1. Introduction
The importance of halogens (X = Cl, Br, and I) as catalysts in the destruction of ozone in the atmosphere is well recognized [
1]. Halogen source gases are emitted at the surface both naturally and from human activities, and eventually can be photolyzed by sunlight or react chemically to convert their halogen content into inorganic forms. Ozone destruction takes place through a number of different chemical cycles in which halogen atoms are the key reactant in the ozone loss step.
ClO radicals are major contributors to stratospheric ozone loss in the Arctic and Antarctic poles [
1,
2]. During polar winter and spring, heterogeneous processes convert relatively inert chlorine reservoir species (ClONO
2 and HCl) into active, photolabile forms (Cl
2 and HOCl). These photolabile species are rapidly photolyzed in sunlight to produce Cl, which reacts with O
3 to form ClO. Subsequent reaction of ClO with another ClO molecule at cold temperatures to form ClOOCl initiates a catalytic reaction cycle [
3,
4], which repeatedly forms Cl in sunlight and subsequently destroys O
3 via the Cl + O
3 reaction. This is one of many catalytic ozone-loss cycles at work in the stratosphere, in which a single Cl atom can destroy numerous O
3 molecules [
1].
BrO radicals are formed in the atmospheric breakdown of bromine-containing compounds and are similarly involved in ozone destruction occurring in the polar and midlatitude stratosphere [
1,
2]. Ozone-destroying Br is often formed in catalytic cycles that involve interhalogen reactions. The BrO + ClO reaction cycle in particular produces both Br and Cl atoms to react with O
3. The reactivity of bromine on a per atom basis is approximately 70 times more destructive to stratospheric ozone than is chlorine [
1,
5,
6].
The iodine-catalyzed destruction of O
3 is through IO, formed directly in the reaction between I atoms and O
3. Recent work suggested that iodine plays a more significant role than previously believed in stratospheric ozone chemistry and, moreover, that iodine is approximately 400–1000 times more effective at destroying ozone than stratospheric chlorine in the lower stratosphere. In the future, the share of halogen-induced ozone loss in the stratosphere due to reactions of iodine will likely be greater than it is today [
7,
8].
Of significant recent interest in the literature, and providing additional motivation for this work, is the potential for co-injection of inorganic halogens along with sulfate into the stratosphere from explosive volcanic eruptions [
1,
9]. While once believed to be unimportant due to hydrometeor scavenging, it is now well established that halogens from volcanoes are transported to the stratosphere, with recent enhancements in chlorine [
10], bromine [
11], and iodine [
12] all being reported.
Gas-phase reactions involved in ozone loss cycles can have significant temperature dependences [
13]. Hence, kinetic studies must be carried out over a wide range of temperatures to fully describe the behavior of these processes. In particular, extremely cold temperatures are possible in the upper troposphere and lower stratosphere. Largely because of their importance in stratospheric chemistry, the kinetics of O
3 reactions with halogen atoms have been the subject of numerous experimental studies (e.g., Cl + O
3, Br + O
3, and I + O
3) [
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37]. On the theoretical side, however, the number of kinetics studies of O
3 with halogen atoms are relatively sparse [
38,
39,
40,
41,
42,
43,
44,
45,
46].
Farantos et al., 1978 [
38] have studied the classical dynamics of the O
3 reaction with Cl and derived the rate coefficients from 220–300 K. Tyrrell et al., 2001 [
40] have used B3 LYP/-311 + G* theories and found that the reaction between ozone and the Cl atom proceeds via an early transition state and a late transition state. Castillo et al., 2011 [
41] have studied the dynamics of O
3 + Cl by means of quasi-classical trajectory (QCT) calculations using the UQCISD/aug-cc-pVDZ level of theory over the temperature range of 200–400 K. None of these studies used dual level calculations, which are known for more accurate kinetic parameters [
47,
48], especially at low temperatures. Very few groups have studied the reaction mechanism of O
3 with Br using theoretical methods [
44,
45,
46], and theoretical rate coefficients have not been reported in the literature. While there are theoretical studies [
49,
50] of the reaction mechanism of O
3 with iodine oxides, there are no theoretical studies of the reaction mechanism and low temperature kinetics of O
3 with the I atom. To the best of our knowledge, the present study represents the first report of the theoretical temperature-dependent rate coefficients for the reactions of ozone with the bromine atom and the iodine atom.
The temperature ranges used to determine the recommended values in the most recent NASA/JPL [
13] compendium for the reactions of O
3 with Cl, Br, and I atoms are 205–300 K, 195–422 K, and 230–370 K, respectively. The IUPAC [
51]-recommended temperature ranges for the reactions of O
3 with Cl, Br, and I atoms are 180–300 K, 190–430 K, and 230–370 K, respectively. (Hereafter, NASA/JPL and IUPAC recommendations refer to [
13,
51], respectively, with superscript reference numbers not shown.) As the heterogeneous activation of inorganic chlorine primarily occurs when temperatures drop below ~195 K in the polar vortex, rate coefficients for the title reactions in the lower temperature range (especially < 200 K) are very important. The recommended temperature-dependent rate coefficients by both the NASA/JPL and IUPAC evaluations are limited, particularly for iodine. Both experimental and theoretical studies play important roles in improving our understanding of the chemical reactions that are occurring in the stratosphere, with theoretical calculations in particular providing valuable mechanistic insight. Our focus here is to improve the theoretical understanding of the title reactions and extend the temperature range beyond what is experimentally available, from 180 to 400 K for all three halogens with a primary focus on atmospherically relevant conditions.
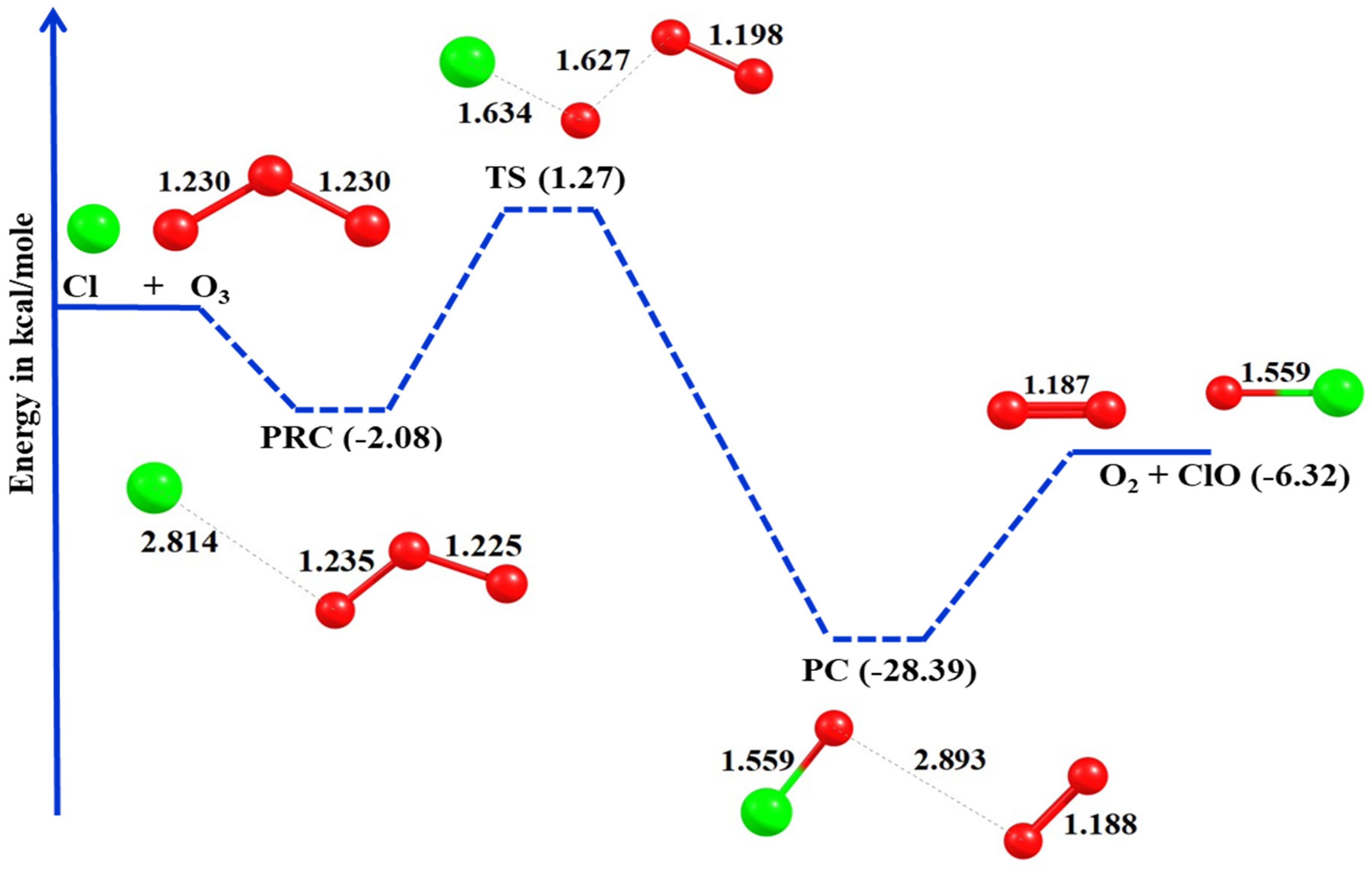
In the present investigation, the kinetics of O3 with halogen atom reactions have been studied using canonical variational transition state theory including small curvature tunneling corrections (CVT/SCT) over the temperature range of 180–400 K. To get more constrained and accurate kinetics parameters, dual level calculations were performed at the MRCI + Q/aug-ano-pVTZ//MP2/aug-cc-pV(T + d)Z and MRCI + Q/aug-ano-RCC-VTZP//MP2/aug-cc-pV(T + d)Z levels of theory with ZORA (zeroth-order regular approximation) using the ORCA program. Thermodynamic properties were studied at the CCSD(T)/aug-cc-pV(T + d)Z level of theory. Reactivity trends of O3 with all the halogens (F, Cl, Br, and I) and reaction pathways are discussed.
2. Computational Methods
The geometries and harmonic frequency calculations of reactants (halogens and O
3), pre-reactive complexes (PRCs), transition states (TSs), product complexes (PCs), and products were performed at the Møller−Plesset level of theory with second order perturbation (MP2) in combination with the augmented correlation consistent polarized triple-ζ (aug-cc-pV(T + d)Z) basis set [
52,
53]. Møller−Plesset perturbation theory assumes Hartee-Fock Hamiltonian as the zero-order perturbation. The combination of the MP2 method with the aug-cc-pV(T + d)Z basis set is extensively used for atmospheric reactions in the literature [
54,
55]. Transition states were identified with one imaginary frequency. Reactants, pre-reactive complexes, product complexes, and products were identified with zero imaginary frequencies. All the electronic structure calculations were performed using Gaussian 09 program suite [
56], and all the normal modes and structures were viewed in Gauss view [
57]. Intrinsic reaction coordinates (IRCs) calculations were carried out at the MP2/aug-cc-pV(T + d)Z level of theory for all the transition states to verify that the transition states are connected to the reactants and products. Thermodynamic properties were studied using Coupled-cluster with single, double, and triple excitation (CCSD(T)) with the aug-cc-pV(T + d)Z level of theory. To obtain more refrained and accurate energies, single point energy calculations were performed. Dual level calculations were carried out at the Multireference configuration interaction (MRCI) method. For better accuracy, the Davidson correction, core-valence correlation, and spin-orbit coupling effects are included (+Q). MRCI + Q in combination with aug-ano-pVTZ, aug-ano-RCC-VTZP, and ZORA (zeroth-order regular approximation) were used for refining energies [
58,
59]. These dual level calculations were performed using the ORCA program, which is one of the most versatile quantum chemistry packages available [
60,
61]. DFT, single-reference correlation, and multi-reference correlation methods can be executed using ORCA. The lowest spin-orbit states, A
1 for O
3 and
2P
3/2 for Cl, Br, and I atoms, were used throughout the calculations in this study.
3. Kinetics
The temperature-dependent rate coefficients for the reactions of halogen atoms with O
3 were calculated with CVT/SCT using the POLYRATE 2016A program and GAUSSRATE [
62,
63]. The POLYRATE program is a well-known software package for calculating kinetics parameters for gas-phase reactions; a detailed procedure was given in our previous articles [
64,
65]. To get the canonical variational transition state rate coefficient, the generalized rate coefficients can be minimized by varying the transition state dividing surface along the reaction coordinate using the following expressions:
Here, kCVT/SCT(T) is the tunneling corrected rate coefficient, which is obtained by multiplying kCVT and a temperature-dependent transmission coefficient CVT/SCT, σ is reaction path degeneracy, kB is Boltzmann’s constant, T is temperature in Kelvin, h is Planck’s constant. QGT is the canonical partition function of the generalized transition state at “s”, and “s” is a reaction coordinate parameter that determines the location of the generalized transition dividing surface. is the partition function of the reactant. VMEP(s) is the potential along the reaction path at “s”, and the minimum energy pathway (MEP) was constructed with a gradient step size of 0.01 Å. The canonical variational transition state is located by maximizing the free energy of activation with respect to “s”. The minimum energy pathway is obtained using direct dynamics for a small range of the reaction path with the mass scaled reaction coordinate “s” from –1.0 to 1.0 Å by using the Page-McIver integrator with a step size of 0.01 Å.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}