
The Role of Emission Sources and Atmospheric Sink in the Seasonal Cycle of CH4 and δ13-CH4: Analysis Based on the Atmospheric Chemistry Transport Model TM5

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. The TM5 Atmospheric Chemistry Transport Model
2.2. CH4 and 13CH4 Flux Fields
2.2.1. Anthropogenic CH4 Flux Data
2.2.2. Natural CH4 Flux Data
2.3. Isotopic Signature
Isotopic Signatures in Spin-Up Simulations
2.4. Atmospheric CH4 and δ13C Observations
2.5. Simulation Setups
3. Results
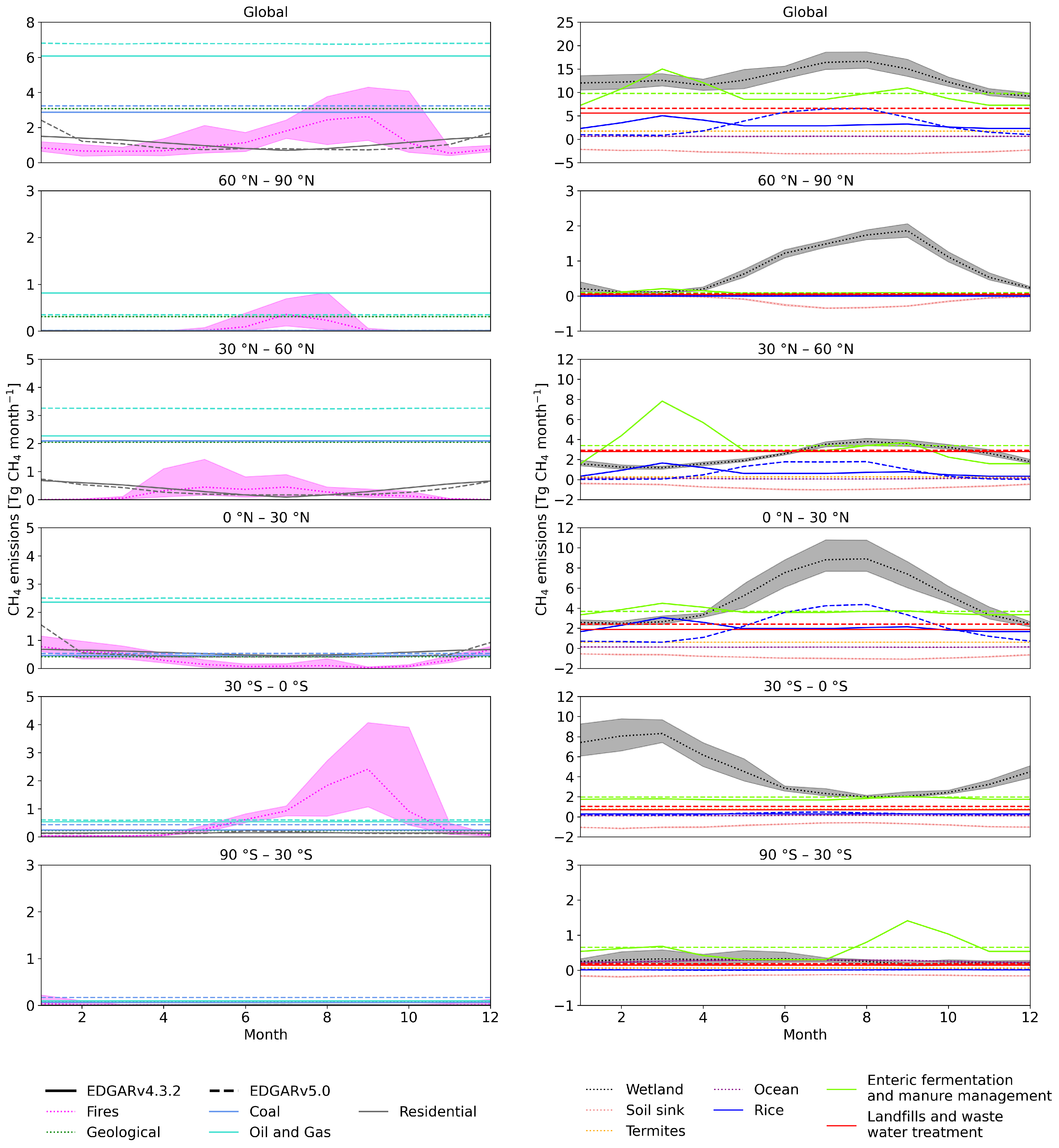
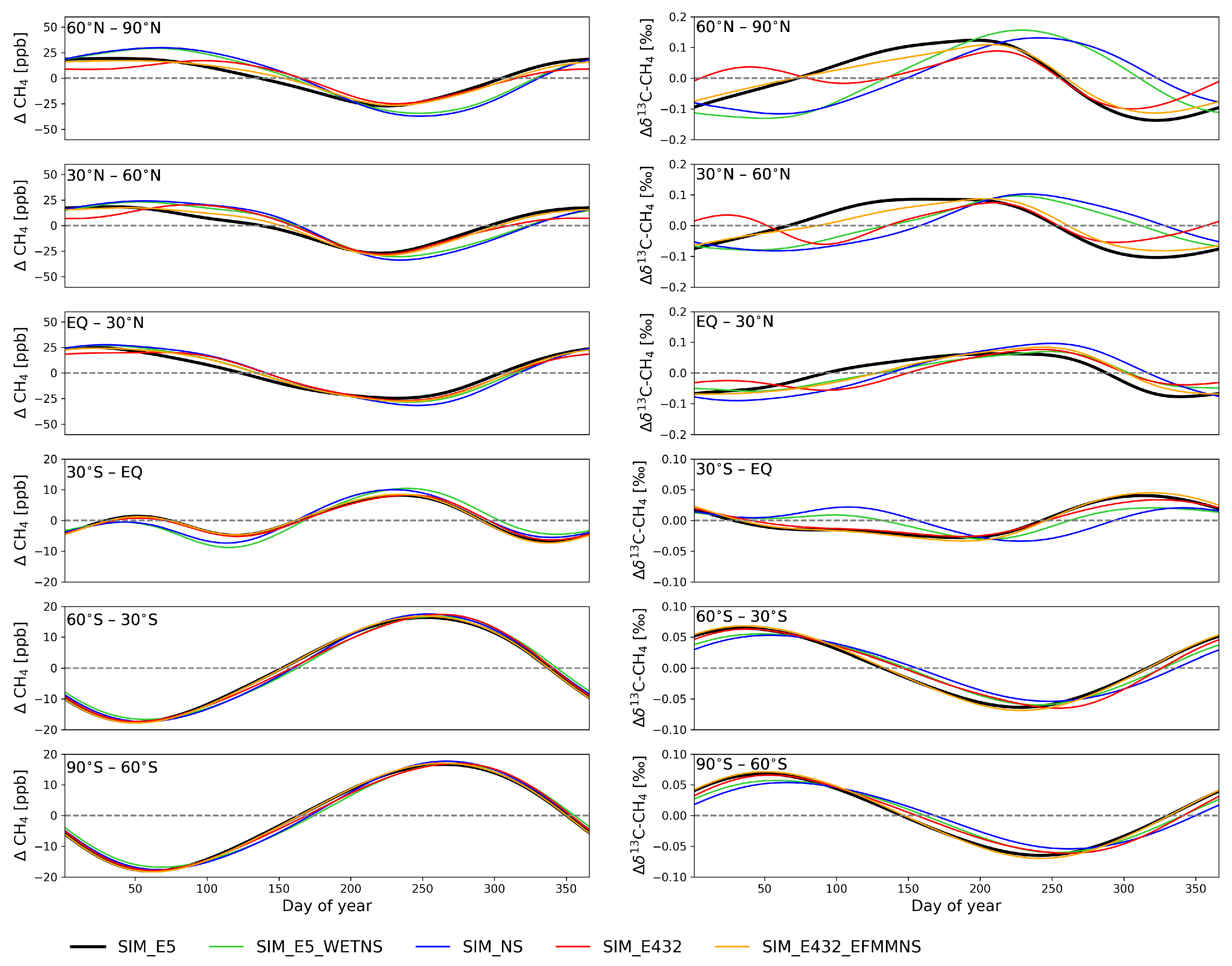
3.1. Zonal Means near the Surface
3.1.1. Peak-to-Peak Amplitude and Shape of C Seasonal Cycle
3.1.2. Phase Ellipses
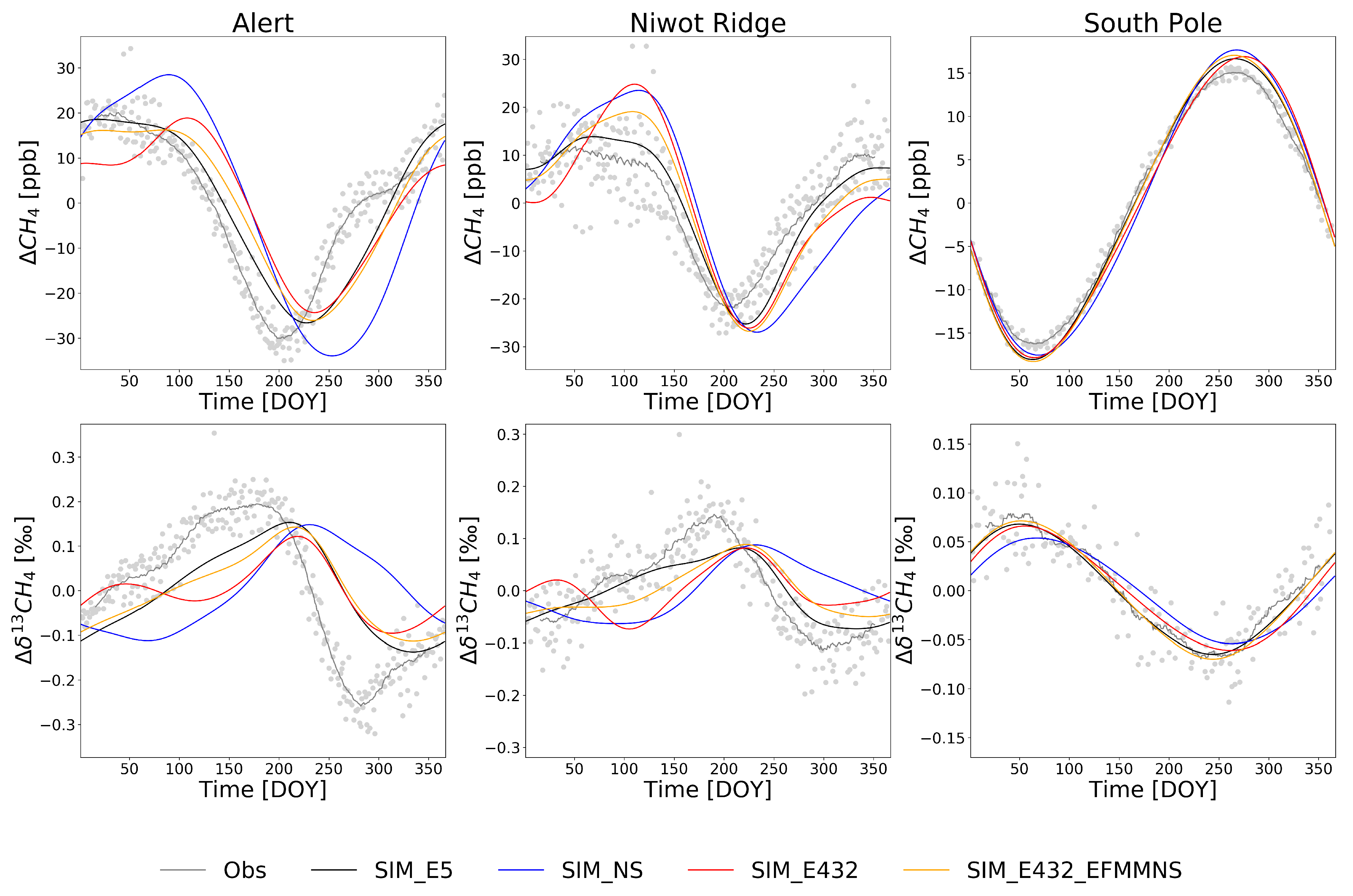
3.2. Comparison to Surface Observations
4. Discussion
4.1. Isotope Signatures
4.2. Seasonal Cycle of CH4 Emissions
4.3. Atmospheric Sinks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EDGAR | Emissions Database for Global Atmospheric Research |
| EFMM | Enteric Fermentation and Manure Management |
| KIE | Kinetic isotopic effect |
| NH | Northern Hemisphere |
| NOAA/GML | National Oceanic and Atmospheric Administration Global Monitoring Laboratory |
| INSTAAR | Institute of Arctic and Alpine Research |
| ECMWF | European Centre for Medium-Range Weather Forecasts |
| IPCC | Intergovernmental Panel on Climate Change |
| LWW | Landfills and wastewater treatment |
| SH | Southern Hemisphere |
| ALT | Alert |
| NWR | Niwot Ridge |
| SPO | South Pole |
| BL | Boundary Layer |
References
- The Core Writing Team; Rajendra, K.P.; Leo, M. (Eds.) IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2015; p. 87. Available online: https://www.ipcc.ch/site/assets/uploads/2018/02/AR5_SYR_FINAL_Front_matters.pdf (accessed on 20 March 2022).
- Hartmann, D.; Klein Tank, A.; Rusticucci, M.; Alexander, L.; Brönnimann, S.; Charabi, Y.; Dentener, F.; Dlugokencky, E.; Easterling, D.; Kaplan, A.; et al. Observations: Atmosphere and Surface. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T., Qin, D., Plattner, G.K., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; Book Section 2; pp. 159–254. [Google Scholar] [CrossRef] [Green Version]
- Saunois, M.; Stavert, A.R.; Poulter, B.; Bousquet, P.; Canadell, J.G.; Jackson, R.B.; Raymond, P.A.; Dlugokencky, E.J.; Houweling, S.; Patra, P.K.; et al. The Global Methane Budget 2000–2017; Earth System Science Data, Copernicus Publications: Göttingen, Germany, 2020; Volume 12, pp. 1561–1623. [Google Scholar] [CrossRef]
- Crippa, M.; Solazzo, E.; Huang, G.; Guizzardi, D.; Koffi, E.; Muntean, M.; Schieberle, C.; Friedrich, R.; Janssens-Maenhout, G. High resolution temporal profiles in the Emissions Database for Global Atmospheric Research. Sci. Data 2020, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, P.; Karstens, U.; Manning, A.J.; Saunois, M.; Tsuruta, A.; Berchet, A.; Vermeulen, A.T.; Arnold, T.; Janssens-Maenhout, G.; Hammer, S.; et al. Inverse modelling of European CH4 emissions during 2006–2012 using different inverse models and reassessed atmospheric observations. Atmos. Chem. Phys. 2018, 18, 901–920. [Google Scholar] [CrossRef] [Green Version]
- Bloom, A.A.; Bowman, K.W.; Lee, M.; Turner, A.J.; Schroeder, R.; Worden, J.R.; Weidner, R.; McDonald, K.C.; Jacob, D.J. A global wetland methane emissions and uncertainty dataset for atmospheric chemical transport models (WetCHARTs version 1.0). Geosci. Model Dev. 2017, 10, 2141–2156. [Google Scholar] [CrossRef] [Green Version]
- Basso, L.S.; Gatti, L.V.; Gloor, M.; Miller, J.B.; Domingues, L.G.; Correia, C.S.C.; Borges, V.F. Seasonality and interannual variability of CH4 fluxes from the eastern Amazon Basin inferred from atmospheric mole fraction profiles. J. Geophys. Res. Atmos. 2016, 121, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Giglio, L.; Randerson, J.T.; van der Werf, G.R. Analysis of daily, monthly, and annual burned area using the fourth-generation global fire emissions database (GFED4). J. Geophys. Res. Biogeosci. 2013, 118, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Kuze, A.; Kikuchi, N.; Kataoka, F.; Suto, H.; Shiomi, K.; Kondo, Y. Detection of Methane Emission from a Local Source Using GOSAT Target Observations. Remote. Sens. 2020, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.; Gautam, R.; Houweling, S.; van der Gon, H.D.; Sadavarte, P.; Borsdorff, T.; Hasekamp, O.; Landgraf, J.; Tol, P.; van Kempen, T.; et al. Satellite observations reveal extreme methane leakage from a natural gas well blowout. Proc. Natl. Acad. Sci. USA 2019, 116, 26376–26381. [Google Scholar] [CrossRef]
- Delwiche, K.B.; Knox, S.H.; Malhotra, A.; Fluet-Chouinard, E.; McNicol, G.; Feron, S.; Ouyang, Z.; Papale, D.; Trotta, C.; Canfora, E.; et al. FLUXNET-CH4: A global, multi-ecosystem dataset and analysis of methane seasonality from freshwater wetlands. Earth Syst. Sci. Data Discuss. 2021, 13, 3607–3689. [Google Scholar] [CrossRef]
- Villarreal, S.; Vargas, R. Representativeness of FLUXNET Sites Across Latin America. J. Geophys. Res. Biogeosci. 2021, 126, e2020JG006090. [Google Scholar] [CrossRef]
- Parker, R.J.; Wilson, C.; Bloom, A.A.; Comyn-Platt, E.; Hayman, G.; McNorton, J.; Boesch, H.; Chipperfield, M.P. Exploring constraints on a wetland methane emission ensemble (WetCHARTs) using GOSAT observations. Biogeosciences 2020, 17, 5669–5691. [Google Scholar] [CrossRef]
- Xu, X.; Riley, W.J.; Koven, C.D.; Billesbach, D.P.; Chang, R.Y.W.; Commane, R.; Euskirchen, E.S.; Hartery, S.; Harazono, Y.; Iwata, H.; et al. A multi-scale comparison of modeled and observed seasonal methane emissions in northern wetlands. Biogeosciences 2016, 13, 5043–5056. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jacob, D.J.; Lu, X.; Maasakkers, J.D.; Scarpelli, T.R.; Sheng, J.X.; Shen, L.; Qu, Z.; Sulprizio, M.P.; Chang, J.; et al. Attribution of the accelerating increase in atmospheric methane during 2010–2018 by inverse analysis of GOSAT observations. Atmos. Chem. Phys. 2021, 21, 3643–3666. [Google Scholar] [CrossRef]
- Warwick, N.J.; Cain, M.L.; Fisher, R.; France, J.L.; Lowry, D.; Michel, S.E.; Nisbet, E.G.; Vaughn, B.H.; White, J.W.C.; Pyle, J.A. Using δ13C-CH4 and δD-CH4 to constrain Arctic methane emissions. Atmos. Chem. Phys. 2016, 16, 14891–14908. [Google Scholar] [CrossRef] [Green Version]
- Tsuruta, A.; Aalto, T.; Backman, L.; Krol, M.C.; Peters, W.; Lienert, S.; Joos, F.; Miller, P.A.; Zhang, W.; Laurila, T.; et al. Methane budget estimates in Finland from the CarbonTracker Europe-CH4 data assimilation system. Tellus B Chem. Phys. Meteorol. 2019, 71, 1565030. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Akiyama, H.; Yagi, K.; Akimoto, H. Global estimations of the inventory and mitigation potential of methane emissions from rice cultivation conducted using the 2006 Intergovernmental Panel on Climate Change Guidelines. Glob. Biogeochem. Cycles 2009, 23, GB2002. [Google Scholar] [CrossRef]
- Elsgaard, L.; Olsen, A.B.; Petersen, S.O. Temperature response of methane production in liquid manures and co-digestates. Sci. Total Environ. 2016, 539, 78–84. [Google Scholar] [CrossRef]
- Nisbet, E.G.; Dlugokencky, E.J.; Manning, M.R.; Lowry, D.; Fisher, R.E.; France, J.L.; Michel, S.E.; Miller, J.B.; White, J.W.C.; Vaughn, B.; et al. Rising atmospheric methane: 2007–2014 growth and isotopic shift. Glob. Biogeochem. Cycles 2016, 30, 1356–1370. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, O.A.; Schwietzke, S.; Arling, V.A.; Etiope, G. Global Inventory of Gas Geochemistry Data from Fossil Fuel, Microbial and Burning Sources, version 2017. Earth Syst. Sci. Data 2017, 9, 639–656. [Google Scholar] [CrossRef] [Green Version]
- Schwietzke, S.; Sherwood, O.A.; Bruhwiler, L.M.P.; Miller, J.B.; Etiope, G.; Dlugokencky, E.J.; Michel, S.E.; Arling, V.A.; Vaughn, B.H.; White, J.W.C.; et al. Upward revision of global fossil fuel methane emissions based on isotope database. Nature 2016, 538, 88–91. [Google Scholar] [CrossRef]
- Thompson, R.L.; Nisbet, E.G.; Pisso, I.; Stohl, A.; Blake, D.; Dlugokencky, E.J.; Helmig, D.; White, J.W.C. Variability in Atmospheric Methane From Fossil Fuel and Microbial Sources Over the Last Three Decades. Geophys. Res. Lett. 2018, 45, 11499–11508. [Google Scholar] [CrossRef] [Green Version]
- Monteil, G.; Houweling, S.; Dlugockenky, E.J.; Maenhout, G.; Vaughn, B.H.; White, J.W.C.; Rockmann, T. Interpreting methane variations in the past two decades using measurements of CH4 mixing ratio and isotopic composition. Atmos. Chem. Phys. 2011, 11, 9141–9153. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Basu, S.; Schwietzke, S.; Bruhwiler, L.M.P.; Dlugokencky, E.J.; Michel, S.E.; Sherwood, O.A.; Tans, P.P.; Thoning, K.; Etiope, G.; et al. Improved Constraints on Global Methane Emissions and Sinks Using δ13C-CH4. Glob. Biogeochem. Cycles 2021, 35, e2021GB007000. [Google Scholar] [CrossRef]
- Houweling, S.; Krol, M.; Bergamaschi, P.; Frankenberg, C.; Dlugokencky, E.J.; Morino, I.; Notholt, J.; Sherlock, V.; Wunch, D.; Beck, V.; et al. A multi-year methane inversion using SCIAMACHY, accounting for systematic errors using TCCON measurements. Atmos. Chem. Phys. 2014, 14, 3991–4012. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.L.; Stell, A.C.; Gedney, N.; Comyn-Platt, E.; Hayman, G.; Rigby, M.; Poulter, B.; Hornibrook, E.R.C. Spatially Resolved Isotopic Source Signatures of Wetland Methane Emissions. Geophys. Res. Lett. 2018, 45, 3737–3745. [Google Scholar] [CrossRef]
- Feinberg, A.I.; Coulon, A.; Stenke, A.; Schwietzke, S.; Peter, T. Isotopic source signatures: Impact of regional variability on the δ13CH4 trend and spatial distribution. Atmos. Environ. 2018, 174, 99–111. [Google Scholar] [CrossRef]
- Etiope, G.; Ciotoli, G.; Schwietzke, S.; Schoell, M. Gridded maps of geological methane emissions and their isotopic signature. Earth Syst. Sci. Data 2019, 11, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Brownlow, R.; Lowry, D.; Fisher, R.E.; France, J.L.; Lanoisellé, M.; White, B.; Wooster, M.J.; Zhang, T.; Nisbet, E.G. Isotopic Ratios of Tropical Methane Emissions by Atmospheric Measurement: Tropical Methane δ13 C Source Signatures. Glob. Biogeochem. Cycles 2017, 31, 1408–1419. [Google Scholar] [CrossRef] [Green Version]
- Saueressig, G.; Crowley, J.N.; Bergamaschi, P.; Brühl, C.; Brenninkmeijer, C.A.M.; Fischer, H. Carbon 13 and D kinetic isotope effects in the reactions of CH4 with O(1D) and OH: New laboratory measurements and their implications for the isotopic composition of stratospheric methane. J. Geophys. Res. Atmos. 2001, 106, 23127–23138. [Google Scholar] [CrossRef]
- Cantrell, C.A.; Shetter, R.E.; McDaniel, A.H.; Calvert, J.G.; Davidson, J.A.; Lowe, D.C.; Tyler, S.C.; Cicerone, R.J.; Greenberg, J.P. Carbon kinetic isotope effect in the oxidation of methane by the hydroxyl radical. J. Geophys. Res. Atmos. 1990, 95, 22455–22462. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, P.; Brühl, C.; Brenninkmeijer, C.a.M.; Saueressig, G.; Crowley, J.N.; Grooß, J.U.; Fischer, H.; Crutzen, P.J. Implications of the large carbon kinetic isotope effect in the reaction CH4 + Cl for the 13C/12C ratio of stratospheric CH4. Geophys. Res. Lett. 1996, 23, 2227–2230. [Google Scholar] [CrossRef]
- Allan, W.; Manning, M.R.; Lassey, K.R.; Lowe, D.C.; Gomez, A.J. Modeling the variation of δ13C in atmospheric methane: Phase ellipses and the kinetic isotope effect. Glob. Biogeochem. Cycles 2001, 15, 467–481. [Google Scholar] [CrossRef]
- Bergamaschi, P.; Bräunlich, M.; Marik, T.; Brenninkmeijer, C.A.M. Measurements of the carbon and hydrogen isotopes of atmospheric methane at Izaña, Tenerife: Seasonal cycles and synoptic-scale variations. J. Geophys. Res. Atmos. 2000, 105, 14531–14546. [Google Scholar] [CrossRef]
- Tyler, S.C.; Rice, A.L.; Ajie, H.O. Stable isotope ratios in atmospheric CH4: Implications for seasonal sources and sinks. J. Geophys. Res. Atmos. 2007, 112, D03303. [Google Scholar] [CrossRef]
- Fujita, R.; Morimoto, S.; Umezawa, T.; Ishijima, K.; Patra, P.K.; Worthy, D.E.J.; Goto, D.; Aoki, S.; Nakazawa, T. Temporal Variations of the Mole Fraction, Carbon, and Hydrogen Isotope Ratios of Atmospheric Methane in the Hudson Bay Lowlands, Canada. J. Geophys. Res. Atmos. 2018, 123, 4695–4711. [Google Scholar] [CrossRef]
- Allan, W.; Struthers, H.; Lowe, D.C. Methane carbon isotope effects caused by atomic chlorine in the marine boundary layer: Global model results compared with Southern Hemisphere measurements. J. Geophys. Res. Atmos. 2007, 112, D04306. [Google Scholar] [CrossRef]
- Hossaini, R.; Chipperfield, M.P.; Saiz-Lopez, A.; Fernandez, R.; Monks, S.; Feng, W.; Brauer, P.; Glasow, R.v. A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation. J. Geophys. Res. Atmos. 2016, 121, 14271–14297. [Google Scholar] [CrossRef] [Green Version]
- Gromov, S.; Brenninkmeijer, C.A.M.; Jöckel, P. A very limited role of tropospheric chlorine as a sink of the greenhouse gas methane. Atmos. Chem. Phys. 2018, 18, 9831–9843. [Google Scholar] [CrossRef] [Green Version]
- Heimann, M. Technical Report No.10: The Global Atmospheric Tracer Model TM2 Technical Report/Deutsches Klimarechenzentrum, Modellbetreuungsgruppe. Hamburg, Germany. October 1996. Available online: https://pure.mpg.de/rest/items/item_3337960_3/component/file_3337962/content (accessed on 20 March 2012).
- Krol, M.; Houweling, S.; Bregman, B.; Broek, M.v.d.; Segers, A.; Velthoven, P.v.; Peters, W.; Dentener, F.; Bergamaschi, P. The two-way nested global chemistry-transport zoom model TM5: Algorithm and applications. Atmos. Chem. Phys. 2005, 5, 417–432. [Google Scholar] [CrossRef] [Green Version]
- Huijnen, V.; Williams, J.; van Weele, M.; van Noije, T.; Krol, M.; Dentener, F.; Segers, A.; Houweling, S.; Peters, W.; de Laat, J.; et al. The global chemistry transport model TM5: Description and evaluation of the tropospheric chemistry version 3.0. Geosci. Model Dev. 2010, 3, 445–473. [Google Scholar] [CrossRef] [Green Version]
- Janssens-Maenhout, G.; Crippa, M.; Guizzardi, D.; Muntean, M.; Schaaf, E.; Dentener, F.; Bergamaschi, P.; Pagliari, V.; Olivier, J.G.J.; Peters, J.A.H.W.; et al. EDGAR v4.3.2 Global Atlas of the three major greenhouse gas emissions for the period 1970–2012. Earth Syst. Sci. Data 2019, 11, 959–1002. [Google Scholar] [CrossRef] [Green Version]
- Tsuruta, A.; Aalto, T.; Backman, L.; Hakkarainen, J.; Laan-Luijkx, I.T.v.d.; Krol, M.C.; Spahni, R.; Houweling, S.; Laine, M.; Dlugokencky, E.; et al. Global methane emission estimates for 2000–2012 from CarbonTracker Europe-CH4 v1.0. Geosci. Model Dev. 2017, 10, 1261–1289. [Google Scholar] [CrossRef] [Green Version]
- Gregory, D.; Morcrette, J.J.; Jakob, C.; Beljaars, A.C.M.; Stockdale, T. Revision of convection, radiation and cloud schemes in the ECMWF integrated forecasting system. Q.J.R. Meteorol. Soc. 2000, 126, 1685–1710. [Google Scholar] [CrossRef]
- de Laeter, J.R.; Böhlke, J.K.; Bièvre, P.D.; Hidaka, H.; Peiser, H.S.; Rosman, K.J.R.; Taylor, P.D.P. Atomic weights of the elements. Review 2000 (IUPAC Technical Report). De Gruyter Sect. Pure Appl. Chem. 2003, 75, 683–800. [Google Scholar] [CrossRef]
- Spivakovsky, C.M.; Logan, J.A.; Montzka, S.A.; Balkanski, Y.J.; Foreman-Fowler, M.; Jones, D.B.A.; Horowitz, L.W.; Fusco, A.C.; Brenninkmeijer, C.a.M.; Prather, M.J.; et al. Three-dimensional climatological distribution of tropospheric OH: Update and evaluation. J. Geophys. Res. Atmos. 2000, 105, 8931–8980. [Google Scholar] [CrossRef]
- Jöckel, P.; Tost, H.; Pozzer, A.; Brühl, C.; Buchholz, J.; Ganzeveld, L.; Hoor, P.; Kerkweg, A.; Lawrence, M.G.; Sander, R.; et al. The atmospheric chemistry general circulation model ECHAM5/MESSy1: Consistent simulation of ozone from the surface to the mesosphere. ATmospheric Chem. Phys. 2006, 6, 5067–5104. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, H.; Fletcher, S.E.M.; Veidt, C.; Lassey, K.R.; Brailsford, G.W.; Bromley, T.M.; Dlugokencky, E.J.; Michel, S.E.; Miller, J.B.; Levin, I.; et al. A 21st-century shift from fossil-fuel to biogenic methane emissions indicated by 13CH4. Science 2016, 352, 80–84. [Google Scholar] [CrossRef]
- Montzka, S.A.; Krol, M.; Dlugokencky, E.; Hall, B.; Jöckel, P.; Lelieveld, J. Small Interannual Variability of Global Atmospheric Hydroxyl. Science 2011, 331, 67–69. [Google Scholar] [CrossRef] [Green Version]
- Lelieveld, J.; Gromov, S.; Pozzer, A.; Taraborrelli, D. Global tropospheric hydroxyl distribution, budget and reactivity. Atmos. Chem. Phys. 2016, 16, 12477–12493. [Google Scholar] [CrossRef] [Green Version]
- Crowley, J.N.; Saueressig, G.; Bergamaschi, P.; Fischer, H.; Harris, G.W. Carbon kinetic isotope effect in the reaction CH4+Cl: A relative rate study using FTIR spectroscopy. Chem. Phys. Lett. 1999, 303, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Spahni, R.; Wania, R.; Neef, L.; van Weele, M.; Pison, I.; Bousquet, P.; Frankenberg, C.; Foster, P.N.; Joos, F.; Prentice, I.C.; et al. Constraining global methane emissions and uptake by ecosystems. Biogeosciences 2011, 8, 1643–1665. [Google Scholar] [CrossRef] [Green Version]
- Lienert, S.; Joos, F. A Bayesian ensemble data assimilation to constrain model parameters and land-use carbon emissions. Biogeosciences 2018, 15, 2909–2930. [Google Scholar] [CrossRef] [Green Version]
- Snover, A.K.; Quay, P.D. Hydrogen and carbon kinetic isotope effects during soil uptake of atmospheric methane. Glob. Biogeochem. Cycles 2000, 14, 25–39. [Google Scholar] [CrossRef]
- Röckmann, T.; Brass, M.; Borchers, R.; Engel, A. The isotopic composition of methane in the stratosphere: High-altitude balloon sample measurements. Atmos. Chem. Phys. 2011, 11, 13287–13304. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; McElroy, M.B.; Spivakovsky, C.M.; Jones, D.B.A. On the contribution of anthropogenic Cl to the increase in δ13C of atmospheric methane. Glob. Biogeochem. Cycles 2002, 16, 20-1–20-11. [Google Scholar] [CrossRef]
- Houghton, J.; Meira Filho, L.; Lim, K.; Trennton, I.; Mamaty, I.; Bonduki, Y.; Griggs, D.; Callander, B. Revised 1996 IPCC Guidelines for National Greenhouse Gas Inventories; Intergovernmental Panel on Climate Change, Meteorological Office: Bracknell, UK, 1997; Volumes 1–3.
- Fisher, R.E.; France, J.L.; Lowry, D.; Lanoisellé, M.; Brownlow, R.; Pyle, J.A.; Cain, M.; Warwick, N.; Skiba, U.M.; Drewer, J.; et al. Measurement of the 13C isotopic signature of methane emissions from northern European wetlands. Glob. Biogeochem. Cycles 2017, 31, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Schmidt, S. Reevaluation of the oceanic flux of methane: Uncertainties and long term variations. Chemosphere 1993, 26, 579–589. [Google Scholar] [CrossRef]
- Dee, D.P.; Uppala, S.M.; Simmons, A.J.; Berrisford, P.; Poli, P.; Kobayashi, S.; Andrae, U.; Balmaseda, M.A.; Balsamo, G.; Bauer, P.; et al. The ERA-Interim reanalysis: Configuration and performance of the data assimilation system. Q. J. R. Meteorol. Soc. 2011, 137, 553–597. [Google Scholar] [CrossRef]
- Ito, A.; Inatomi, M. Use of a process-based model for assessing the methane budgets of global terrestrial ecosystems and evaluation of uncertainty. Biogeosciences 2012, 9, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Hmiel, B.; Petrenko, V.V.; Dyonisius, M.N.; Buizert, C.; Smith, A.M.; Place, P.F.; Harth, C.; Beaudette, R.; Hua, Q.; Yang, B.; et al. Preindustrial 14 CH4 indicates greater anthropogenic fossil CH4 emissions. Nature 2020, 578, 409–412. [Google Scholar] [CrossRef]
- Still, C.J.; Berry, J.A.; Collatz, G.J.; DeFries, R.S. Global distribution of C3 and C4 vegetation: Carbon cycle implications. Glob. Biogeochem. Cycles 2003, 17, 6–1–6–14. [Google Scholar] [CrossRef]
- Sherwood, O.; Schwietzke, S.; Arling, V.; Etiope, G. Methane δ13C Source Signature Measurements for Improved Atmospheric. 2016. Available online: http://www.esrl.noaa.gov/gmd/ccgg/d13C-src-inv/ (accessed on 23 May 2022).
- Guidelines for the Measurement of Methane and Nitrous Oxide and their Quality Assurance; WMO/TD-No. 1478, GAW Report No. 185; World Meteorological Organization: Geneva, Switzerland, 2009.
- Miller, J.B.; Mack, K.A.; Dissly, R.; White, J.W.C.; Dlugokencky, E.J.; Tans, P.P. Development of analytical methods and measurements of 13C/12C in atmospheric CH4 from the NOAA Climate Monitoring and Diagnostics Laboratory Global Air Sampling Network. J. Geophys. Res. Atmos. 2002, 107, ACH 11-1–ACH 11-15. [Google Scholar] [CrossRef]
- Thoning, K.W.; Tans, P.P.; Komhyr, W.D. Atmospheric carbon dioxide at Mauna Loa Observatory: 2. Analysis of the NOAA GMCC data, 1974–1985. J. Geophys. Res. Atmos. 1989, 94, 8549–8565. [Google Scholar] [CrossRef]
- Nisbet, E.G.; Manning, M.R.; Dlugokencky, E.J.; Fisher, R.E.; Lowry, D.; Michel, S.E.; Myhre, C.L.; Platt, S.M.; Allen, G.; Bousquet, P.; et al. Very Strong Atmospheric Methane Growth in the 4 Years 2014–2017: Implications for the Paris Agreement. Glob. Biogeochem. Cycles 2019, 33, 318–342. [Google Scholar] [CrossRef]
- Dlugokencky, E.; Masarie, K.; Tans, P.; Conway, T.; Xiong, X. Is the amplitude of the methane seasonal cycle changing? Atmos. Environ. 1997, 31, 21–26. [Google Scholar] [CrossRef]
- Javadinejad, S.; Eslamian, S.; Ostad-Ali-Askari, K. Investigation of monthly and seasonal changes of methane gas with respect to climate change using satellite data. Appl. Water Sci. 2019, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Kivimäki, E.; Lindqvist, H.; Hakkarainen, J.; Laine, M.; Sussmann, R.; Tsuruta, A.; Detmers, R.; Deutscher, N.M.; Dlugokencky, E.J.; Hase, F.; et al. Evaluation and Analysis of the Seasonal Cycle and Variability of the Trend from GOSAT Methane Retrievals. Remote Sens. 2019, 11, 882. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.a.K.; Rasmussen, R.A. Sources, sinks, and seasonal cycles of atmospheric methane. J. Geophys. Res. Ocean. 1983, 88, 5131–5144. [Google Scholar] [CrossRef]
- Hein, R.; Crutzen, P.J.; Heimann, M. An inverse modeling approach to investigate the global atmospheric methane cycle. Glob. Biogeochem. Cycles 1997, 11, 43–76. [Google Scholar] [CrossRef]
- Lowe, D.C.; Koshy, K.; Bromley, T.; Allan, W.; Struthers, H.; Mani, F.; Maata, M. Seasonal cycles of mixing ratio and 13C in atmospheric methane at Suva, Fiji. J. Geophys. Res. Atmos. 2004, 109, D23308. [Google Scholar] [CrossRef]
- Hornibrook, E.R.C. The Stable Carbon Isotope Composition of Methane Produced and Emitted from Northern Peatlands. In Carbon Cycling in Northern Peatlands; American Geophysical Union (AGU): Washington, DC, USA, 2009; pp. 187–203. [Google Scholar] [CrossRef]
- Chang, J.; Peng, S.; Ciais, P.; Saunois, M.; Dangal, S.R.S.; Herrero, M.; Havlík, P.; Tian, H.; Bousquet, P. Revisiting enteric methane emissions from domestic ruminants and their δ 13 C CH4 source signature. Nat. Commun. 2019, 10, 3420. [Google Scholar] [CrossRef]
- Levin, I.; Bergamaschi, P.; Dörr, H.; Trapp, D. Stable isotopic signature of methane from major sources in Germany. Chemosphere 1993, 26, 161–177. [Google Scholar] [CrossRef]
- Zhang, G.; Ma, J.; Yang, Y.; Yu, H.; Shi, Y.; Xu, H. Variations of Stable Carbon Isotopes of CH4 Emission from Three Typical Rice Fields in China. Pedosphere 2017, 27, 52–64. [Google Scholar] [CrossRef]
- France, J.L.; Fisher, R.E.; Lowry, D.; Allen, G.; Andrade, M.F.; Bauguitte, S.J.B.; Bower, K.; Broderick, T.J.; Daly, M.C.; Forster, G.; et al. δ13C methane source signatures from tropical wetland and rice field emissions. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2022, 380, 20200449. [Google Scholar] [CrossRef]
- Sriskantharajah, S.; Fisher, R.E.; Lowry, D.; Aalto, T.; Hatakka, J.; Aurela, M.; Laurila, T.; Lohila, A.; Kuitunen, E.; Nisbet, E.G. Stable carbon isotope signatures of methane from a Finnish subarctic wetland. Tellus B Chem. Phys. Meteorol. 2012, 64, 18818. [Google Scholar] [CrossRef]
- Tyler, S.C.; Brailsford, G.W.; Yagi, K.; Minami, K.; Cicerone, R.J. Seasonal variations in methane flux and δl3CH4 values for rice paddies in Japan and their implications. Glob. Biogeochem. Cycles 1994, 8, 1–12. [Google Scholar] [CrossRef]
- Bergamaschi, P. Seasonal variations of stable hydrogen and carbon isotope ratios in methane from a Chinese rice paddy. J. Geophys. Res. Atmos. 1997, 102, 25383–25393. [Google Scholar] [CrossRef]
- Marik, T.; Fischer, H.; Conen, F.; Smith, K. Seasonal variations in stable carbon and hydrogen isotope ratios in methane from rice fields. Glob. Biogeochem. Cycles 2002, 16, 41-1–41-11. [Google Scholar] [CrossRef]
- Lopez, M.; Sherwood, O.A.; Dlugokencky, E.J.; Kessler, R.; Giroux, L.; Worthy, D.E.J. Isotopic signatures of anthropogenic CH4 sources in Alberta, Canada. Atmos. Environ. 2017, 164, 280–288. [Google Scholar] [CrossRef]
- Kelly, C.A.; Dise, N.B.; Martens, C.S. Temporal variations in the stable carbon isotopic composition of methane emitted from Minnesota peatlands. Glob. Biogeochem. Cycles 1992, 6, 263–269. [Google Scholar] [CrossRef]
- Chanton, J.P.; Rutkowski, C.M.; Schwartz, C.C.; Ward, D.E.; Boring, L. Factors influencing the stable carbon isotopic signature of methane from combustion and biomass burning. J. Geophys. Res. Atmos. 2000, 105, 1867–1877. [Google Scholar] [CrossRef]
- Zazzeri, G.; Lowry, D.; Fisher, R.E.; France, J.L.; Lanoisellé, M.; Kelly, B.F.J.; Necki, J.M.; Iverach, C.P.; Ginty, E.; Zimnoch, M.; et al. Carbon isotopic signature of coal-derived methane emissions to the atmosphere: From coalification to alteration. Atmos. Chem. Phys. 2016, 16, 13669–13680. [Google Scholar] [CrossRef] [Green Version]
- Tyler, S.C.; Crill, P.M.; Brailsford, G.W. 13C12C Fractionation of methane during oxidation in a temperate forested soil. Geochim. Cosmochim. Acta 1994, 58, 1625–1633. [Google Scholar] [CrossRef]
- Reeburgh, W.S.; Hirsch, A.I.; Sansone, F.J.; Popp, B.N.; Rust, T.M. Carbon kinetic isotope effect accompanying microbial oxidation of methane in boreal forest soils. Geochim. Cosmochim. Acta 1997, 61, 4761–4767. [Google Scholar] [CrossRef] [Green Version]
- Team, M.; Nisbet, E.G.; Allen, G.; Fisher, R.E.; France, J.L.; Lee, J.D.; Lowry, D.; Andrade, M.F.; Bannan, T.J.; Barker, P.; et al. Isotopic signatures of methane emissions from tropical fires, agriculture and wetlands: The MOYA and ZWAMPS flights. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2022, 380, 20210112. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sasakawa, M.; Machida, T.; Aalto, T.; Worthy, D.; Lavric, J.V.; Lund Myhre, C.; Stohl, A. Methane fluxes in the high northern latitudes for 2005–2013 estimated using a Bayesian atmospheric inversion. Atmos. Chem. Phys. 2017, 17, 3553–3572. [Google Scholar] [CrossRef] [Green Version]
- Bousquet, P.; Ringeval, B.; Pison, I.; Dlugokencky, E.J.; Brunke, E.G.; Carouge, C.; Chevallier, F.; Fortems-Cheiney, A.; Frankenberg, C.; Hauglustaine, D.A.; et al. Source attribution of the changes in atmospheric methane for 2006–2008. Atmos. Chem. Phys. 2011, 11, 3689–3700. [Google Scholar] [CrossRef] [Green Version]
- Melton, J.R.; Wania, R.; Hodson, E.L.; Poulter, B.; Ringeval, B.; Spahni, R.; Bohn, T.; Avis, C.A.; Beerling, D.J.; Chen, G.; et al. Present state of global wetland extent and wetland methane modelling: Conclusions from a model inter-comparison project (WETCHIMP). Biogeosciences 2013, 10, 753–788. [Google Scholar] [CrossRef] [Green Version]
- Aalto, T.; Tsuruta, A.; Mäkelä, J.; Mueller, J.; Tenkanen, M.; Burke, E.; Chadburn, S.; Gao, Y.; Kangasaho, V.; Kleinen, T.; et al. Air temperature and precipitation constraining the modelled wetland methane emissions in a boreal region in Northern Europe. Biogeosciences 2022. to be submitted. [Google Scholar]
- Fung, I.; John, J.; Lerner, J.; Matthews, E.; Prather, M.; Steele, L.P.; Fraser, P.J. Three-dimensional model synthesis of the global methane cycle. J. Geophys. Res. Atmos. 1991, 96, 13033–13065. [Google Scholar] [CrossRef]
- Tenkanen, M.; Tsuruta, A.; Rautiainen, K.; Kangasaho, V.; Ellul, R.; Aalto, T. Utilizing Earth Observations of Soil Freeze/Thaw Data and Atmospheric Concentrations to Estimate Cold Season Methane Emissions in the Northern High Latitudes. Remote Sens. 2021, 13, 5059. [Google Scholar] [CrossRef]
- Rosentreter, J.A.; Borges, A.V.; Deemer, B.R.; Holgerson, M.A.; Liu, S.; Song, C.; Melack, J.; Raymond, P.A.; Duarte, C.M.; Allen, G.H.; et al. Half of global methane emissions come from highly variable aquatic ecosystem sources. Nat. Geosci. 2021, 14, 225–230. [Google Scholar] [CrossRef]
- Oh, Y.; Zhuang, Q.; Liu, L.; Welp, L.R.; Lau, M.C.Y.; Onstott, T.C.; Medvigy, D.; Bruhwiler, L.; Dlugokencky, E.J.; Hugelius, G.; et al. Reduced net methane emissions due to microbial methane oxidation in a warmer Arctic. Nat. Clim. Chang. 2020, 10, 317–321. [Google Scholar] [CrossRef]
- Arndt, C.; Leytem, A.; Hristov, A.; Zavala-Araiza, D.; Cativiela, J.; Conley, S.; Daube, C.; Faloona, I.; Herndon, S. Short-term methane emissions from 2 dairy farms in California estimated by different measurement techniques and US Environmental Protection Agency inventory methodology: A case study. J. Dairy Sci. 2018, 101, 11461–11479. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Griffis, T.J.; Baker, J.M.; Millet, D.B.; Wood, J.D.; Dlugokencky, E.J.; Andrews, A.E.; Sweeney, C.; Hu, C.; Kolka, R.K. Source Partitioning of Methane Emissions and its Seasonality in the U.S. Midwest. J. Geophys. Res. Biogeosci. 2018, 123, 646–659. [Google Scholar] [CrossRef]
- VanderZaag, A.; Flesch, T.; Desjardins, R.; Baldé, H.; Wright, T. Measuring methane emissions from two dairy farms: Seasonal and manure-management effects. Agric. For. Meteorol. 2014, 194, 259–267. [Google Scholar] [CrossRef]
- Cárdenas, A.; Ammon, C.; Schumacher, B.; Stinner, W.; Herrmann, C.; Schneider, M.; Weinrich, S.; Fischer, P.; Amon, T.; Amon, B. Methane emissions from the storage of liquid dairy manure: Influences of season, temperature and storage duration. Waste Manag. 2021, 121, 393–402. [Google Scholar] [CrossRef]
- Husted, S. Seasonal Variation in Methane Emission from Stored Slurry and Solid Manures. J. Environ. Qual. 1994, 23, 585–592. [Google Scholar] [CrossRef]
- Ulyatt, M.J.; Lassey, K.R.; Shelton, I.D.; Walker, C.F. Seasonal variation in methane emission from dairy cows and breeding ewes grazing ryegrass/white clover pasture in New Zealand. N. Z. J. Agric. Res. 2002, 45, 217–226. [Google Scholar] [CrossRef]
- Mapfumo, L.; Grobler, S.M.; Mupangwa, J.F.; Scholtz, M.M.; Muchenje, V. Enteric methane output from selected herds of beef cattle raised under extensive arid rangelands. Pastoralism 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Gregson, K.; Marshall, S.; Dent, J.B.; Heal, O.W. Global Methane Emissions from Rice Paddies. Chemosphere 1996, 33, 879–897. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, H.; Ren, W.; Tao, B.; Lu, C.; Yang, J.; Banger, K.; Pan, S. Methane emissions from global rice fields: Magnitude, spatiotemporal patterns, and environmental controls. Glob. Biogeochem. Cycles 2016, 30, 1246–1263. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, F.; Berresheim, H. Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation. Nature 2006, 442, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jacob, D.J.; Eastham, S.D.; Sulprizio, M.P.; Zhu, L.; Chen, Q.; Alexander, B.; Sherwen, T.; Evans, M.J.; Lee, B.H.; et al. The role of chlorine in global tropospheric chemistry. Atmos. Chem. Phys. 2019, 19, 3981–4003. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Emission Source | Signature Value (‰) | Signature Value (‰) |
|---|---|---|
| (Used in This Study) | [24] | |
| Enteric Fermentation and Manure Management (EFMM) | [−67.9, −54.5] 1, −66.8 2 | −62 |
| Landfills and Wastewater Treatment (LWW) | −55.6 2 | −55 |
| Rice (RICE) | −62.1 2 | −63 |
| Coal | [−64.1, −36.1] 1, −40 2 | −35 |
| Oil and Gas | [−56.6, −29.1] 1, −40 2 | −40 |
| Residential | −40 2 | −38 |
| Wetlands | [−74.9, −50] 3, −61.3 2 | −59 |
| Fires | [−25, −12] 1, −22.2 2 | −21.8 |
| Ocean | −47 2 | −59 |
| Termites | −65.2 2 | −57 |
| Geological | [−68, −24.3] 4, −40 2 | −40 |
| Data Source | Component | Emission | Amplitude | MIN | MAX |
|---|---|---|---|---|---|
| EDGAR v4.3.2 | Enteric Fermentation and Manure Management (EFMM) | 109.5 [102.1, 115.8] | 7.50 [7.19, 7.75] | Nov | Mar |
| Landfills and Wastewater Treatment (LWW) | 63.8 [59.3, 68.5] | * | * | * | |
| Rice (RICE) | 35.0 [32.4, 37.6] | 2.61 [2.39, 2.74] | Jan, Dec | Mar. | |
| Coal | 29.8 [21.9, 38.2] | 0.03 [0.001, 0.11] | Jul | Jan | |
| Oil and Gas | 68.2 [61.3, 77.1] | 0.07 [0.01, 0.23] | Jul | Nov | |
| Residential | 12.9 [12.1, 13.5] | 0.78 [0.75, 0.81] | Jul | Jan | |
| Total | 319.0 [291.3, 350.8] | 10.05 [9.55, 10.41] | Nov | Mar | |
| EDGAR v5.0 | Enteric Fermentation and Manure Management (EFMM) | 108.5 [102.0, 114.8] | * | * | * |
| Landfills and Wastewater Treatment (LWW) | 71.4 [66.6, 76.5] | * | * | * | |
| Rice | 35.1 [32.6, 37.3] | 5.51 [5.1, 5.83] | Mar | Aug | |
| Coal | 29.5 [21.4, 38.2] | * | * | * | |
| Oil and Gas | 67.5 [61.1, 77.4] | 0.10 [0.04, 0.25] | Sept | Jan | |
| Residential | 12.5 [12.1, 12.8] | 1.65 [1.58, 1.72] | Sept | Jan | |
| Total | 324.5 [297.7, 356.8] | 5.13 [4.72, 5.47] | Mar | Aug | |
| LPX-Bern v1.4 | Wetland | 157.3 [150.6, 165.7] | 8.1 [6.12, 10.0] | Nov, Dec | Jul, Aug |
| LPX-Bern v1.4 | Soil sink | 33.0 [32.6, 33.7] | 0.98 [0.89, 1.03] | Jul | Jan, Feb |
| GFED v4.2 | Fires | 14.4 [11.3, 19.7] | 2.49 [1.07, 3.95] | Feb–Apr, Nov | Jun–Aug |
| Tsuruta et al. (2017) | Ocean | 7.72 [7.52, 8.1] | 0.08 [0.06, 0.1] | Feb, Apr–Jun, Nov | Jan, Mar, Jul, Aug, Oct, Nov |
| Etiope et al. (2019) | Geological | 5.0 | * | * | * |
| Ito and Inatomi (2012) | Termites | 20.8 [20.8, 20.9] | * | * | * |
| Station | Station Code | Country | Latitude | Longitude | Elevation (m a.s.l.) | Inatke Height (m a. g.) |
|---|---|---|---|---|---|---|
| Alert | ALT | Nunavut, Canada | 82.4508° N | 62.5072° W | 195 | 5 |
| Niwot Ridge | NWR | Colorado, USA | 40.0531° N | 105.5864° W | 3526 | 3 |
| South Pole | SPO | Antarctica | 89.98° S | 24.8° W | 2821.3 | 3–11.3 |
| Simulation | Anthropogenic Emission Fields | Removed Seasonal Cycle |
|---|---|---|
| SIM_E5 | EDGAR v5.0 | - |
| SIM_E5_WETNS | EDGAR v5.0 | Wetlands |
| SIM_NS | EDGAR v5.0 | All emissions |
| SIM_E432 | EDGAR v4.3.2 | - |
| SIM_E432_EFMMNS | EDGAR v4.3.2 | EFMM |
| Species | Station Code | Observation | SIM_E5 | SIM_NS | SIM_E432 | SIM_E432_EFMMNS |
|---|---|---|---|---|---|---|
| C (‰) | ALT | 0.45 | 0.29 | 0.26 | 0.22 | 0.26 |
| NWR | 0.26 | 0.15 | 0.15 | 0.16 | 0.14 | |
| SPO | 0.15 | 0.13 | 0.11 | 0.13 | 0.14 | |
| CH4 (ppb) | ALT | 50.1 | 45.2 | 62.4 | 43.2 | 42.3 |
| NWR | 33.7 | 39.1 | 50.5 | 50.9 | 45.9 | |
| SPO | 31.3 | 34.7 | 35.2 | 34.7 | 35.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kangasaho, V.; Tsuruta, A.; Backman, L.; Mäkinen, P.; Houweling, S.; Segers, A.; Krol, M.; Dlugokencky, E.J.; Michel, S.; White, J.W.C.; et al. The Role of Emission Sources and Atmospheric Sink in the Seasonal Cycle of CH4 and δ13-CH4: Analysis Based on the Atmospheric Chemistry Transport Model TM5. Atmosphere 2022, 13, 888. https://doi.org/10.3390/atmos13060888
Kangasaho V, Tsuruta A, Backman L, Mäkinen P, Houweling S, Segers A, Krol M, Dlugokencky EJ, Michel S, White JWC, et al. The Role of Emission Sources and Atmospheric Sink in the Seasonal Cycle of CH4 and δ13-CH4: Analysis Based on the Atmospheric Chemistry Transport Model TM5. Atmosphere. 2022; 13(6):888. https://doi.org/10.3390/atmos13060888
Chicago/Turabian StyleKangasaho, Vilma, Aki Tsuruta, Leif Backman, Pyry Mäkinen, Sander Houweling, Arjo Segers, Maarten Krol, Edward J. Dlugokencky, Sylvia Michel, James W. C. White, and et al. 2022. "The Role of Emission Sources and Atmospheric Sink in the Seasonal Cycle of CH4 and δ13-CH4: Analysis Based on the Atmospheric Chemistry Transport Model TM5" Atmosphere 13, no. 6: 888. https://doi.org/10.3390/atmos13060888