
Sr1-xKxFeO3 Perovskite Catalysts with Enhanced RWGS Reactivity for CO2 Hydrogenation to Light Olefins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Perovskite Preparation
2.2. Catalyst Characterization
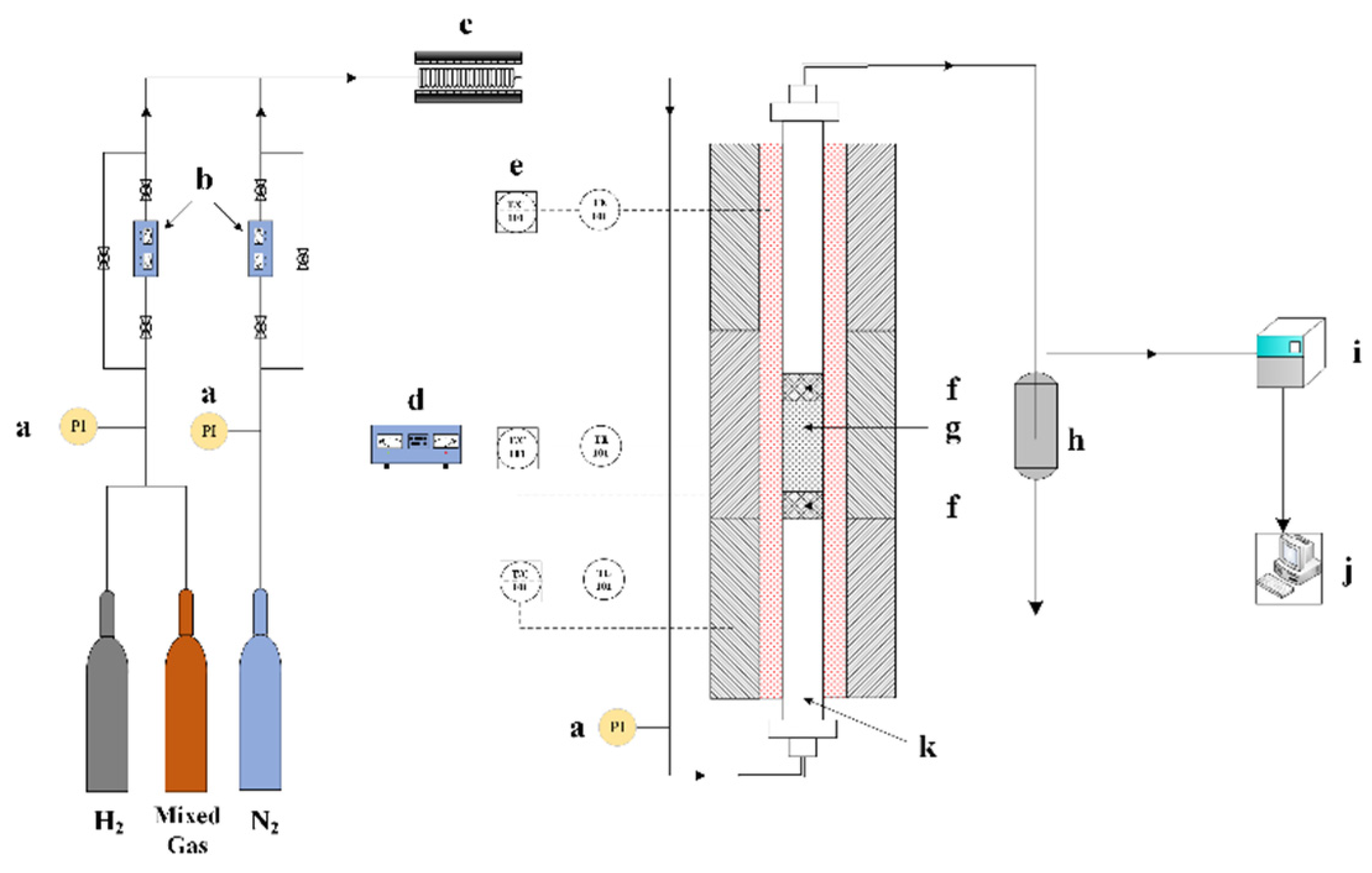
2.3. Catalytic Performance Testing
3. Results and Discussion
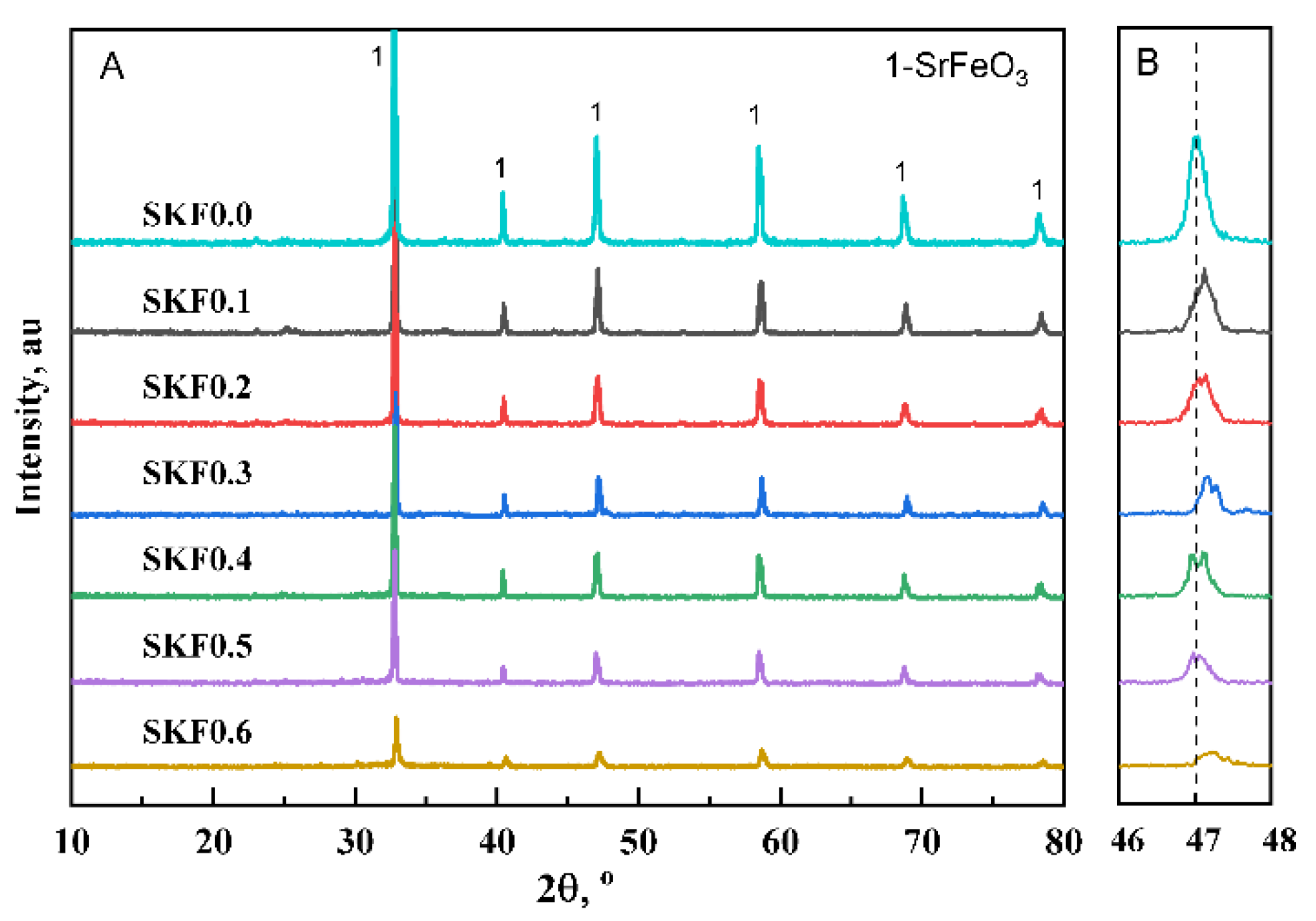
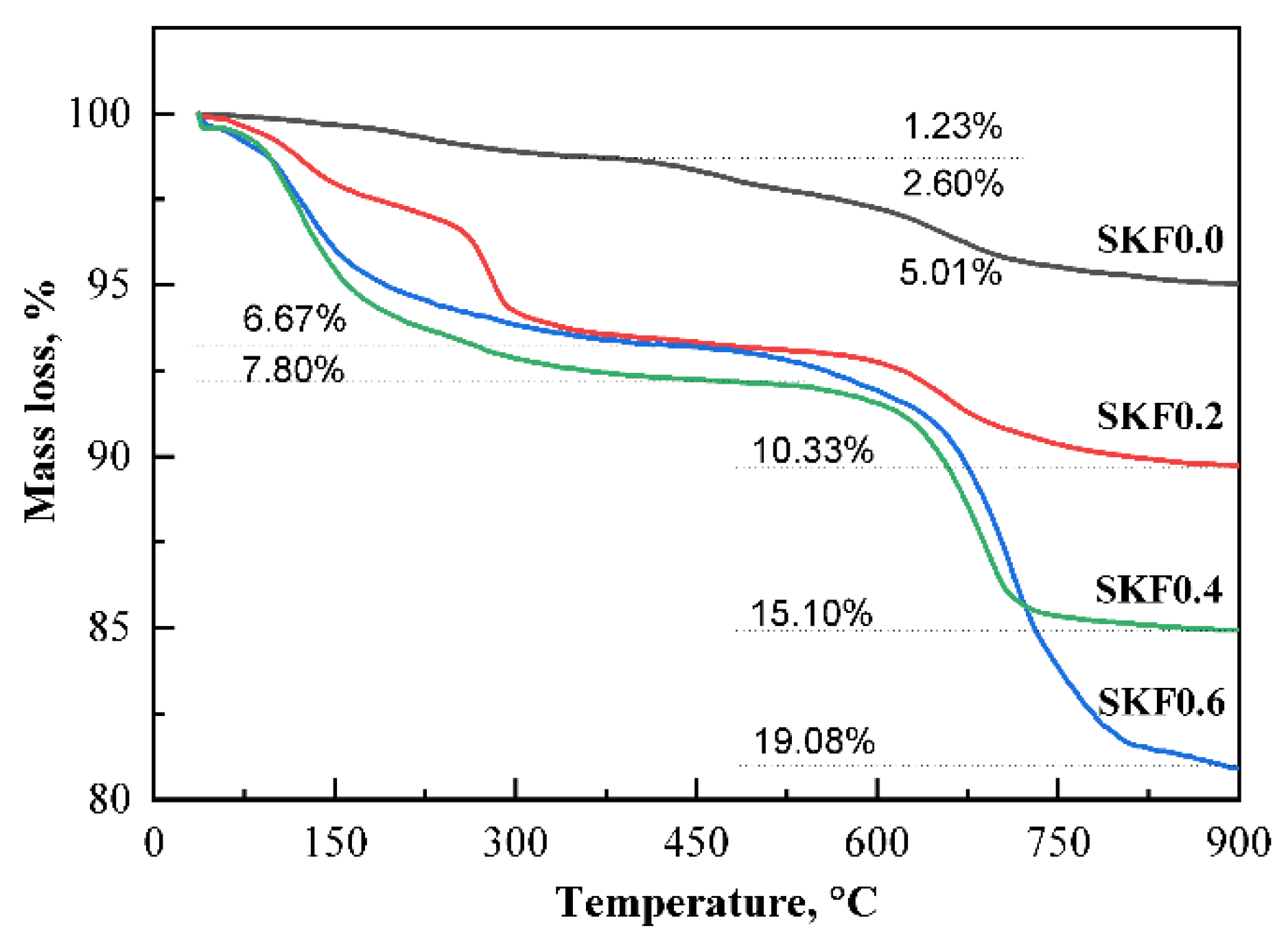
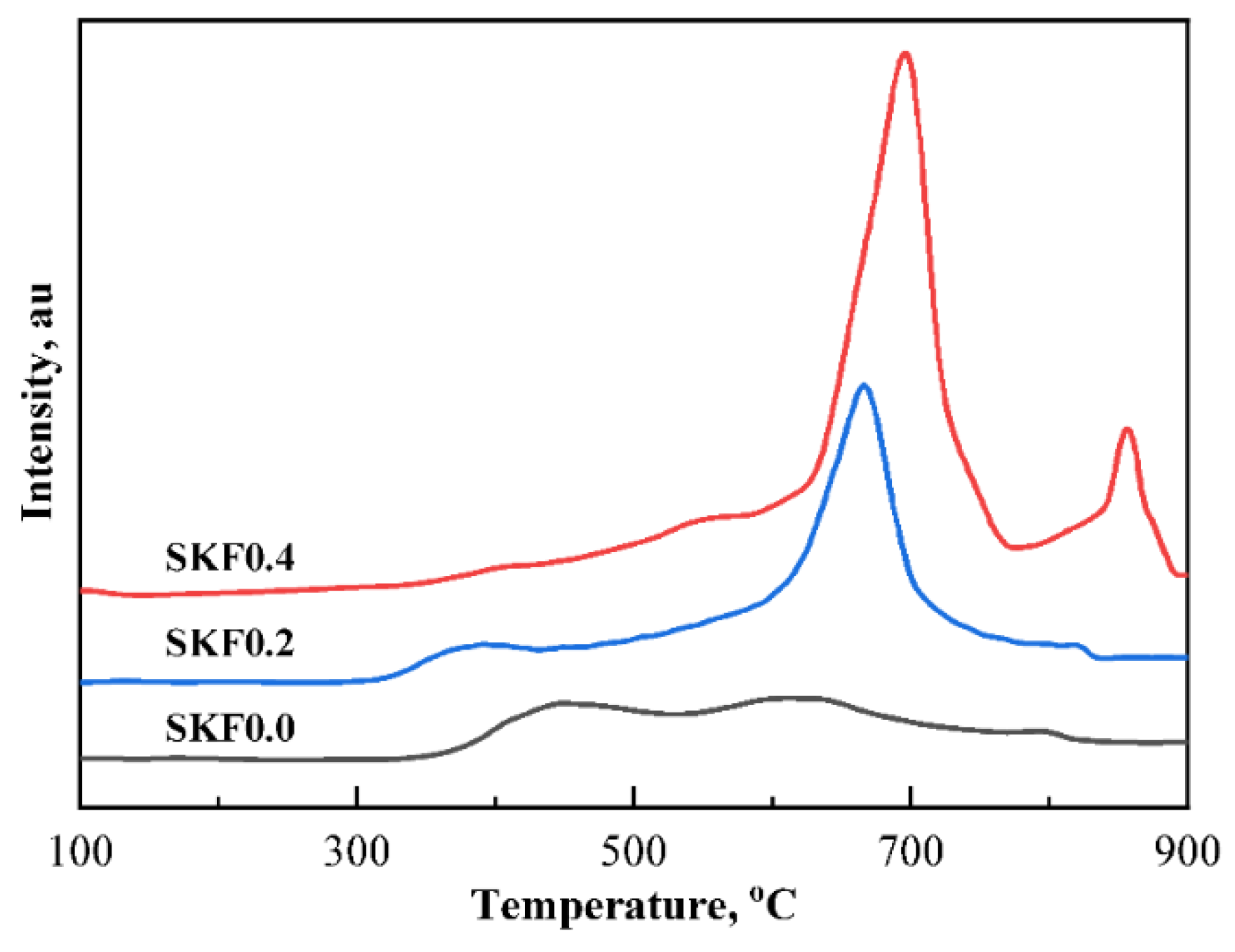
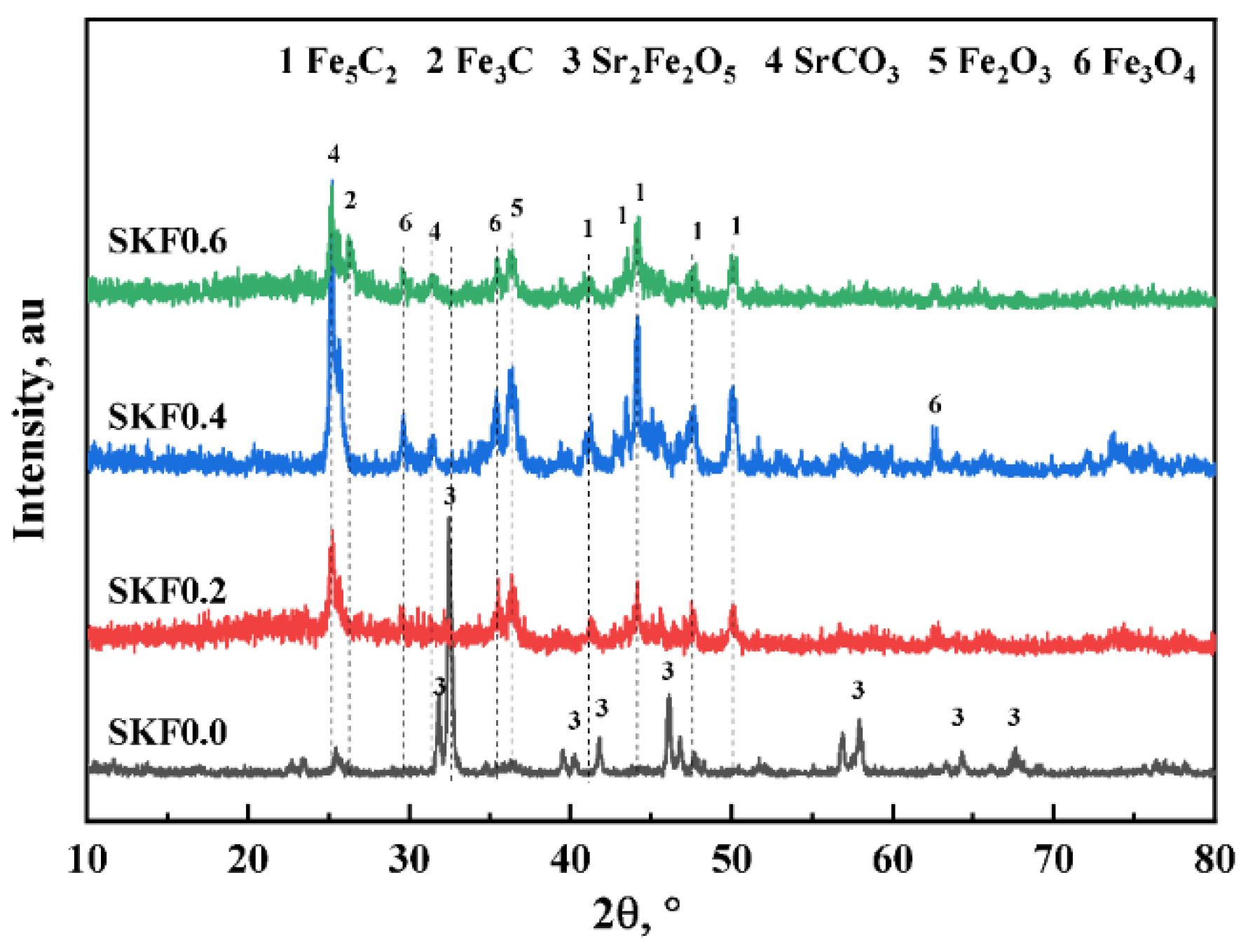
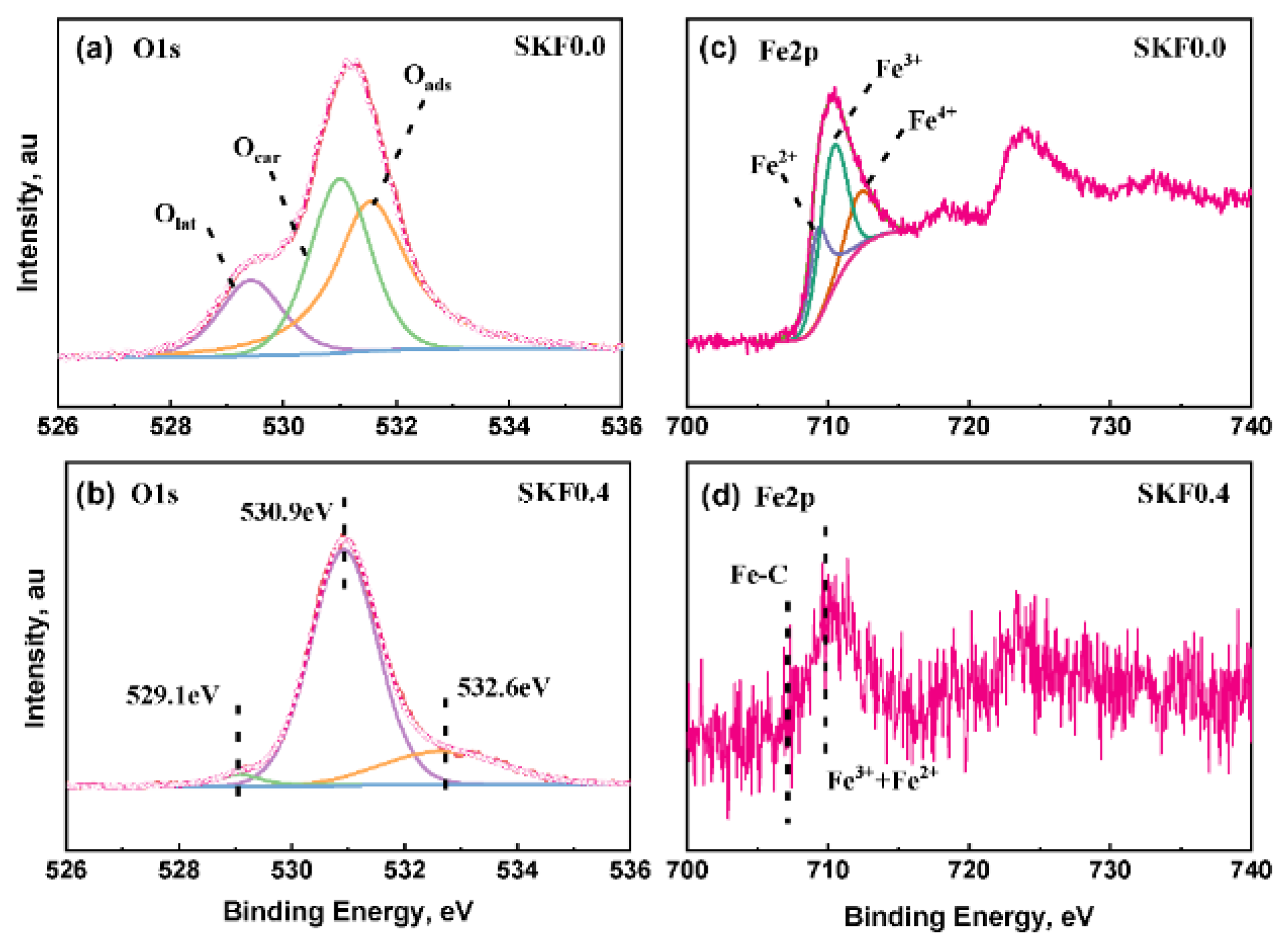
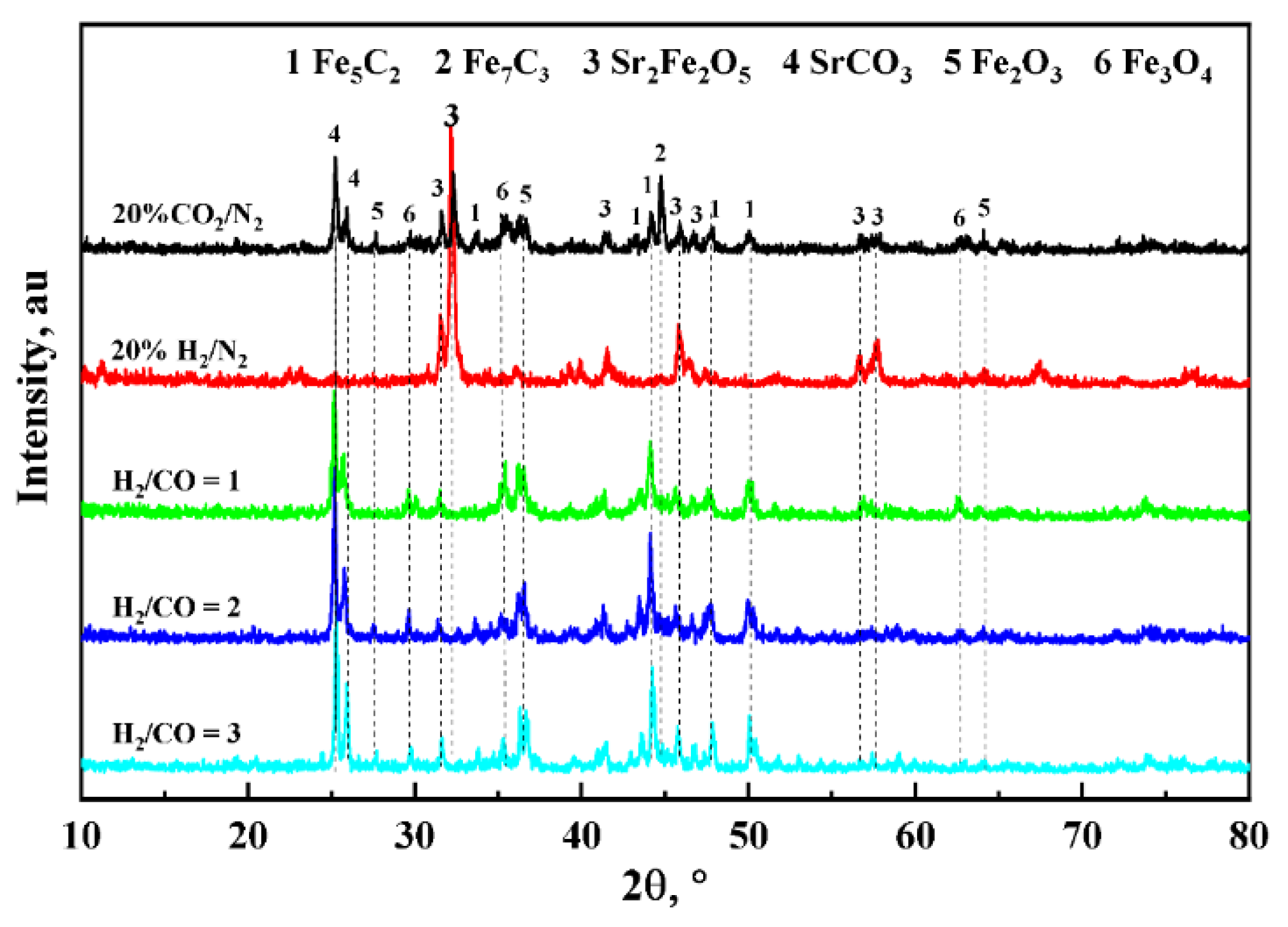
3.1. Characterization of As-Prepared Catalysts
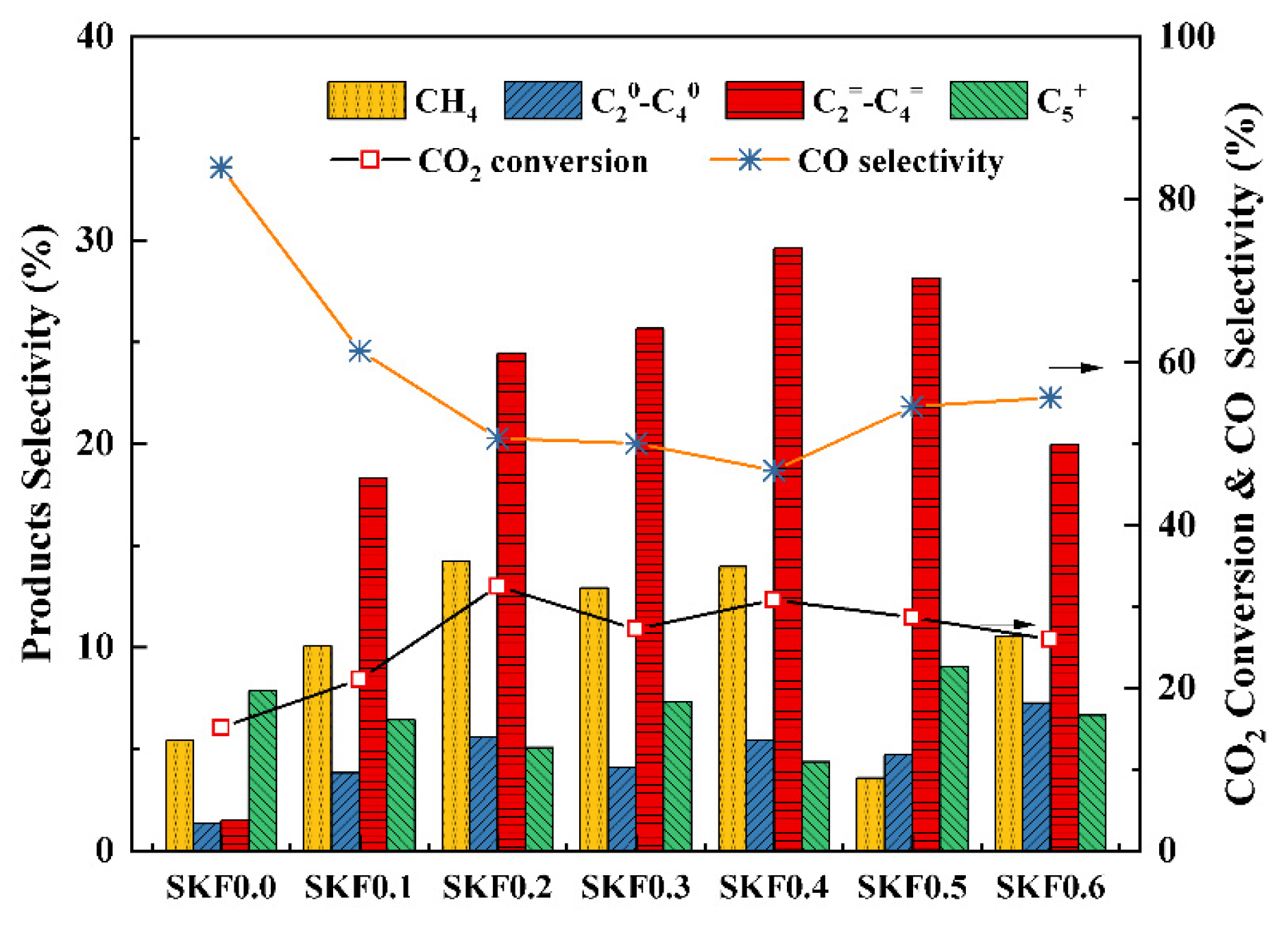
3.2. Catalytic Performance of SKFx Catalysts
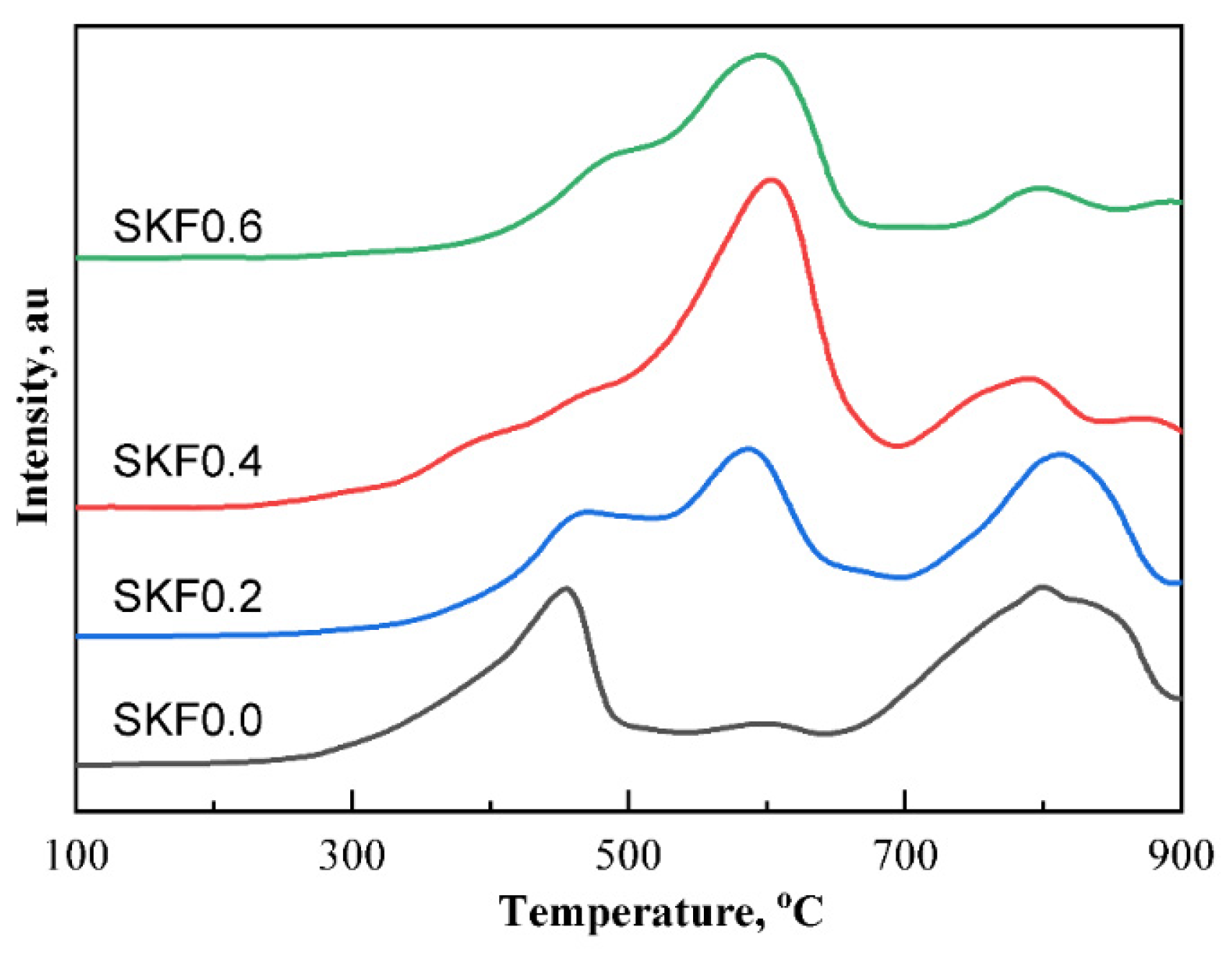
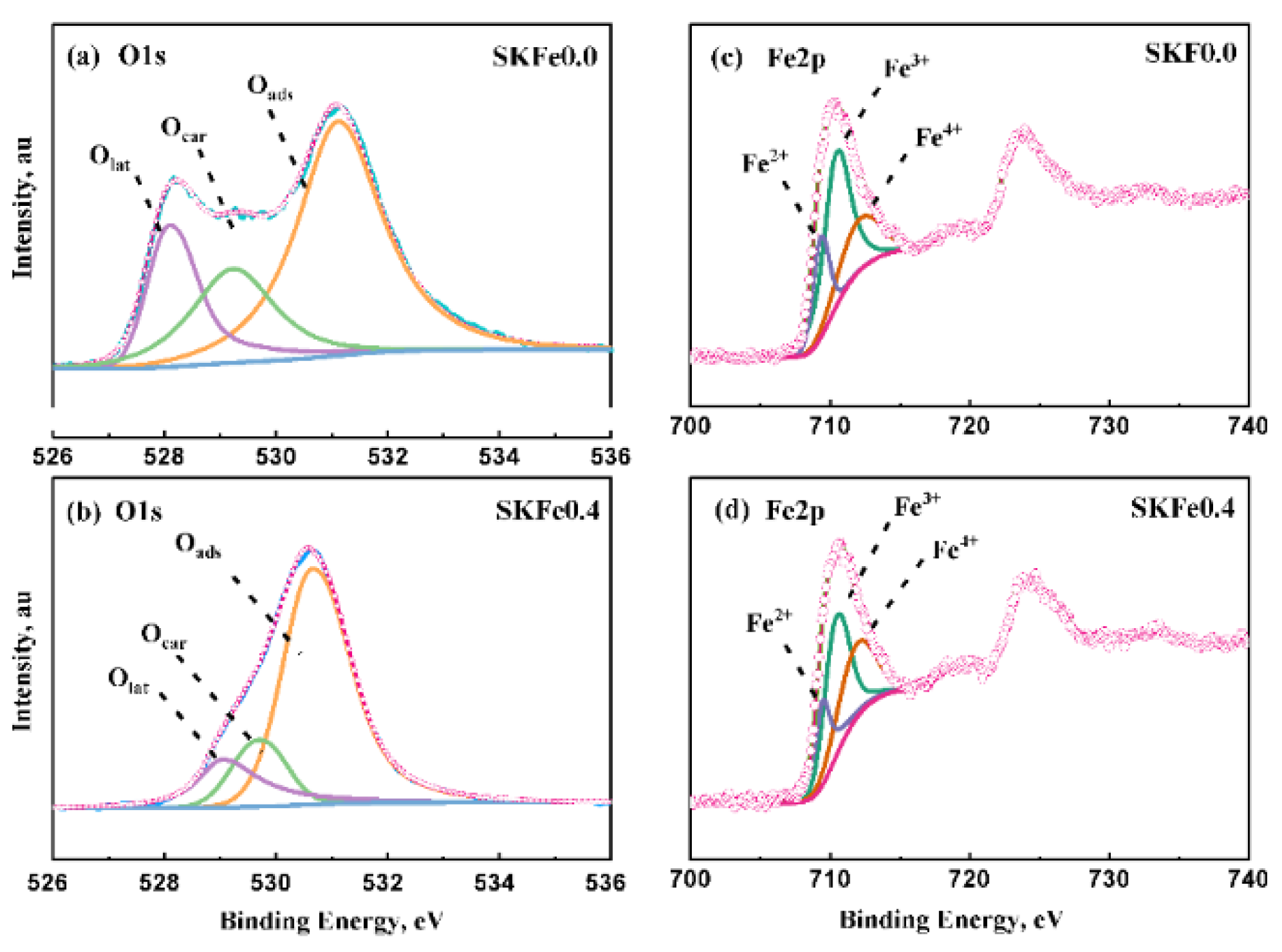
3.3. Redox Performance of Catalysts
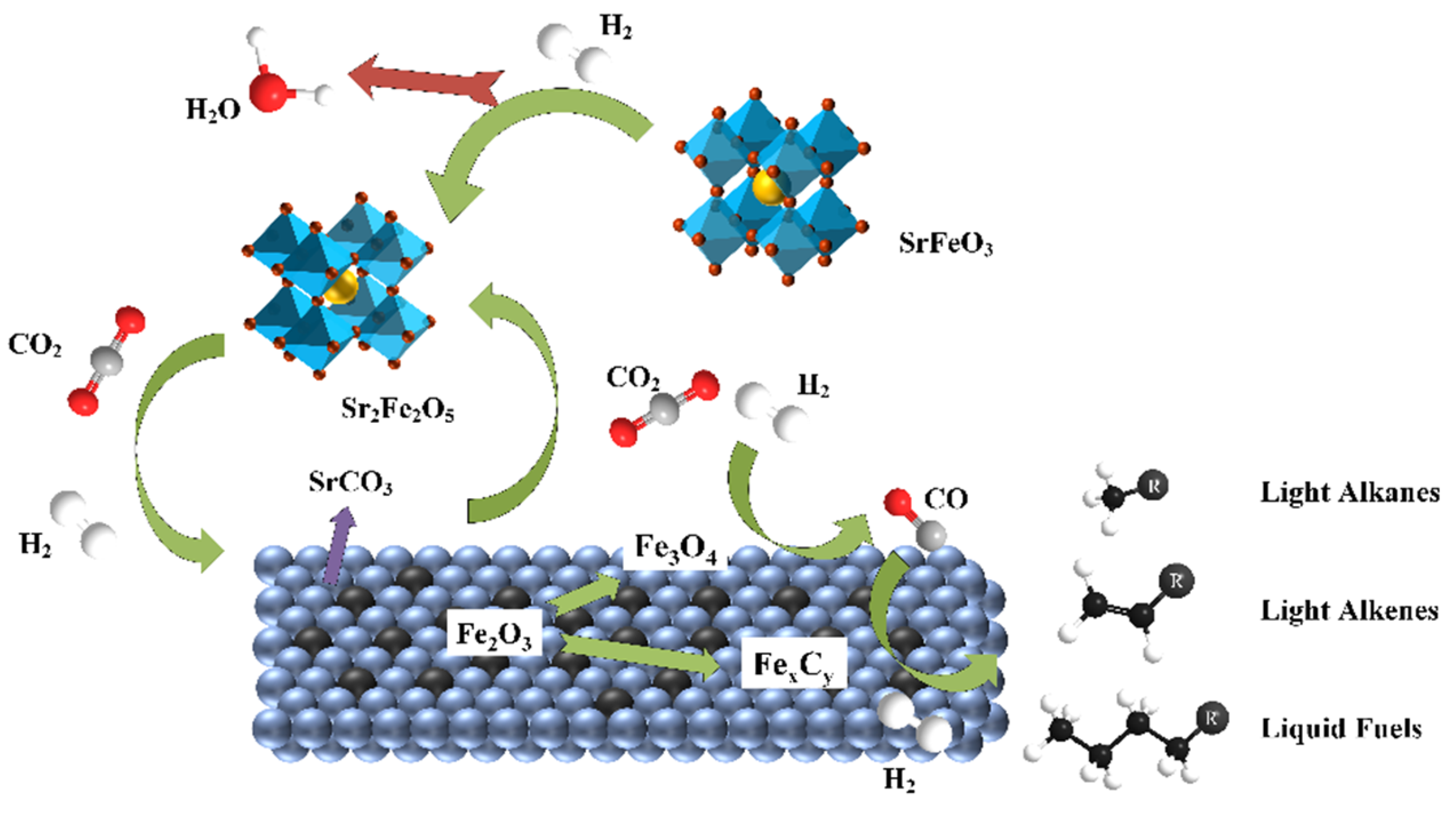
3.4. Probable Reaction Mechanism over K-Substituting Catalysts
4. Conclusions
- (1)
- K-substitution using SKFx perovskite catalysts could promote the production of active-phase Fe3O4 for the RWGS and Fe5C2 for the FTS. SKF0.4 displayed the optimal CO2 conversion of 30.82% and light olefin selectivity of 29.61%.
- (2)
- Apart from the catalytic properties of the acknowledged Fe3O4, both the redox effect of the Sr2Fe2O5 on the CO2 splitting and the formation of SrCO3 carbonates were responsible for the enhanced RWGS.
- (3)
- The reversible reaction Sr2Fe2O5 + 2CO2 = 2SrCO3 + Fe2O3 ensured the structural stability and high dispersion of the active phase. As the rate-determining step of the CO2 hydrogenation reaction to light olefins using SKFx perovskite catalysts, the FTS should be enhanced in the future by partially substituting the B-site of perovskite-type catalysts.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Tackett, B.M.; Gomez, E.; Chen, J.G. Net reduction of CO2 via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes. Nat. Catal. 2019, 2, 381–386. [Google Scholar] [CrossRef]
- Ojelade, O.A.; Zaman, S.F. A review on CO2 hydrogenation to lower olefins: Understanding the structure-property relationships in heterogeneous catalytic systems. J. CO2 Util. 2021, 47, 101506. [Google Scholar] [CrossRef]
- Wang, D.; Xie, Z.; Porosoff, M.D.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to produce olefins and aromatics. Chem 2021, 7, 2277–2311. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Park, S.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Numpilai, T.; Cheng, C.K.; Limtrakul, J.; Witoon, T. Recent advances in light olefins production from catalytichydrogenation of carbon dioxide. Process. Saf. Environ. 2021, 151, 401–427. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Qu, Y.; Liu, H.; Tang, C.; Miao, S.; Feng, Z.; An, H.; Li, C. Highly selective conversion of carbon dioxide to lower olefins. ACS Catal. 2017, 7, 8544–8548. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Boreriboon, N.; Jiang, X.; Song, C.; Prasassarakich, P. Higher hydrocarbons synthesis from CO2 hydrogenation over K- and La-promoted Fe–Cu/TiO2 catalysts. Top. Catal. 2018, 61, 1551–1562. [Google Scholar] [CrossRef]
- Wei, J.; Sun, J.; Wen, Z.; Fang, C.; Ge, Q.; Xu, H. New insights into the effect of sodium on Fe3O4-based nanocatalysts for CO2 hydrogenation to light olefins. Catal. Sci. Technol. 2016, 6, 4786–4793. [Google Scholar] [CrossRef]
- Liu, R.; Ma, Z.; Sears, J.D.; Juneau, M.; Neidig, M.L.; Porosoff, M.D. Identifying correlations in Fischer-Tropsch synthesis and CO2 hydrogenation over Fe-based ZSM-5 catalysts. J. CO2 Util. 2020, 41, 101290. [Google Scholar] [CrossRef]
- Numpilai, T.; Chanlek, N.; Poo-Arporn, Y.; Cheng, C.K.; Siri-Nguan, N.; Sornchamni, T.; Chareonpanich, M.; Kongkachuichay, P.; Yigit, N.; Rupprechter, G. Tuning interactions of surface-adsorbed species over Fe-Co/K-Al2O3 catalyst by different K contents: Selective CO2 hydrogenation to light olefins. ChemCatChem 2020, 12, 3306–3320. [Google Scholar] [CrossRef]
- Zhu, M.; Tian, P.; Ford, M.E.; Chen, J.; Xu, J.; Han, Y.; Wachs, I.E. Nature of reactive oxygen intermediates on copper-promoted iron-chromium oxide catalysts during CO2 activation. ACS Catal. 2020, 10, 7857–7863. [Google Scholar] [CrossRef]
- Witoon, T.; Numpilai, T.; Dolsiririttigul, N.; Chanlek, N.; Poo-arporn, Y.; Cheng, C.K.; Ayodele, B.V.; Chareonpanich, M.; Limtrakul, J. Enhanced activity and stability of SO42−/ZrO2 by addition of Cu combined with CuZnOZrO2 for direct synthesis of dimethyl ether from CO2 hydrogenation. Int. J. Hydrogen Energy 2022, in press. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, S.; Su, X.; Fan, S.; Ma, Q.; Zhao, T. Selective formation of light olefins from CO2 hydrogenation over Fe–Zn– K catalysts. J. CO2 Util. 2015, 12, 95–100. [Google Scholar] [CrossRef]
- Witoon, T.; Chaipraditgul, N.; Numpilai, T.; Lapkeatseree, V.; Ayodele, B.V.; Cheng, C.K.; Siri-Nguan, N.; Sornchamni, T.; Limtrakul, J. Highly active Fe-Co-Zn/K-Al2O3 catalysts for CO2 hydrogenation to light olefins. Chem. Eng. Sci. 2021, 233, 116428. [Google Scholar] [CrossRef]
- Numpilai, T.; Chanlek, N.; Poo-Arporn, Y.; Wannapaiboon, S.; Cheng, C.K.; Siri-Nguan, N.; Sornchamni, T.; Kongkachuichay, P.; Chareonpanich, M.; Rupprechter, G.; et al. Pore size effects on physicochemical properties of Fe-Co/K-Al2O3 catalysts and their catalytic activity in CO2 hydrogenation to light olefins. Appl. Surf. Sci. 2019, 483, 581–592. [Google Scholar] [CrossRef]
- Rafati, M.; Wang, L.; Shahbazi, A. Effect of silica and alumina promoters on coprecipitated Fe–Cu–K based catalysts for the enhancement of CO2 utilization during Fischer–Tropsch synthesis. J. CO2 Util. 2015, 12, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Price, C.A.H.; Reina, T.R.; Liu, J. Engineering heterogenous catalysts for chemical CO2 utilization: Lessons from thermal catalysis and advantages of yolk@shell structured nanoreactors. J. Energy Chem. 2021, 57, 304–324. [Google Scholar] [CrossRef]
- Ren, J.; Mebrahtu, C.; van Koppen, L.; Martinovic, F.; Hofmann, J.P.; Hensen, E.J.M.; Palkovits, R. Enhanced CO2 methanation activity over La2-xCexNiO4 perovskite-derived catalysts: Understanding the structure-performance relationships. Chem. Eng. J. 2021, 426, 131760. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, G.; Liu, Y. Perovskite-Type Oxides as the Catalyst Precursors for Preparing Supported Metallic Nanocatalysts: A Review. Ind. Eng. Chem. Res. 2018, 57, 1–17. [Google Scholar] [CrossRef]
- Lau, C.Y.; Dunstan, M.T.; Hu, W.; Grey, C.P.; Scott, S.A. Large scale in silico screening of materials for carbon capture through chemical looping. Energy Environ. Sci. 2017, 10, 818. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Imtiaz, Q.; Donat, F.; Müller, C.R.; Li, F. Chemical looping beyond combustion—A perspective. Energy Environ. Sci. 2020, 13, 772–804. [Google Scholar] [CrossRef] [Green Version]
- Pour, N.M.; Azimi, G.; Leion, H.; Rydén, M.; Lyngfelt, A. Production and examination of oxygen-carrier materials based on manganese ores and Ca(OH)2 in chemical looping with oxygen uncoupling. AIChE J. 2014, 60, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Ezbiri, M.; Allen, K.M.; Gàlvez, M.E.; Michalsky, R.; Steinfeld, A. Design principles of perovskites for thermochemical oxygen separation. ChemSusChem 2015, 8, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihai, O.; Chen, D.; Holmen, A. Catalytic consequence of oxygen of lanthanum ferrite perovskite in chemical looping reforming of methane. Ind. Eng. Chem. Res. 2011, 50, 2613–2621. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Wang, X.; Ji, X.; Wei, J.; Zhang, J. Orthogonal Preparation of SrFeO3-δ Nanocomposites as Effective Oxygen Transfer Agents for Chemical-Looping Steam Methane Reforming. Energy Fuels 2021, 35, 17848–17860. [Google Scholar] [CrossRef]
- Lindenthal, L.; Popovic, J.; Rameshan, R.; Huber, J.; Schrenk, F.; Ruh, T.; Nenning, A.; Loffler, S.; Opitz, A.K.; Rameshan, C. Novel perovskite catalysts for CO2 utilization—Exsolution enhanced reverse water-gas shift activity. Appl. Catal. B Environ. 2021, 292, 120183. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kent, R.A.; Yung, M.M.; Kuhn, J.N. Carbon Dioxide Conversion by Reverse Water−Gas Shift Chemical Looping on Perovskite-Type Oxides. Ind. Eng. Chem. Res. 2014, 53, 5828–5837. [Google Scholar] [CrossRef]
- Ao, M.; Pham, G.H.; Sage, V.; Pareek, V.; Liu, S. Perovskite-derived trimetallic Co-Ni-Cu catalyst for higher alcohol synthesis from syngas. Fuel Process. Technol. 2019, 193, 141–148. [Google Scholar] [CrossRef]
- Liu, Z.; Jia, G.; Zhao, C.; Xing, Y. Selective Iron Catalysts for Direct Fischer−Tropsch Synthesis to Light Olefins. Ind. Eng. Chem. Res. 2021, 60, 6137–6146. [Google Scholar] [CrossRef]
- Ma, L.; Gao, X.; Ma, J.; Hu, X.; Zhang, J.; Guo, Q. K/LaFeMnO3 Perovskite-Type Oxide Catalyst for the Production of C2–C4 Olefins via CO Hydrogenation. Catal. Lett. 2022, 152, 1451–1460. [Google Scholar] [CrossRef]
- Utsis, N.; Vidruk-Nehemya, R.; Landau, M.V.; Herskowitz, M. Novel bifunctional catalysts based on crystalline multi-oxide matrices containing iron ions for CO2 hydrogenation to liquid fuels and chemicals. Faraday Discuss. 2016, 188, 545. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xu, X.; Fan, G.; Yang, L.; Li, F. Unveiling the roles of Fe-Co interactions over ternary spinel-type ZnCoxFe2−xO4 catalysts for highly efficient CO2 hydrogenation to produce light olefins. J. Catal. 2021, 400, 355–366. [Google Scholar] [CrossRef]
- Yu, W.; Wang, X.; Liu, Y.; Wei, J.; Zhang, J. Effect of Composition on the Redox Performance of Strontium Ferrite Nanocomposite. Energy Fuels 2020, 34, 8644–8652. [Google Scholar] [CrossRef]
- Marek, E.; Hu, W.; Gaultoisd, M.; Grey, C.; Scott, S.A. The use of strontium ferrite in chemical looping systems. Appl. Energy 2018, 223, 369–382. [Google Scholar] [CrossRef]
- Wang, X.; Du, X.; Yu, W.; Zhang, J.; Wei, J. Coproduction of Hydrogen and Methanol from Methane by Chemical Looping Reforming. Ind. Eng. Chem. Res. 2019, 58, 10296–10306. [Google Scholar] [CrossRef]
- Meiri, N.; Dinburg, Y.; Amoyal, M.; Koukouliev, V.; Nehemya, R.V.; Landau, M.V.; Herskowitz, M. Novel process and catalytic materials for converting CO2 and H2 containing mixtures to liquid fuels and chemicals. Faraday Discuss. 2015, 183, 197. [Google Scholar] [CrossRef]
- Takeda, Y.; Kanno, K.; Takada, T.; Yamamoto, O.; Takano, M.; Nakayama, N.; Bando, Y. Phase relation in the oxygen nonstoichiometric system, SrFeOx (2.5 ≤ x ≤ 3.0). J. Solid State Chem. 1986, 63, 237–249. [Google Scholar] [CrossRef]
- Ikeda, H.; Nikata, S.; Hirakawa, E.; Tsuchida, A.; Miura, N. Oxygen sorption/desorption behavior and crystal structural change for SrFeO3-δ. Chem. Eng. Sci. 2016, 147, 166–172. [Google Scholar] [CrossRef]
- Gallagher, P.K.; Sanders, J.P.; Woodward, P.M.; Lokuhewa, I.N. Reactions within the system, 2SrCO3–Fe2O3 Part I. CO2 atmosphere, J. Therm. Anal. Calorim. 2005, 80, 217–223. [Google Scholar] [CrossRef]
- Ngantsoue-Hoc, W.; Zhang, Y.; O’Brien, R.; Luo, M.; Davis, B.H. Fischer-Tropsch synthesis: Activity and selectivity for Group I alkali promoted iron-based catalysts. Appl. Catal. A-Gen. 2002, 236, 77–89. [Google Scholar] [CrossRef]
- Tian, X.; Zheng, C.; Zhao, H. Ce-modified SrFeO3-δ for ethane oxidative dehydrogenation coupled with CO2 splitting via a chemical looping scheme. Appl. Catal. B Environ. 2022, 303, 120894. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Jia, L.; Sun, C.; Chen, B.; Liu, R.; Tan, Y.; Tu, W. Effects of the reducing gas atmosphere on performance of FeCeNa catalyst for the hydrogenation of CO2 to olefins. Chem. Eng. J. 2022, 428, 131388. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Fe2p3/2 | O1s | ||||
|---|---|---|---|---|---|---|
| Fe2+/% | Fe3+/% | Fe4+/% | Olat/% | Ocar/% | Oads/% | |
| SKF0.0 (fresh) | 20.41 | 53.85 | 25.74 | 20.81 | 21.63 | 57.56 |
| SKF0.4 (fresh) | 22.24 | 44.6 | 33.16 | 15.36 | 14.74 | 69.9 |
| SKF0.0 (used) | 28.02 | 49.23 | 22.75 | 15.31 | 32.64 | 41.35 |
| Conditions | H/C | Conversion (%) | Selectivity (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CO2 | CO | CO | CO2 | CH4 | S/C5+ | ||||
| CO2+N2 | - | 6.54 | - | 86.22 | - | - | - | - | - |
| CO2+H2 | 1 | 16.89 | - | 68.56 | - | 10.39 | 2.19 | 17.14 | 1.72 |
| 2 | 28.74 | - | 61.52 | - | 11.21 | 4.04 | 15.25 | 7.98 | |
| 3 | 30.82 | - | 46.68 | - | 13.96 | 5.40 | 29.61 | 4.35 | |
| 4 | 31.54 | - | 45.57 | - | 19.30 | 7.34 | 25.91 | 1.88 | |
| CO+H2 | 2 | - | 86.36 | - | 35.02 | 22.57 | 2.96 | 22.10 | 17.35 |
| 3 | - | 89.57 | - | 41.42 | 21.75 | 3.82 | 24.69 | 8.32 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Wang, X.; Chen, M.; Gao, X.; Liu, Y.; Guo, Q. Sr1-xKxFeO3 Perovskite Catalysts with Enhanced RWGS Reactivity for CO2 Hydrogenation to Light Olefins. Atmosphere 2022, 13, 760. https://doi.org/10.3390/atmos13050760
Hou Y, Wang X, Chen M, Gao X, Liu Y, Guo Q. Sr1-xKxFeO3 Perovskite Catalysts with Enhanced RWGS Reactivity for CO2 Hydrogenation to Light Olefins. Atmosphere. 2022; 13(5):760. https://doi.org/10.3390/atmos13050760
Chicago/Turabian StyleHou, Yuanhao, Xinyu Wang, Ming Chen, Xiangyu Gao, Yongzhuo Liu, and Qingjie Guo. 2022. "Sr1-xKxFeO3 Perovskite Catalysts with Enhanced RWGS Reactivity for CO2 Hydrogenation to Light Olefins" Atmosphere 13, no. 5: 760. https://doi.org/10.3390/atmos13050760