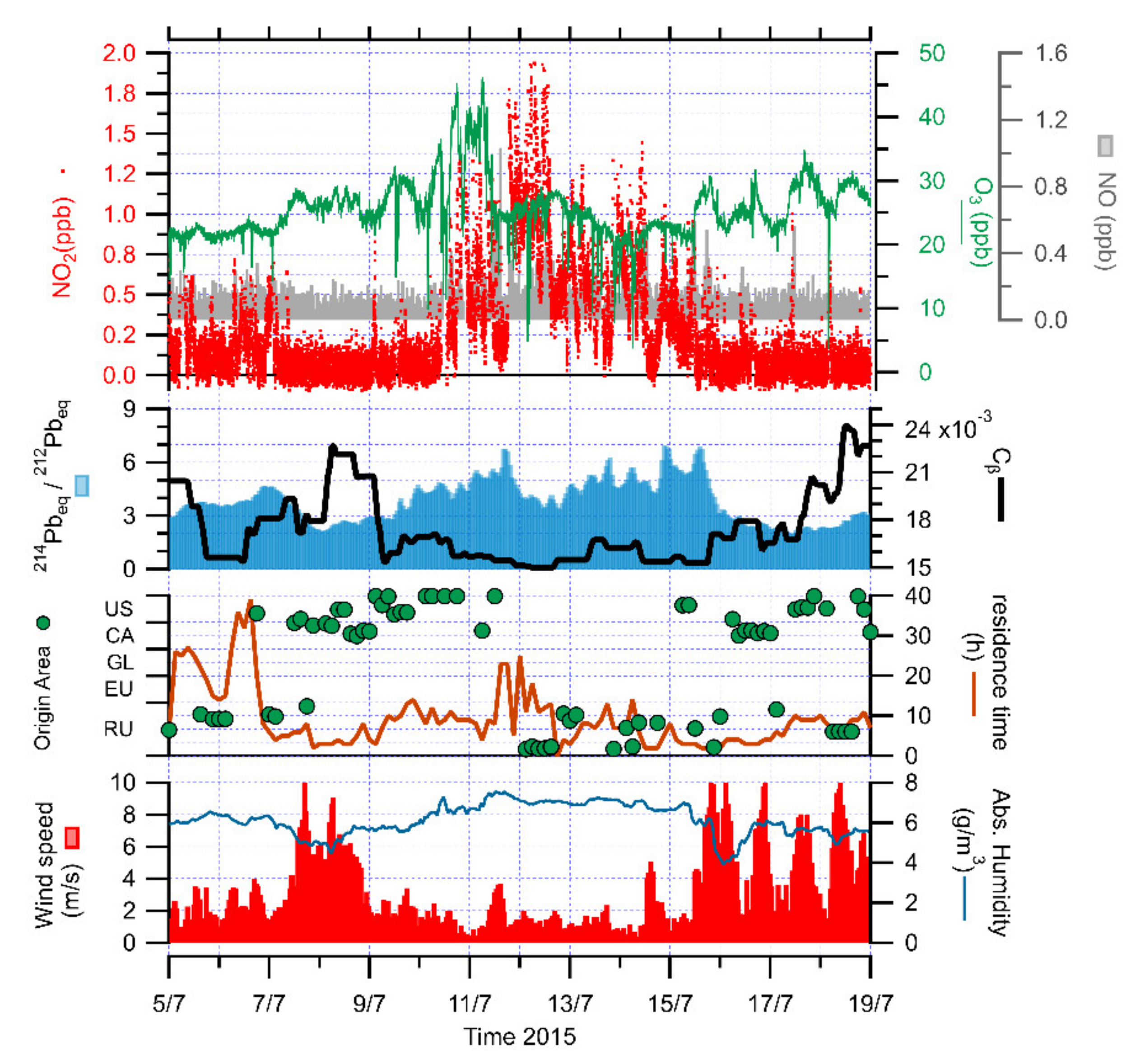
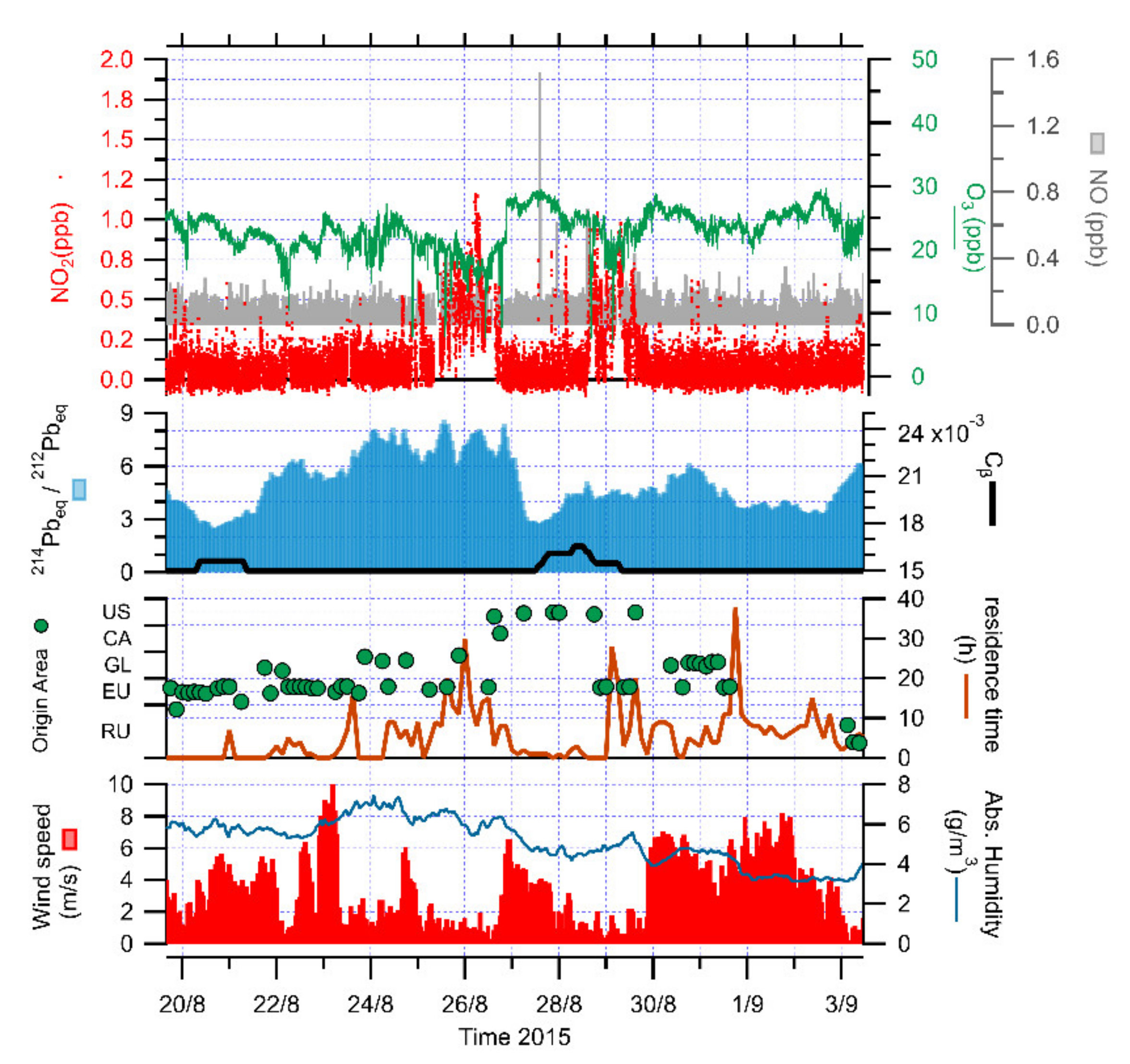
3.1. Identification of Peaks
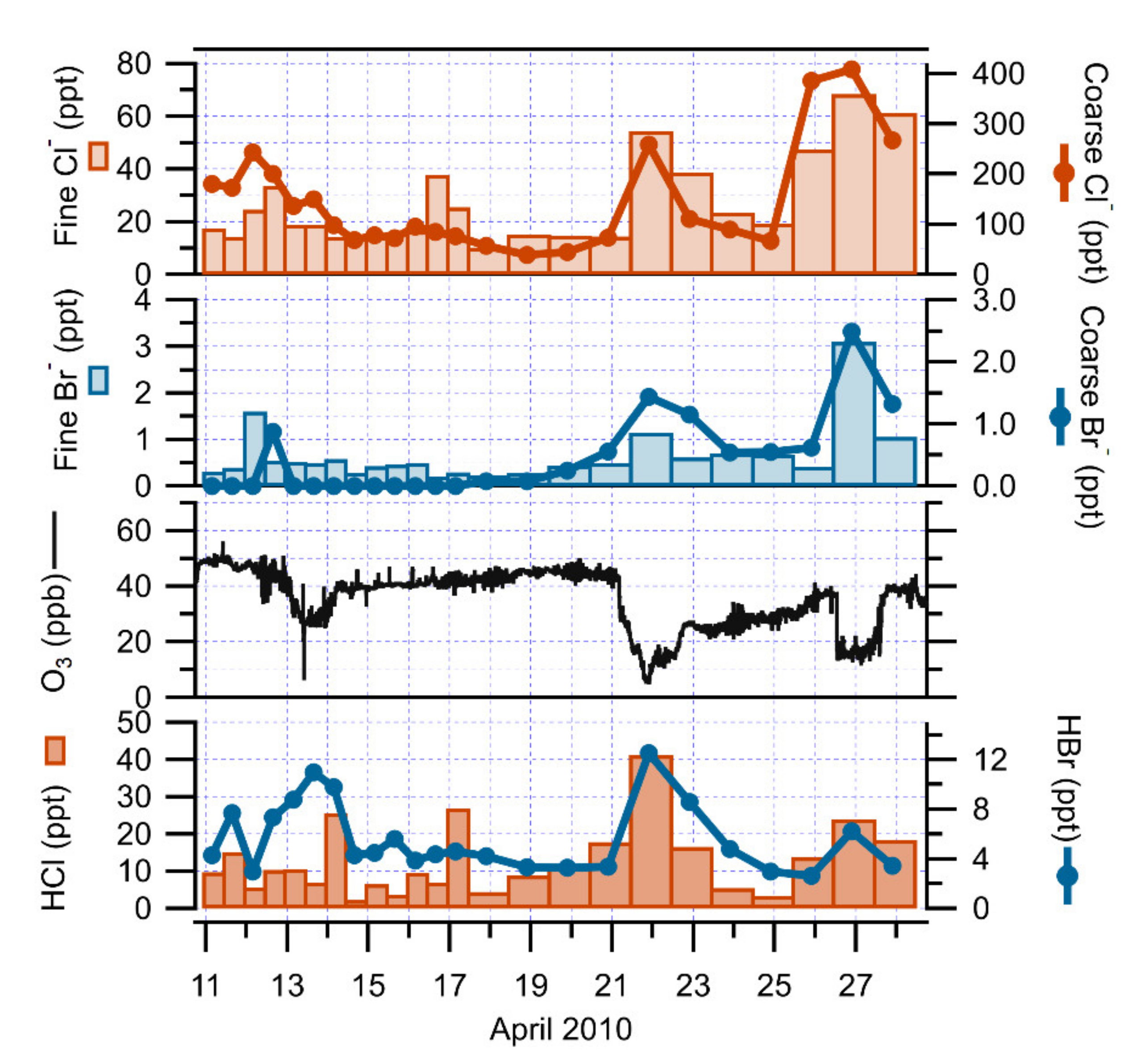
3.2. The Role of Halogens in Ozone Depletion
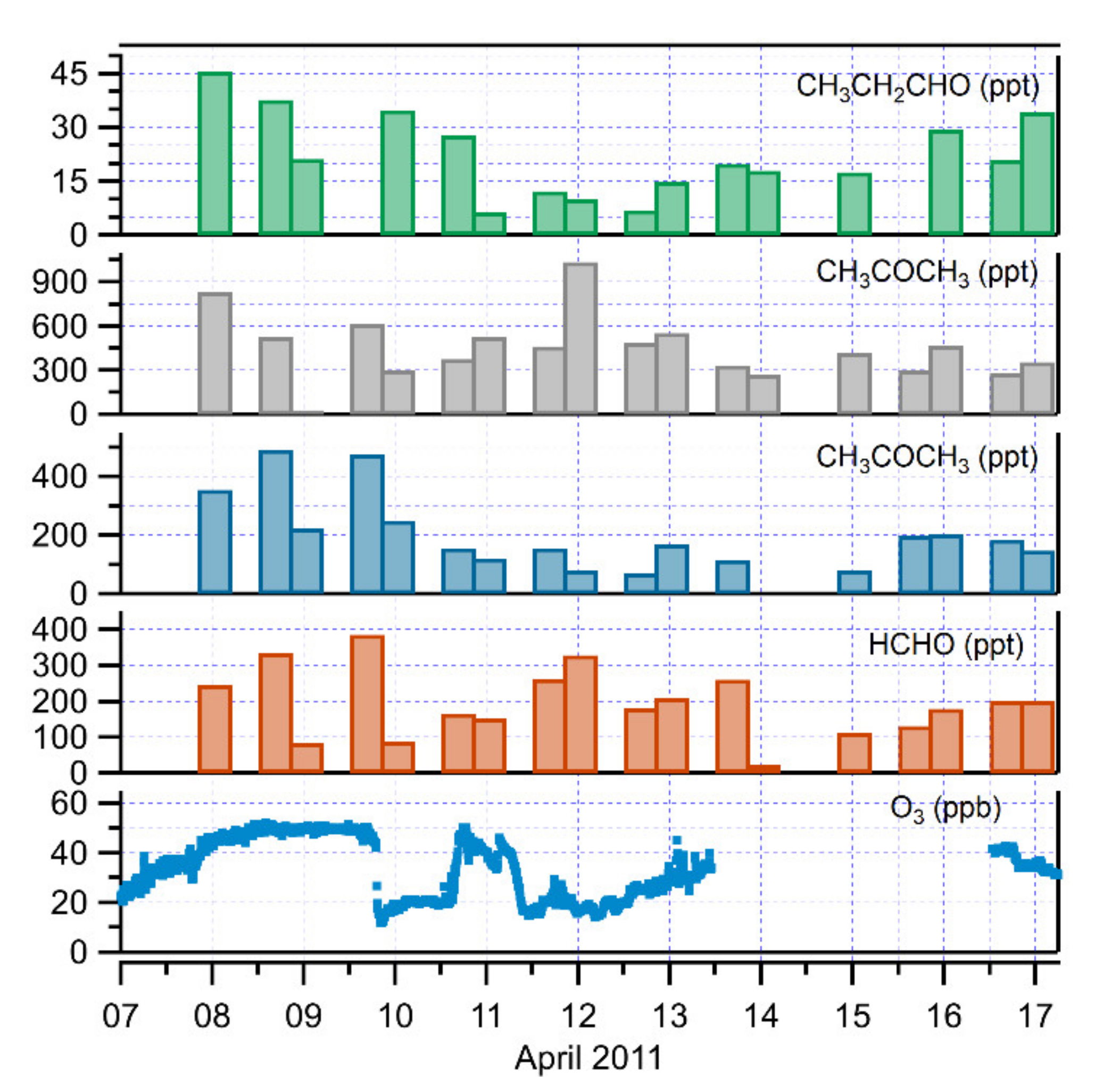
3.3. The Role of Carbonyl Compounds in Ozone Depletion
3.4. The Role of Atmospheric Stability
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fowler, D.; Coyle, M.; Skiba, U.; Sutton, M.A.; Cape, J.N.; Reis, S.; Sheppard, L.J.; Jenkins, A.; Grizzetti, B.; Galloway, J.N.; et al. The global nitrogen cycle in the twenty-first century. Philos. Trans. R. Soc. B 2013, 368, 20130164. [Google Scholar] [CrossRef]
- Law, K.S.; Roiger, A.; Thomas, J.L.; Marelle, L.; Raut, J.-C.; Dalsøren, S.; Fuglestvedt, J.; Tuccella, P.; Weinzierl, B.; Schlager, H. Local Arctic air pollution: Sources and impacts. Ambio 2017, 46, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Schmale, J.; Arnold, S.R.; Law, K.S.; Thorp, T.; Anenberg, S.; Simpson, W.R.; Mao, J.; Pratt, K.A. Local Arctic Air Pollution: A Neglected but Serious Problem. Earth’s Future 2018, 6, 1385–1412. [Google Scholar] [CrossRef]
- Grannas, A.M.; Jones, A.E.; Dibb, J.; Ammann, M.; Anastasio, C.; Beine, H.J.; Bergin, M.; Bottenheim, J.; Boxe, C.S.; Carver, G.; et al. An overview of snow photochemistry: Evidence, mechanisms and impacts. Atmos. Chem. Phys. 2007, 7, 4329–4373. [Google Scholar] [CrossRef]
- Shindell, D.; Faluvegi, G.; Lacis, A.; Hansen, J.; Ruedy, R.; Aguilar, E. Role of tropospheric ozone increases in 20th-century climate change. J. Geophys. Res. 2006, 111. [Google Scholar] [CrossRef]
- Honrath, R.E.; Jaffe, D.A. The seasonal cycle of nitrogen oxides in the Arctic troposphere at Barrow, Alaska. J. Geophys. Res. 1992, 97, 20615. [Google Scholar] [CrossRef]
- Beine, H.J.; Engardt, M.; Jaffe, D.A.; Hov, Ø.; Holme´n, K.; Stordal, F. Measurements of NOx and aerosol particles at the NY-A lesund Zeppelin mountain station on Svalbard: Influence of regional and local pollution sources. Atmos. Environ. 1996, 30, 1067–1079. [Google Scholar] [CrossRef]
- Beine, H.J.; Jaffe, D.A.; Solberg, S.; Holmén, K.; Stordal, F.; Engardt, M.; Schmidbauer, N. NOx during ozone depletion events in the arctic troposphere at Ny-Alesund, Svalbard. Tellus 1997, 49, 556–565. [Google Scholar] [CrossRef][Green Version]
- Beine, H.J.; Honrath, R.E.; Dominé, F.; Simpson, W.; Fuentes, J.D. NOx during background and ozone depletion periods at Alert: Fluxes above the snow surface. J. Geophys. Res. 2002, 107, ACH-7. [Google Scholar] [CrossRef]
- Solberg, S.; Krognes, T.; Stordal, F.; Hov, O.; Beine, H.J.; Jaffe, D.A.; Clemitshaw, K.C.; Penkett, S.A. Reactive Nitrogen Compounds at Spitsbergen in the Norwegian Arctic. J. Atmos. Chem. 1997, 28, 209–225. [Google Scholar] [CrossRef]
- Amoroso, A.; Domine, F.; Esposito, G.; Morin, S.; Savarino, J.; Nardino, M.; Montagnoli, M.; Bonneville, J.-M.; Clement, J.-C.; Ianniello, A.; et al. Microorganisms in Dry Polar Snow Are Involved in the Exchanges of Reactive Nitrogen Species with the Atmosphere. Environ. Sci. Technol. 2010, 44, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Ianniello, A.; Spataro, F.; Salvatori, R.; Valt, M.; Nardino, M.; Björkman, M.P.; Esposito, G.; Montagnoli, M. Air-snow exchange of reactive nitrogen species at Ny-Ålesund, Svalbard (Arctic). Rend. Lincei 2016, 27 (Suppl. 1), 33–45. [Google Scholar] [CrossRef]
- Fontijn, A.; Sabadell, A.J.; Ronco, R.J. Homogeneous chemiluminescent measurement of nitric oxide with ozone. Implications for continuous selective monitoring of gaseous air pollutants. Anal. Chem. 1970, 42, 575–579. [Google Scholar] [CrossRef]
- Villena, G.; Bejan, I.G.; Kurtenbach, R.; Wiesen, P.; Kleffmann, J. Interferences of commercial NO2 instruments in the urban atmosphere and in a smog chamber. Atmos. Meas. Tech. 2012, 5, 149–159. [Google Scholar] [CrossRef]
- Reed, C.; Evans, M.J.; Di Carlo, P.; Lee, J.D.; Carpenter, L.J. Interferences in photolytic NO2 measurements: Explanation for an apparent missing oxidant? Atmos. Chem. Phys. 2016, 16, 4707–4724. [Google Scholar] [CrossRef]
- Beine, H.J.; Allegrini, I.; Sparapani, R.; Ianniello, A.; Valentini, F. Three years of springtime trace gas and particle measurements at Ny-Ålesund, Svalbard. Atmos. Environ. 2001, 35, 3645–3658. [Google Scholar] [CrossRef]
- Ianniello, A.; Beine, H.J.; Sparapani, R.; Di Bari, F.; Allegrini, I.; Fuentes, J.D. Denuder measurements of gas and aerosol species above Arctic snow surfaces at Alert 2000. Atmos. Environ. 2002, 36, 5299–5309. [Google Scholar] [CrossRef]
- Dunlea, E.J.; Herndon, S.C.; Nelson, D.D.; Volkamer, R.M.; Martini, F.S.; Sheehy, P.M.; Zahniser, M.S.; Shorter, J.H.; Wormhoudt, J.C.; Lamb, B.K.; et al. Evaluation of nitrogen dioxide chemiluminescence monitors in a polluted urban environment. Atmos. Chem. Phys. 2007, 7, 2691–2704. [Google Scholar] [CrossRef]
- Salzano, R.; Pasini, A.; Ianniello, A.; Mazzola, M.; Traversi, R.; Udisti, R. High time-resolved radon progeny measurements in the Arctic region (Svalbard islands, Norway): Results and potentialities. Atmos. Chem. Phys. 2018, 18, 6959–6969. [Google Scholar] [CrossRef]
- Mazzola, M.; Viola, A.P.; Lanconelli, C.; Vitale, V. Atmospheric observations at the Amundsen-Nobile Climate Change Tower in Ny-Ålesund, Svalbard. Rend. Lincei 2016, 27, 7–18. [Google Scholar] [CrossRef]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Esau, I.; Repina, I. Wind Climate in Kongsfjorden, Svalbard, and Attribution of Leading Wind Driving Mechanisms through Turbulence-Resolving Simulations. Adv. Meteorol. 2012, 2012, 1–16. [Google Scholar] [CrossRef]
- Dekhtyareva, A.; Edvardsen, K.; Holmén, K.; Hermansen, O.; Hansson, H.-C.; Longhurst, J.W.S.; Brebbia, C.A.; Barnes, J. Influence of local and regional air pollution on atmospheric measurements in Ny-Ålesund. Int. J. Sustain. Dev. Plan. 2016, 11, 578–587. [Google Scholar] [CrossRef]
- Dekhtyareva, A.; Holmén, K.; Maturilli, M.; Hermansen, O.; Graversen, R. Effect of seasonal mesoscale and microscale meteorological conditions in Ny-Ålesund on results of monitoring of long-range transported pollution. Polar Res. 2018, 37, 1508196. [Google Scholar] [CrossRef]
- Maturilli, M.; Herber, A.; König-Langlo, G. Climatology and time series of surface meteorology in Ny-Ålesund, Svalbard. Earth Syst. Sci. Data 2013, 5, 155–163. [Google Scholar] [CrossRef]
- Koo, J.-H.; Wang, Y.; Kurosu, T.P.; Chance, K.; Rozanov, A.; Richter, A.; Oltmans, S.J.; Thompson, A.M.; Hair, J.W.; Fenn, M.A.; et al. Characteristics of tropospheric ozone depletion events in the Arctic spring: Analysis of the ARCTAS, ARCPAC, and ARCIONS measurements and satellite BrO observations. Atmos. Chem. Phys. 2012, 12, 9909–9922. [Google Scholar] [CrossRef]
- Barrie, L.A.; Bottenheim, J.W.; Schnell, R.; Crutzen, P.J.; Rasmussen, R.A. Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere. Nature 1988, 334, 138–141. [Google Scholar] [CrossRef]
- Tarasick, D.; Bottenheim, J.W. Surface ozone depletion episodes in the Arctic and Antarctic from historical ozonesonde records. Atmos. Chem. Phys. 2002, 2, 197–205. [Google Scholar] [CrossRef]
- Strong, C.; Fuentes, J.; Davis, R.; Bottenheim, J. Thermodynamic attributes of Arctic boundary layer ozone depletion. Atmos. Environ. 2002, 36, 2641–2652. [Google Scholar] [CrossRef]
- Morin, S.; Hönninger, G.; Staebler, R.M.; Bottenheim, J.W. A high time resolution study of boundary layer ozone chemistry and dynamics over the Arctic Ocean near Alert, Nunavut. Geophys. Res. Lett. 2005, 32, 08809. [Google Scholar] [CrossRef]
- Simpson, W.; von Glasow, R.; Riedel, K.; Anderson, P.; Ariya, P.; Bottenheim, J.; Burrows, J.P.; Carpenter, L.J.; Friess, U.; Goodsite, M.; et al. Halogens and their role in polar boundary-layer ozone depletion. Atmos. Chem. Phys. 2007, 7, 4375–4418. [Google Scholar] [CrossRef]
- Solberg, S.; Schmidbauer, N.; Semb, A.; Stordal, F. Boundary-layer ozone depletion as seen in the Norwegian Arctic in spring. J. Atmos. Chem. 1996, 23, 301–332. [Google Scholar] [CrossRef]
- Eneroth, K.; Holmén, K.; Berg, T.; Schmidbauer, N.; Solberg, S. Springtime depletion of tropospheric ozone, gaseous elemental mercury and non-methane hydrocarbons in the European Arctic, and its relation to atmospheric transport. Atmos. Environ. 2007, 41, 8511–8526. [Google Scholar] [CrossRef]
- Bottenheim, J.W.; Netcheva, S.; Morin, S.; Nghiem, S.V. Ozone in the boundary layer air over the Arctic Ocean: Measurements during the TARA transpolar drift 2006–2008. Atmos. Chem. Phys. 2009, 9, 4545–4557. [Google Scholar] [CrossRef]
- Thompson, C.R.; Shepson, P.B.; Liao, J.; Huey, L.G.; Apel, E.C.; Cantrell, C.A.; Flocke, F.; Orlando, J.; Fried, A.; Hall, S.R.; et al. Interactions of bromine, chlorine, and iodine photochemistry during ozone depletions in Barrow, Alaska. Atmos. Chem. Phys. 2015, 15, 9651–9679. [Google Scholar] [CrossRef]
- Evans, M.J.; Jacob, D.J.; Atlas, E.; Cantrell, C.A.; Eisele, F.; Flocke, F.; Fried, A.; Mauldin, R.L.; Ridley, B.A.; Wert, B.; et al. Coupled evolution of BrOx-ClOx-HOx-NOxchemistry during bromine-catalyzed ozone depletion events in the arctic boundary layer. J. Geophys. Res. 2003, 108, 8368. [Google Scholar] [CrossRef]
- Tas, E.; Peleg, M.; Pedersen, D.U.; Matveev, V.; Pour Biazar, A.; Luria, M. Measurement-based modeling of bromine chemistry in the boundary layer: 1. Bromine chemistry at the Dead Sea. Atmos. Chem. Phys. 2006, 6, 5589–5604. [Google Scholar] [CrossRef]
- Tas, E.; Peleg, M.; Pedersen, D.U.; Matveev, V.; Biazar, A.P.; Luria, M. Measurement-based modeling of bromine chemistry in the Dead Sea boundary layer–Part 2: The influence of NO2 on bromine chemistry at mid-latitude areas. Atmos. Chem. Phys. 2008, 8, 4811–4821. [Google Scholar] [CrossRef]
- Herrmann, M.; Sihler, H.; Frieß, U.; Wagner, T.; Platt, U.; Gutheil, E. Time-dependent 3D simulations of tropospheric ozone depletion events in the Arctic spring using the Weather Research and Forecasting model coupled with Chemistry (WRF-Chem). Atmos. Chem. Phys. 2021, 21, 7611–7638. [Google Scholar] [CrossRef]
- Wilson, T.R.S. Salinity and the Major Elements of Sea Water. In Chemical Oceanography, 2nd ed.; Riley, J.P., Skirrow, G., Eds.; Academic Press: London, UK, 1975; Volume 1, pp. 365–413. [Google Scholar]
- Vogt, R.; Crutzen, P.J.; Sander, R. A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer. Nature 1996, 383, 327–330. [Google Scholar] [CrossRef]
- Langendörfer, U.; Lehrer, E.; Wagenbach, D.; Platt, U. Observation of Filterable Bromine Variabilities During Arctic Tropospheric Ozone Depletion Events in High (1 h) Time Resolution. J. Atmos. Chem. 1999, 34, 39–54. [Google Scholar] [CrossRef]
- Sander, R.; Keene, W.C.; Pszenny, A.A.P.; Arimoto, R.; Ayers, G.P.; Baboukas, E.; Cainey, J.M.; Crutzen, P.J.; Duce, R.A.; Hönninger, G.; et al. Inorganic bromine in the marine boundary layer: A critical review. Atmos. Chem. Phys. 2003, 3, 1301–1336. [Google Scholar] [CrossRef]
- Krnavek, L.; Simpson, W.; Carlson, D.; Domine, F.; Douglas, T.A.; Sturm, M. The chemical composition of surface snow in the Arctic: Examining marine, terrestrial, and atmospheric influences. Atmos. Environ. 2012, 50, 349–359. [Google Scholar] [CrossRef]
- Lehrer, E.; Wagenbach, D.; Platt, U. Aerosol chemical composition during tropospheric ozone depletion at Ny Alesund/Svalbard. Tellus 1997, 49, 486–495. [Google Scholar] [CrossRef]
- Karl, M.; Leck, C.; Rad, F.M.; Bäcklund, A.; Lopez-Aparicio, S.; Heintzenberg, J. New insights in sources of the sub-micrometre aerosol at Mt. Zeppelin observatory (Spitsbergen) in the year 2015. Tellus B Chem. Phys. Meteorol. 2019, 71, 1–29. [Google Scholar] [CrossRef]
- Tuckermann, M.; Ackermann, R.; Gölz, C.; Lorenzen-Schmidt, H.; Senne, T.; Stutz, J.; Trost, B.; Unold, W.; Platt, U. DOAS-observation of halogen radical-catalysed arctic boundary layer ozone destruction during the ARCTOC-campaigns 1995 and 1996 in Ny-Ålesund, Spitsbergen. Tellus B Chem. Phys. Meteorol. 1997, 49, 533–555. [Google Scholar] [CrossRef]
- Martinez, M.; Arnold, T.; Perner, D. The role of bromine and chlorine chemistry for arctic ozone depletion events in Ny-Ålesund and comparison with model calculations. Ann. Geophys. 1999, 17, 941–956. [Google Scholar] [CrossRef]
- Legrand, M.; Yang, X.; Preunkert, S.; Theys, N. Year-round records of sea salt, gaseous, and particulate inorganic bromine in the atmospheric boundary layer at coastal (Dumont d’Urville) and central (Concordia) East Antarctic sites. J. Geophys. Res. Atmos. 2016, 121, 997–1023. [Google Scholar] [CrossRef]
- Mellouki, A.; Wallington, T.J.; Chen, J. Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate. Chem. Rev. 2015, 115, 3984–4014. [Google Scholar] [CrossRef]
- De Serves, C. Gas phase formaldehyde and peroxide measurements in the Arctic atmosphere. J. Geophys. Res. Space Phys. 1994, 99, 25391–25398. [Google Scholar] [CrossRef]
- Notholt, J.; Toon, G.; Stordal, F.; Solberg, S.; Schmidbauer, N.; Becker, E.; Meier, A.; Sen, B. Seasonal variations of atmospheric trace gases in the high Arctic at 79 N. J. Geophys. Res. 1997, 102, 12855–12861. [Google Scholar] [CrossRef]
- Hornbrook, R.S.; Hills, A.J.; Riemer, D.D.; Abdelhamid, A.; Flocke, F.M.; Hall, S.R.; Huey, L.G.; Knapp, D.J.; Liao, J.; Mauldin, R.L.; et al. Arctic springtime observations of volatile organic compounds during the OASIS-2009 campaign. J. Geophys. Res. Atmos. 2016, 121, 9789–9813. [Google Scholar] [CrossRef]
- Sumner, A.L.; Shepson, P.B. Snowpack production of formaldehyde and its effect on the Arctic troposphere. Nature 1999, 398, 230–233. [Google Scholar] [CrossRef]
- Sumner, A.; Shepson, P.; Grannas, A.; Bottenheim, J.; Anlauf, K.; Worthy, D.; Schroeder, W.; Steffen, A.; Dominé, F.; Perrier, S.; et al. Atmospheric chemistry of formaldehyde in the Arctic troposphere at Polar Sunrise, and the influence of the snowpack. Atmos. Environ. 2002, 36, 2553–2562. [Google Scholar] [CrossRef]
- Rudolph, J.; Fu, B.R.; Anlauf, K.; Bottenheim, J.; Thompson, A. Halogen atom concentrations in the Arctic Troposphere derived from hydrocarbon measurements: Impact on the budget of formaldehyde. Geophys. Res. Lett. 1999, 26, 2941–2944. [Google Scholar] [CrossRef]
- Riedel, K.; Allan, W.; Weller, R.; Schrems, O. Discrepancies between formaldehyde measurements and methane oxidation model predictions in the Antarctic troposphere: An assessment of other possible formaldehyde sources. J. Geophys. Res. 2005, 110, 15308. [Google Scholar] [CrossRef]
- Guimbaud, C.; Grannas, A.; Shepson, P.B.; Fuentes, J.D.; Boudries, H.; Bottenheim, J.W.; Dominé, F.; Houdier, S.; Perrier, S.; Biesenthal, T.B.; et al. Snowpack processing of acetaldehyde and acetone in the Arctic atmospheric boundary layer. Atmos. Environ. 2002, 36, 2743–2752. [Google Scholar] [CrossRef]
- Salzano, R.; Lanconelli, C.; Esposito, G.; Giusto, M.; Montagnoli, M.; Salvatori, R. On the Seasonality of the Snow Optical Behaviour at Ny Ålesund (Svalbard Islands, Norway). Geosciences 2021, 11, 112. [Google Scholar] [CrossRef]
- Markowicz, K.; Pakszys, P.; Ritter, C.; Zielinski, T.; Udisti, R.; Cappelletti, D.; Mazzola, M.; Shiobara, M.; Xian, P.; Zawadzka, O.; et al. Impact of North American intense fires on aerosol optical properties measured over the European Arctic in July 2015. J. Geophys. Res. Atmos. 2016, 121, 487. [Google Scholar] [CrossRef]
- Ritter, C.; Burgos, M.A.; Böckmann, C.; Mateos, D.; Lisok, J.; Markowicz, K.; Moroni, B.; Cappelletti, D.; Udisti, R.; Maturilli, M.; et al. Microphysical properties and radiative impact of an intense biomass burning aerosol event measured over Ny-Ålesund, Spitsbergen in July 2015. Tellus B Chem. Phys. Meteorol. 2018, 70, 1–23. [Google Scholar] [CrossRef]
- Moroni, B.; Cappelletti, D.; Crocchianti, S.; Becagli, S.; Caiazzo, L.; Traversi, R.; Udisti, R.; Mazzola, M.; Markowicz, K.; Ritter, C.; et al. Morphochemical characteristics and mixing state of long range transported wildfire particles at Ny-Ålesund (Svalbard Islands). Atmos. Environ. 2017, 156, 135–145. [Google Scholar] [CrossRef]
- Moroni, B.; Ritter, C.; Crocchianti, S.; Markowicz, K.; Mazzola, M.; Becagli, S.; Traversi, R.; Krejci, R.; Tunved, P.; Cappelletti, D. Individual Particle Characteristics, Optical Properties and Evolution of an Extreme Long-Range Transported Biomass Burning Event in the European Arctic (Ny-Ålesund, Svalbard Islands). J. Geophys. Res. Atmos. 2020, 125, e2019JD031535. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Heidt, L.E.; Krasnec, J.P.; Pollock, W.H.; Seiler, W. Biomass burning as a source of atmospheric gases CO, H2, N2O, NO, CH3Cl and COS. Nature 1979, 282, 253–256. [Google Scholar] [CrossRef]
- Arnold, S.R.; Emmons, L.K.; Monks, S.A.; Law, K.; Ridley, D.A.; Turquety, S.; Tilmes, S.; Thomas, J.L.; Bouarar, I.; Flemming, J.; et al. Biomass burning influence on high-latitude tropospheric ozone and reactive nitrogen in summer 2008: A multi-model analysis based on POLMIP simulations. Atmos. Chem. Phys. 2015, 15, 6047–6068. [Google Scholar] [CrossRef]
- Kramer, L.J.; Helmig, D.; Burkhart, J.F.; Stohl, A.; Oltmans, S.; Honrath, R.E. Seasonal variability of atmospheric nitrogen oxides and non-methane hydrocarbons at the GEOSummit station, Greenland. Atmos. Chem. Phys. 2015, 15, 6827–6849. [Google Scholar] [CrossRef]
- Parrington, M.; Palmer, P.I.; Lewis, A.; Lee, J.D.; Rickard, A.R.; Di Carlo, P.; Taylor, J.W.; Hopkins, J.R.; Punjabi, S.; Oram, D.E.; et al. Ozone photochemistry in boreal biomass burning plumes. Atmos. Chem. Phys. 2013, 13, 7321–7341. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All NO | Clean NO | All NO2 | Clean NO2 | O3 | |
|---|---|---|---|---|---|
| 2010 | ppt | ppt | Ppt | ppt | ppb |
| No. of cases | 7306 | 6015 | 7306 | 6015 | 19673 |
| Minimum | 3.3 | 3.3 | 7.0 | 7.0 | 4.7 |
| Maximum | 14,501 | 149.5 | 25,927 | 149.8 | 69.7 |
| Median | 12.6 | 12.2 | 26.0 | 24.7 | 42.1 |
| Mean | 33.5 | 15.2 | 109.9 | 28.6 | 39.0 |
| Standard Dev | 349.8 | 12.1 | 769.5 | 19.3 | 10.1 |
| 2011 | ppt | ppt | ppt | ppt | ppb |
| No. of cases | 5804 | 4791 | 5804 | 4791 | 15,230 |
| Minimum | 3.3 | 3.3 | 7.0 | 7.0 | 11.5 |
| Maximum | 13,887 | 287.6 | 78,000 | 365.2 | 61.6 |
| Median | 11.2 | 11.2 | 32.8 | 33.7 | 43.1 |
| Mean | 35.0 | 16.3 | 227.5 | 48.9 | 39.4 |
| Standard Dev | 276.1 | 23.6 | 1783.8 | 51.6 | 10.7 |
| 2015 | ppt | ppt | ppt | ppt | ppb |
| No. of cases | 205614 | 131749 | 205614 | 131749 | 181308 |
| Minimum | <d. l. 1 | <d. l. 1 | <d. l. 1 | <d. l. 1 | 1.1 |
| Maximum | 94200 | 1500 | 95600 | 3900 | 50.9 |
| Median | 50 | 50 | 100 | 100 | 24.9 |
| Mean | 100 | 60 | 300 | 200 | 25.6 |
| Standard Dev | 700 | 100 | 900 | 200 | 5.8 |
| HCl | HBr | |||||
|---|---|---|---|---|---|---|
| All periods | ppt | ppt | ppt | ppt | ppt | ppt |
| No. of cases | 24 | 24 | 24 | 24 | 24 | 24 |
| Minimum | <d. l. 1 | 2.6 | 38.7 | 10.4 | <d. l. 1 | 0.2 |
| Maximum | 41.2 | 12.5 | 408.2 | 68.5 | 2.5 | 3.1 |
| Median | 10.1 | 4.4 | 96.9 | 18.9 | 0.1 | 0.5 |
| Mean | 12.8 | 5.6 | 144.0 | 26.6 | 0.4 | 0.7 |
| Standard Dev | 9.3 | 2.8 | 102.7 | 16.5 | 0.6 | 0.6 |
| O3-depletion period | ppt | ppt | ppt | ppt | ppt | ppt |
| No. of cases | 5 | 5 | 5 | 5 | 5 | 5 |
| Minimum | 16.6 | 3.5 | 110.5 | 38.8 | 1.2 | 0.6 |
| Maximum | 41.2 | 12.5 | 408.2 | 68.5 | 2.5 | 3.1 |
| Median | 21.2 | 7.4 | 262.9 | 57.9 | 1.4 | 1.1 |
| Mean | 25.1 | 7.7 | 261.1 | 55.8 | 1.6 | 1.5 |
| Standard Dev | 11.2 | 3.8 | 121.6 | 12.7 | 0.6 | 1.1 |
| HCHO | CH3CHO | CH3CH2CHO | CH3COCH3 | |
|---|---|---|---|---|
| All periods | ppt | ppt | ppt | ppt |
| No. of cases | 56 | 54 | 31 | 56 |
| Minimum | 22.8 | 12.5 | <d. l. 1 | 2.5 |
| Maximum | 805.4 | 496.9 | 45.6 | 1039.0 |
| Median | 229.2 | 185.5 | 21.2 | 333.0 |
| Mean | 266.3 | 197.6 | 24.0 | 370.1 |
| Standard Dev | 150.1 | 118.5 | 10.5 | 198.4 |
| Ozone-depletion period | ppt | ppt | ppt | ppt |
| No. of cases | 5 | 5 | 5 | 5 |
| Minimum | 88.2 | 82.2 | 8.0 | 303.6 |
| Maximum | 328.9 | 251.1 | 34.8 | 1039.0 |
| Median | 182.6 | 156.7 | 12.2 | 463.5 |
| Mean | 206.1 | 162.6 | 18.6 | 534.1 |
| Standard Dev | 92.8 | 59.8 | 12.0 | 260.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ianniello, A.; Salzano, R.; Salvatori, R.; Esposito, G.; Spataro, F.; Montagnoli, M.; Mabilia, R.; Pasini, A. Nitrogen Oxides (NOx) in the Arctic Troposphere at Ny-Ålesund (Svalbard Islands): Effects of Anthropogenic Pollution Sources. Atmosphere 2021, 12, 901. https://doi.org/10.3390/atmos12070901
Ianniello A, Salzano R, Salvatori R, Esposito G, Spataro F, Montagnoli M, Mabilia R, Pasini A. Nitrogen Oxides (NOx) in the Arctic Troposphere at Ny-Ålesund (Svalbard Islands): Effects of Anthropogenic Pollution Sources. Atmosphere. 2021; 12(7):901. https://doi.org/10.3390/atmos12070901
Chicago/Turabian StyleIanniello, Antonietta, Roberto Salzano, Rosamaria Salvatori, Giulio Esposito, Francesca Spataro, Mauro Montagnoli, Rosanna Mabilia, and Antonello Pasini. 2021. "Nitrogen Oxides (NOx) in the Arctic Troposphere at Ny-Ålesund (Svalbard Islands): Effects of Anthropogenic Pollution Sources" Atmosphere 12, no. 7: 901. https://doi.org/10.3390/atmos12070901
APA StyleIanniello, A., Salzano, R., Salvatori, R., Esposito, G., Spataro, F., Montagnoli, M., Mabilia, R., & Pasini, A. (2021). Nitrogen Oxides (NOx) in the Arctic Troposphere at Ny-Ålesund (Svalbard Islands): Effects of Anthropogenic Pollution Sources. Atmosphere, 12(7), 901. https://doi.org/10.3390/atmos12070901