

Potential Strong Inhibition on Ozone Production Sensitivity by Particle Uptake


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
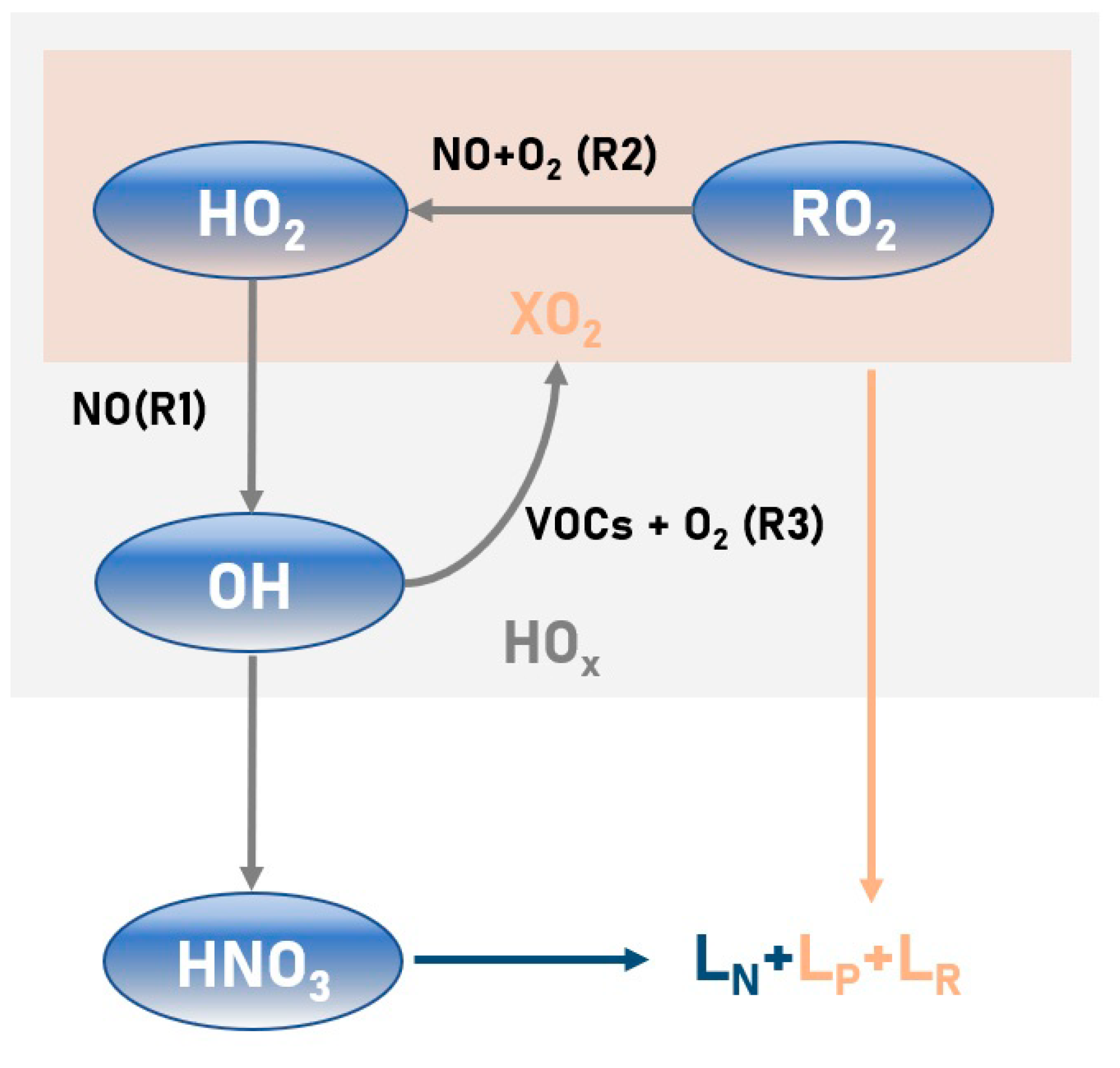
2.1. Ozone Sensitivity Analysis
2.2. Measurements
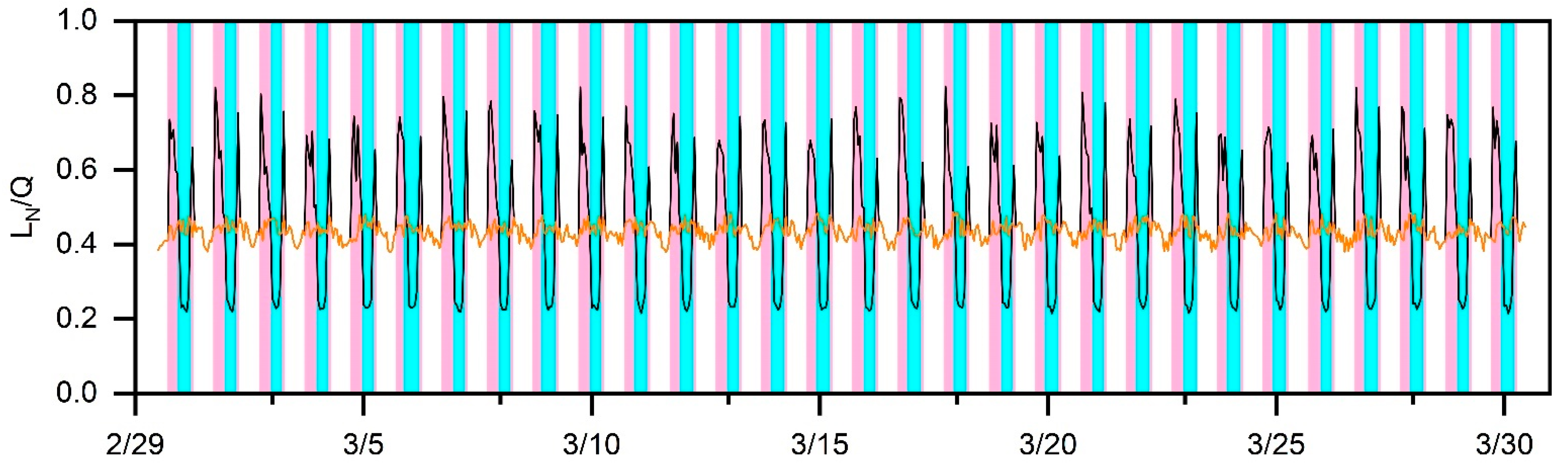
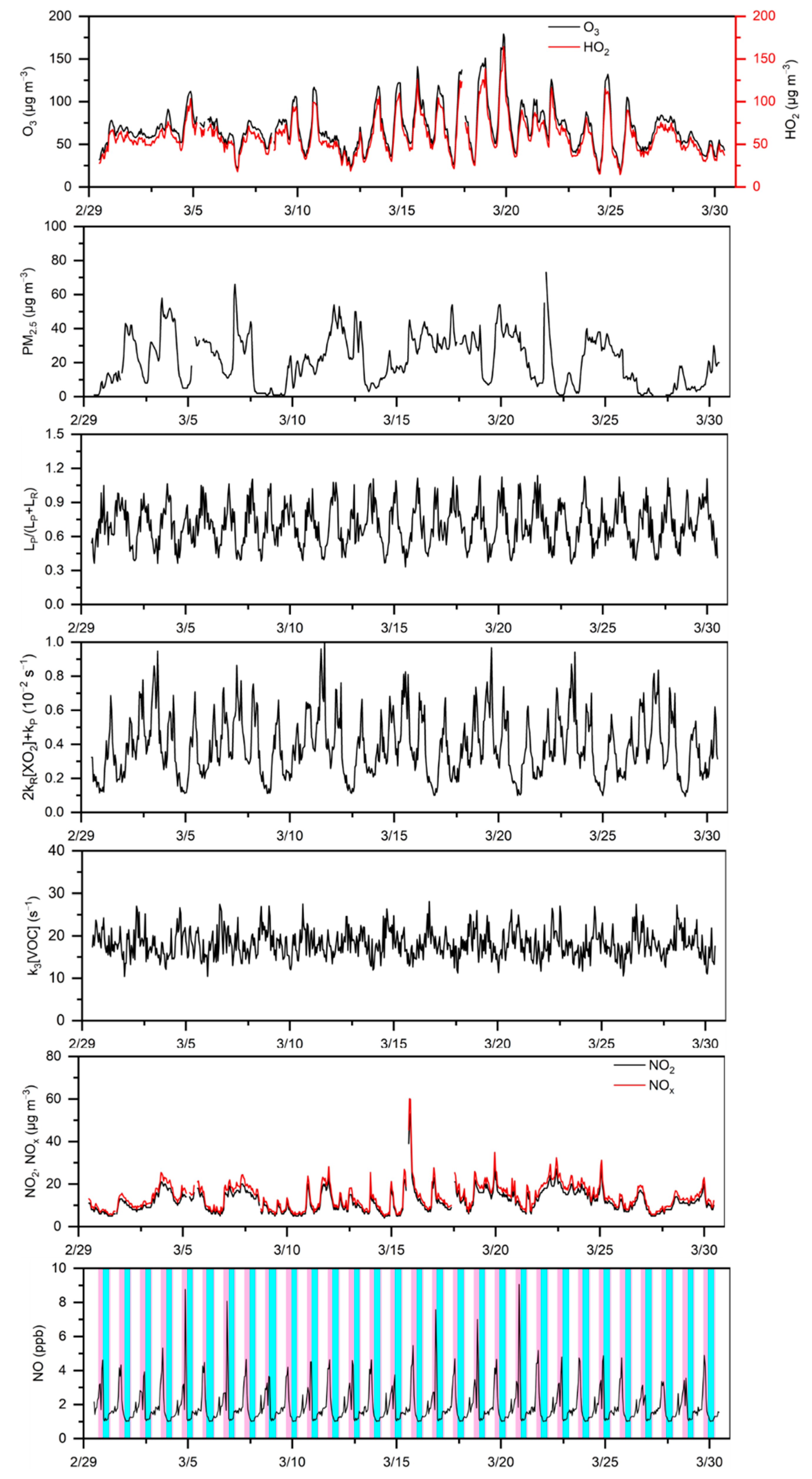
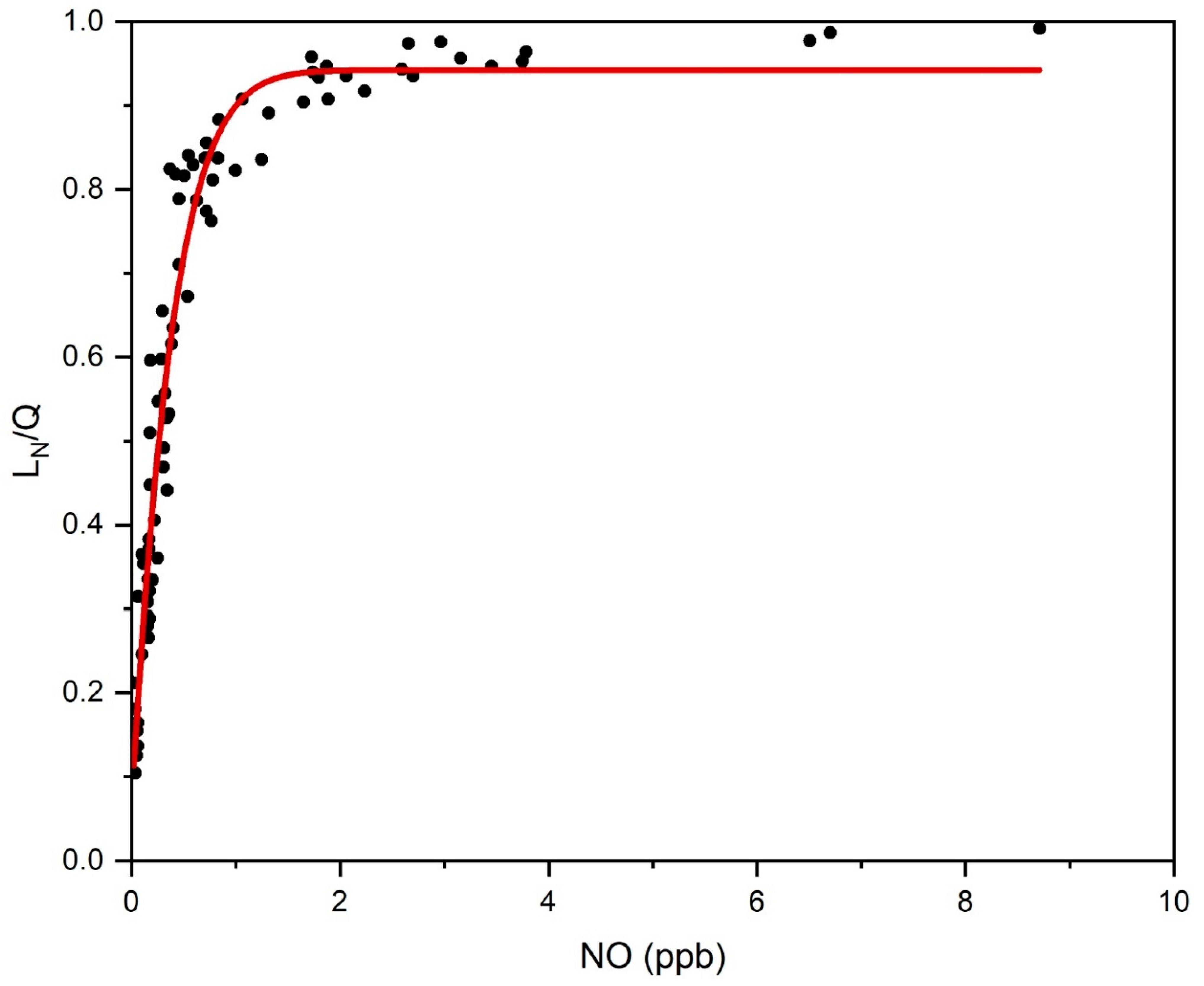
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monks, P.S.; Archibald, A.T.; Colette, A.; Cooper, O.; Coyle, M.; Derwent, R.; Fowler, D.; Granier, C.; Law, K.S.; Mills, G.E.; et al. Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer. Atmos. Chem. Phys. 2015, 15, 8889–8973. [Google Scholar] [CrossRef]
- Mills, G.; Buse, A.; Gimeno, B.; Bermejo, V.; Holland, M.; Emberson, L.; Pleijel, H. A synthesis of AOT40-based response functions and critical levels of ozone for agricultural and horticultural crops. Atmos. Environ. 2007, 41, 2630–2643. [Google Scholar] [CrossRef]
- Novak, K.; Skelly, J.M.; Schaub, M.; Kräuchi, N.; Hug, C.; Landolt, W.; Bleuler, P. Ozone air pollution and foliar injury development on native plants of Switzerland. Environ. Pollut. 2003, 125, 41–52. [Google Scholar] [CrossRef]
- Lefohn, A.S.; Malley, C.S.; Smith, L.; Wells, B.; Hazucha, M.; Simon, H.; Naik, V.; Mills, G.; Schultz, M.G.; Paoletti, E. Tropospheric ozone assessment report: Global ozone metrics for climate change, human health, and crop/ecosystem research. Elem. Sci. Anthr. 2018, 6, 27. [Google Scholar] [CrossRef]
- Sun, G.E.; McLaughlin, S.B.; Porter, J.H.; Uddling, J.; Mulholland, P.J.; Adams, M.B.; Pederson, N. Interactive influences of ozone and climate on streamflow of forested watersheds. Glob. Change Biol. 2012, 18, 3395–3409. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Kleinman, L.I.; Daum, P.H.; Lee, J.H.; Lee, Y.-N.; Nunnermacker, L.J.; Springston, S.R.; Newman, L.; Weinstein-Lloyd, J.; Sillman, S. Dependence of ozone production on NO and hydrocarbons in the troposphere. Geophys. Res. Lett. 1997, 24, 2299–2302. [Google Scholar] [CrossRef]
- Kleinman, L.I.; Daum, P.H.; Lee, Y.-N.; Nunnermacker, L.J.; Springston, S.R.; Weinstein-Lloyd, J.; Rudolph, J. Sensitivity of ozone production rate to ozone precursors. Geophys. Res. Lett. 2001, 28, 2903–2906. [Google Scholar] [CrossRef]
- Kleinman, L.I. The dependence of tropospheric ozone production rate on ozone precursors. Atmos. Environ. 2005, 39, 575–586. [Google Scholar] [CrossRef]
- Kleinman, L.I. Ozone process insights from field experiments—Part II: Observation-based analysis for ozone production. Atmos. Environ. 2000, 34, 2023–2033. [Google Scholar] [CrossRef]
- Taketani, F.; Kanaya, Y.; Pochanart, P.; Liu, Y.; Li, J.; Okuzawa, K.; Kawamura, K.; Wang, Z.; Akimoto, H. Measurement of overall uptake coefficients for HO2 radicals by aerosol particles sampled from ambient air at Mts. Tai and Mang (China). Atmos. Chem. Phys. 2012, 12, 11907–11916. [Google Scholar] [CrossRef] [Green Version]
- George, I.; Matthews, P.; Whalley, L.; Brooks, B.; Goddard, A.; Baeza-Romero, M.; Heard, D. Measurements of uptake coefficients for heterogeneous loss of HO2 onto submicron inorganic salt aerosols. Phys. Chem. Chem. Phys. 2013, 15, 12829–12845. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Song, H.; Tang, M.; Lu, K. Measurements of HO2 uptake coefficient on aqueous (NH4)2SO4 aerosol using aerosol flow tube with LIF system. Chin. Chem. Lett. 2019, 30, 2236–2240. [Google Scholar] [CrossRef]
- Ammann, M.; Cox, R.A.; Crowley, J.N.; Jenkin, M.E.; Mellouki, A.; Rossi, M.J.; Troe, J.; Wallington, T.J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI—Heterogeneous reactions with liquid substrates. Atmos. Chem. Phys. 2013, 13, 8045–8228. [Google Scholar] [CrossRef]
- Kanaya, Y.; Fukuda, M.; Akimoto, H.; Takegawa, N.; Komazaki, Y.; Yokouchi, Y.; Koike, M.; Kondo, Y. Urban photochemistry in central Tokyo: Rates and regimes of oxidant (O3+ NO2) production. J. Geophys. Res. 2008, 113, 8671. [Google Scholar] [CrossRef]
- Xie, M.; Zhu, K.; Wang, T.; Chen, P.; Han, Y.; Li, S.; Zhuang, B.; Shu, L. Temporal characterization and regional contribution to O3 and NOx at an urban and a suburban site in Nanjing, China. Sci. Total Environ. 2016, 551–552, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Krechmer, J.E.; Shutter, J.D.; Barber, V.P.; Li, Y.; Helstrom, E.; Franco, L.J.; Cox, J.L.; Hrdina, A.I.H.; Goss, M.B.; et al. Real-Time Laboratory Measurements of VOC Emissions, Removal Rates, and Byproduct Formation from Consumer-Grade Oxidation-Based Air Cleaners. Environ. Sci. Technol. Lett. 2021, 8, 1020–1025. [Google Scholar] [CrossRef]
- Kaser, L.; Peron, A.; Graus, M.; Striednig, M.; Wohlfahrt, G.; Juráň, S.; Karl, T. Interannual variability of terpenoid emissions in an alpine city. Atmos. Chem. Phys. 2022, 22, 5603–5618. [Google Scholar] [CrossRef]
- Hammer, M.U. Findings on H2O2/HNO3 as an indicator of ozone sensitivity in Baden-Württemberg, Berlin-Brandenburg, and the Po valley based on numerical simulations. J. Geophys. Res. 2002, 107, 211. [Google Scholar]
- Peng, Y.P.; Chen, K.S.; Lai, C.H.; Lu, P.J.; Kao, J.H. Concentrations of H2O2 and HNO3 and O3–VOC–NOx sensitivity in ambient air in southern Taiwan. Atmos. Environ. 2006, 40, 6741–6751. [Google Scholar] [CrossRef]
- Sillman, S.; He, D.; Pippin, M.R.; Daum, P.H.; Imre, D.G.; Kleinman, L.I.; Lee, J.H.; Weinstein-Lloyd, J. Model correlations for ozone, reactive nitrogen, and peroxides for Nashville in comparison with measurements: Implications for O3-NOx-hydrocarbon chemistry. J. Geophys. Res. Atmos. 1998, 103, 22629–22644. [Google Scholar] [CrossRef]
- Tonnesen, G.S.; Dennis, R.L. Analysis of radical propagation efficiency to assess ozone sensitivity to hydrocarbons and NOx: Local indicators of instantaneous odd oxygen production sensitivity. J. Geophys. Res. Atmos. 2000, 105, 9213–9225. [Google Scholar] [CrossRef]
- Sillman, S.; He, D.; Cardelino, C.; Imhoff, R.E. The Use of Photochemical Indicators to Evaluate Ozone-NOx-Hydrocarbon Sensitivity: Case Studies from Atlanta, New York, and Los Angeles. J. Air Waste Manag. Assoc. 1997, 47, 1030–1040. [Google Scholar] [CrossRef]
- Kanaya, Y.; Pochanart, P.; Liu, Y.; Li, J.; Tanimoto, H.; Kato, S.; Suthawaree, J.; Inomata, S.; Taketani, F.; Okuzawa, K. Rates and regimes of photochemical ozone production over Central East China in June 2006: A box model analysis using comprehensive measurements of ozone precursors. Atmos. Chem. Phys. 2009, 9, 7711–7723. [Google Scholar] [CrossRef]
- Sillman, S. The relation between ozone, NOx and hydrocarbons in urban and polluted rural environments. Atmos. Environ. 1999, 33, 1821–1845. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Sadanaga, Y.; Li, J.; Matsuoka, K.; Takemura, M.; Fujii, T.; Nakagawa, M.; Kohno, N.; Nakashima, Y.; Sato, K.; et al. Relative and Absolute Sensitivity Analysis on Ozone Production in Tsukuba, a City in Japan. Environ. Sci. Technol. 2019, 53, 13629–13635. [Google Scholar] [CrossRef]
- Pöschl, U.; Rudich, Y.; Ammann, M. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—Part 1: General equations, parameters, and terminology. Atmos. Chem. Phys. 2007, 7, 5989–6023. [Google Scholar] [CrossRef]
- Fuchs, N.; Sutugin, A.G. High-dispersed aerosols. In Topics in Current Aerosol Research; Elsevier: Amsterdam, The Netherlands, 1971; p. 1. [Google Scholar]
- Sadanaga, Y. The importance of NO2 and volatile organic compounds in the urban air from the viewpoint of the OH reactivity. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Sadanaga, Y.; Yoshino, A.; Watanabe, K.; Yoshioka, A.; Wakazono, Y.; Kanaya, Y.; Kajii, Y. Development of a measurement system of OH reactivity in the atmosphere by using a laser-induced pump and probe technique. Rev. Sci. Instrum. 2004, 75, 2648–2655. [Google Scholar] [CrossRef]
- Sadanaga, Y.; Yoshino, A.; Kato, S.; Kajii, Y. Measurements of OH reactivity and photochemical ozone production in the urban atmosphere. Environ. Sci. Technol. 2005, 39, 8847–8852. [Google Scholar] [CrossRef] [PubMed]
- Griffith, S.M.; Hansen, R.F.; Dusanter, S.; Michoud, V.; Gilman, J.B.; Kuster, W.C.; Veres, P.R.; Graus, M.; Gouw, J.A.; Roberts, J.; et al. Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets. J. Geophys. Res. Atmos. 2016, 121, 4211–4232. [Google Scholar] [CrossRef]
- Lou, S.; Holland, F.; Rohrer, F.; Lu, K.; Bohn, B.; Brauers, T.; Chang, C.C.; Fuchs, H.; Häseler, R.; Kita, K.; et al. Atmospheric OH reactivities in the Pearl River Delta–China in summer 2006: Measurement and model results. Atmos. Chem. Phys. 2010, 10, 11243–11260. [Google Scholar] [CrossRef] [Green Version]
- Stadtler, S.; Simpson, D.; Schröder, S.; Taraborrelli, D.; Bott, A.; Schultz, M. Ozone impacts of gas–aerosol uptake in global chemistry transport models. Atmos. Chem. Phys. 2018, 18, 3147–3171. [Google Scholar] [CrossRef]
- Yoshino, A.; Sadanaga, Y.; Watanabe, K.; Kato, S.; Miyakawa, Y.; Matsumoto, J.; Kajii, Y. Measurement of total OH reactivity by laser-induced pump and probe technique—comprehensive observations in the urban atmosphere of Tokyo. Atmos. Environ. 2006, 40, 7869–7881. [Google Scholar] [CrossRef]
- Mazzuca, G.M.; Ren, X.; Loughner, C.P.; Estes, M.; Crawford, J.H.; Pickering, K.E.; Weinheimer, A.J.; Dickerson, R.R. Ozone production and its sensitivity to NO2 and VOCs: Results from the DISCOVER-AQ field experiment, Houston. Atmos. Chem. Phys. 2016, 16, 14463–14474. [Google Scholar] [CrossRef]
- Burkholder, J.; Sander, S.; Abbatt, J.; Barker, J.; Cappa, C.; Crounse, J.; Dibble, T.; Huie, R.; Kolb, C.; Kurylo, M. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies; Evaluation Number 19; Jet Propulsion Laboratory, National Aeronautics and Space: Pasadena, CA, USA, 2020. [Google Scholar]
- Atkinson, R.; Baulch, D.; Cox, R.; Hampson, R., Jr.; Kerr, J.; Rossi, M.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry, organic species: Supplement VII. J. Phys. Chem. Ref. Data 1999, 28, 191–393. [Google Scholar] [CrossRef]
- Kanno, N.; Tonokura, K.; Koshi, M. Equilibrium constant of the HO2-H2O complex formation and kinetics of HO2 + HO2-H2O: Implications for tropospheric chemistry. J. Geophys. Res. Atmos. 2006, 111, D20. [Google Scholar] [CrossRef]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.; Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef]
- Sadanaga, Y.; Kondo, S.; Hashimoto, K.; Kajii, Y. Measurement of the rate coefficient for the OH+ NO2 reaction under the atmospheric pressure: Its humidity dependence. Chem. Phys. Lett. 2006, 419, 474–478. [Google Scholar] [CrossRef]
- Kanaya, Y.; Cao, R.; Akimoto, H.; Fukuda, M.; Komazaki, Y.; Yokouchi, Y.; Koike, M.; Tanimoto, H.; Takegawa, N.; Kondo, Y. Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004. J. Geophys. Res. Atmos. 2007, 112, D21. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Wang, L.; Fang, L.; Chen, S.; Zhou, X.; Ma, J.; Pan, Y.; Li, P. Potential Strong Inhibition on Ozone Production Sensitivity by Particle Uptake. Atmosphere 2022, 13, 1558. https://doi.org/10.3390/atmos13101558
Cheng X, Wang L, Fang L, Chen S, Zhou X, Ma J, Pan Y, Li P. Potential Strong Inhibition on Ozone Production Sensitivity by Particle Uptake. Atmosphere. 2022; 13(10):1558. https://doi.org/10.3390/atmos13101558
Chicago/Turabian StyleCheng, Xinliang, Liqiang Wang, Lijuan Fang, Shiyan Chen, Xin Zhou, Jingjun Ma, Yuqing Pan, and Pengfei Li. 2022. "Potential Strong Inhibition on Ozone Production Sensitivity by Particle Uptake" Atmosphere 13, no. 10: 1558. https://doi.org/10.3390/atmos13101558
APA StyleCheng, X., Wang, L., Fang, L., Chen, S., Zhou, X., Ma, J., Pan, Y., & Li, P. (2022). Potential Strong Inhibition on Ozone Production Sensitivity by Particle Uptake. Atmosphere, 13(10), 1558. https://doi.org/10.3390/atmos13101558