Abstract
Recent research on atmospheric particle formation has shown substantial discrepancies between observed and modeled atmospheric sulfate levels. This is because models mostly consider sulfate originating from oxidation by •OH radicals in mechanisms catalyzed by solar radiation while ignoring other pathways of non-radical oxidation that would substantially alter atmospheric sulfate levels. Herein, we use high-level quantum chemical calculations based on density functional theory and coupled cluster theory to show that monoethanolamine (MEA), a typical alkanolamine pollutant released from capture technology, can facilitate the conversion of atmospheric to sulfate in a non•OHradical oxidation mechanism. The initial process is the MEA-induced hydrolysis leading to the formation of •. The latter entity is thereafter oxidized by ozone () and nitrogen dioxide () to form •, which is an identified stabilizing entity in sulfate-based aerosol formation. Results show that the • reaction with is kinetically and thermodynamically more feasible than the reaction with . The presence of an additional water molecule further promotes the • reaction with , which occurs in a barrierless process, while it instead favors HONO formation in the reaction with . The investigated pathway highlights the potential role alkanolamines may play in oxidation to sulfate, especially under conditions that are not favorable for •OH production, thereby providing an alternative sulfate source for aerosol modeling. The studied mechanism is not only relevant to sulfate formation and may effectively compete with reactions with sulfur dioxide and hydroxyl radicals under heavily polluted and highly humid conditions such as haze events, but also an important pathway in MEA removal processes.
1. Introduction
Secondary atmospheric aerosol particles, formed from gas-to-particle conversion, are primarily composed of sulfate, which is one of the major water-soluble inorganic species in the atmosphere [1,2]. These particles are of great concern for their ability to affect human health, reduce visibility, acidify rainwater, and to alter the radiation balance of the atmosphere [3,4,5]. The main source of atmospheric sulfate is from the photooxidation of sulfur dioxide () [6] and although a large number of studies have connected the formation rate of secondary atmospheric aerosol particles to atmospheric sulfate concentration [7,8,9,10], numerical models still fail to reproduce observed atmospheric sulfate concentrations [11]. This has led to considerable debate regarding the mechanisms responsible for sulfate formation, especially during winter haze events where sunlight radiation is weakened [3,12,13]. The incomplete understanding of the sulfate formation mechanism substantially hinders the efficient prediction of haze events, climate change, air quality monitoring, and the development and implementation of measures to mitigate air pollution [14].
Recently, numerous researchers have focused on atmospheric sulfate formation mechanisms and, most importantly, on how they alter aerosol formation rates under severe pollution conditions. Sulfate is formed from the oxidation of sulfur dioxide (), primarily by hydroxyl radicals (•OH) in the gas phase and ozone (), nitrogen oxides (), hydrogen peroxide (), and catalyzed by transition metal ions in aqueous-phase and cloud droplets [15,16,17]. Multiphase oxidation of on solid or aqueous particles is considered a potentially important source of sulfate in the atmosphere as well [18]. Although various oxidation pathways have been identified, the sources of sulfate and the relative importance of the various oxidation pathways for sulfate formation in the atmosphere are yet to be clarified. For example, significant increases in sulfate concentrations were observed during haze events, but their sources remain elusive [19].
The primary gas-phase oxidant, •OH radical, is produced from excited oxygen and water under solar ultraviolet radiation. Despite the fast oxidation of by •OH that initially forms sulfur trioxide (), which is further hydrolyzed to form sulfate, many observations have indicated that there is insufficient •OH in the atmosphere to account for the generally observed increasing sulfate formation in polluted environments [12,20]. Such low solar ultraviolet radiation conditions are particularly pronounced during haze events. Although the abundance of other oxidants (, , ) is relatively high, their reactions with are generally hindered by high energy barriers. This suggests that important alternative pathways for atmospheric sulfate formation do exist, yet they have not been extensively investigated.
Atmospheric bases, including ammonia and amines, are other important components of secondary aerosols. Besides alkylamines such as methylamine and dimethylamine that have been identified as key species in sulfate aerosol formation [21], monoethanolamine (MEA, ), a typical alkanolamine, has been found to enhance new particle formation as well and to stabilize acidic particles [22,23]. MEA is the most widely used baseline solvent in amine-based post-combustion capture (PCC) technology [24,25]. Considering the potential large-scale implementation of amine-based PCC, relatively large amounts of MEA and possibly other alkanolamines may be emitted into the atmosphere from PCC units due to their relatively high vapor pressure [26,27]. It is estimated that nearly 80 tons of MEA may be released into the atmosphere from a capture unit that removes 1 million tons of per year [21,28] thereby increasing the environmental risk that MEA may potentially pose. Many studies have shown that atmospheric conversion can be facilitated by the degradation of amines despite the lack of proper mechanism and kinetics of the driving process [29,30]. Xie et al. showed that MEA can effectively cluster with sulfuric acid and significantly enhance new particle formation [23]. They showed that the removal rate of MEA due to this process was comparable to the rate of MEA oxidation by •OH at 217 K. Nevertheless, the complete role of MEA in environmental chemistry and aerosol chemistry remains unclear.
This study investigates the importance of direct reactions in sulfate aerosol formation in the absence of •OH radicals. Long-term measurements have continually revealed the presence of abundant ammonia and amines in ambient air, including in the marine environment [31,32], and their role has mainly been limited to stabilizing sulfate clusters. Herein, we have investigated sulfate formation from oxidation assisted by MEA using density functional theory calculations and kinetic modeling. The investigated oxidation assisted by MEA is not only a potential source for atmospheric sulfate, but also a removal pathway for MEA. The kinetics of the studied reaction were evaluated and its implication in sulfate aerosol modeling was assessed.
2. Methods
2.1. Quantum Chemical Calculations
Geometric optimizations of all stationary states of the studied reactions were conducted in Gaussian 09 using the ωB97X-D functional [33] in conjunction with the 6-311++G (3df,3pd) basis set. Vibrational frequency analysis and zero-point energies of ωB97XD/6-311++G (3df,3pd) structures were performed at the same level of theory under the harmonic oscillator-rigid rotor approximation at 298.15 K and 1 atm. This level of theory has been shown in previous studies to be sufficient for calculating the geometries and thermochemistry of atmospherically relevant systems [25,34,35,36,37,38]. Transition state configurations were determined using the synchronous transit quasi-Newton method [39] at the same level of theory, while single-point energy corrections on ωB97XD/6-311++G (3df,3pd) structures were performed at the DLPNO-CCSD(T)/aug-cc-pVTZ level of theory [40,41] using the ORCA 4.2.1 program package [42].
2.2. Reaction Kinetics Computation
The reaction rate constants were evaluated using the transition-state theory with the Wigner tunneling correction [43,44,45]. Previous studies have shown that the formation of a ternary complex from three separate reactants first proceeds through formation of a binary complex that interacts with the third species thereafter [46,47]. Given the high atmospheric concentration of water relative to those of other reactants, it is most likely that initial collisions between the three separate reactants (, and MEA) will preferably form the hydrates of and MEA prior to the formation of the ternary complex, ••. Based on these considerations, and • will be determinant in computing the total rate constant of the MEAassisted hydrolysis. According to our calculations (see Table 1), • is the most abundant binary complex among possible binary complexes and its interaction with is likely to lead the process of MEA mediated hydrolysis according to the following equation:
Assuming equilibrium between and reactants and based on the pseudo steady-state approximation, the overall rate of reaction (R1) can be written as:
where MEA•H2O•SO2 is the reaction rate constant written as
k1 is the collision frequency of and to form , k−1 is the decomposition rate constant of back to initial reactants, Keq is the equilibrium constant of formation of the reactant complex, kuni is the unimolecular rate constant of the reaction of the reactant complex to the product. The equilibrium constant is expressed as:
where ∆Geq is the Gibbs free energy change for the formation of the reactant complex, R is the gas constant, and T is the absolute temperature. kuni is expressed as
with being the activation Gibbs free energy change separating the reactant complex from the product state. is the tunnelling effect factor, given by Wigner tunnelling correction:
where h is the Planck constant, ν± the imaginary frequency of the transition state, kB is the Boltzmann constant, T is the absolute temperature.
3. Results and Discussion
3.1. MEAAssisted Hydrolysis
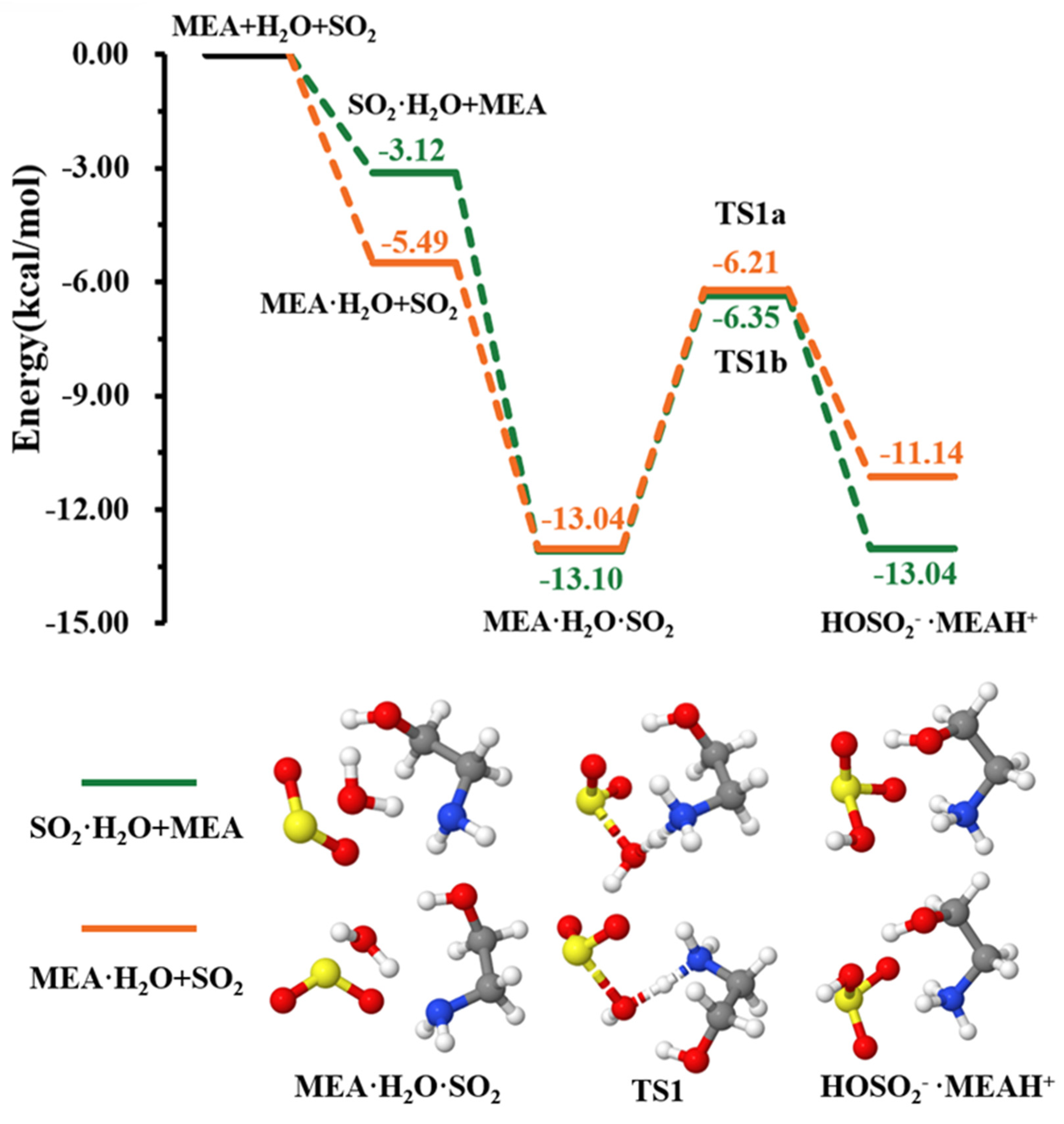
As explained above, the formation of the ternary complex would proceed through the prior formation of the binary complex, followed by interaction with the third species [46,47]. According to our calculations, the formation of and is more favorable than that of as can be seen from the equilibrium constants of their formation presented in Table 1. The two pathways of formation from + and + MEA interactions were readily assessed (see Figure 1). While the formation of is energetically more favorable (electronic energy change of −5.49 kcal mol−1 relative to precursor reactants) than (electronic energy change of −3.12 kcal mol−1 relative to precursor reactants) due to higher ability to form hydrogen bonds, is formed from both + and + MEA interactions with nearly identical electronic energy changes, −13.04 kcal mol−1 and −13.10 kcal mol−1, respectively. The formation Gibbs free energies of these isomers at 298.15 K and 1 atm are similar within 1.70 kcal mol−1, with the formation from + interaction being more favorable. Though both interactions will cumulatively form at standard atmospheric conditions, for simplicity, only the numerical results from + interaction are reported in Table 1.
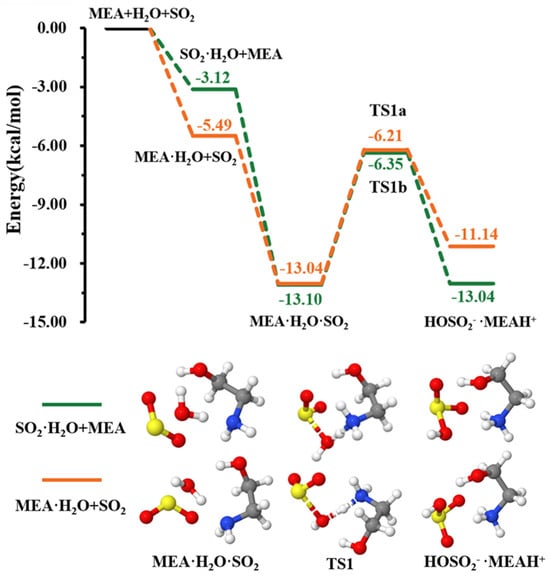
Figure 1.
Energy surface for the MEAcatalyzed hydrolysis. Color coding is yellow for sulfur atoms, red for oxygen atoms, white for hydrogen atoms, and blue for nitrogen atoms. Electronic energy values of all stationary states are indicated, and corresponding Gibbs-free energy values are presented in Table 1.

Table 1.
Electronic energy change (ΔE in kcal mol−1) and Gibbs free energy change (ΔG in kcal mol−1), equilibrium constants (Keq, cm3 molecule−1) of relevant reactions and equilibrium concentration (in molecule cm−3) for some binary complexes in the MEAassisted hydrolysis, all calculated at 298.15 K and 1 atm. These electronic energy changes are plotted in Figures 1 and 3–6.
Table 1.
Electronic energy change (ΔE in kcal mol−1) and Gibbs free energy change (ΔG in kcal mol−1), equilibrium constants (Keq, cm3 molecule−1) of relevant reactions and equilibrium concentration (in molecule cm−3) for some binary complexes in the MEAassisted hydrolysis, all calculated at 298.15 K and 1 atm. These electronic energy changes are plotted in Figures 1 and 3–6.
| ΔE | ΔG | Keq | Concentration | |
|---|---|---|---|---|
| −5.49 | 5.90 | 1.92 10−24 | 3.63 109 | |
| −3.12 | 4.94 | 9.85 10−24 | 7.66 106 | |
| −2.78 | 5.12 | 7.20 10−24 | 4.35 1012 | |
| −4.01 | 6.40 | 8.37 10−25 | 2.03 103 | |
| −13.04 | 8.98 | |||
| TS1a | −6.21 | 16.86 | ||
| • | −11.14 | 15.77 | ||
| • | −37.05 | −3.61 | ||
| TS5 | −16.99 | −1.29 | ||
| • (PD1) | −75.84 | −5.85 | ||
| TS6 | −67.21 | −2.78 | ||
| • | −84.71 | −0.76 | ||
| • | −2.76 | 5.79 | ||
| TS3 | 22.82 | 37.96 | ||
| • | −34.15 | −3.42 | ||
| With an additional water molecule | ||||
| −13.14 | 9.67 | |||
| −18.28 | 15.41 | |||
| TS2 | −14.61 | 18.09 | ||
| −23.35 | 15.47 | |||
| ++ | −93.89 | −6.83 | ||
| −15.74 | 5.43 | |||
| TS4a | 30.35 | 51.96 | ||
| −46.46 | −23.30 | |||
| TS4b | 11.24 | 33.33 | ||
| −15.24 | 9.19 | |||
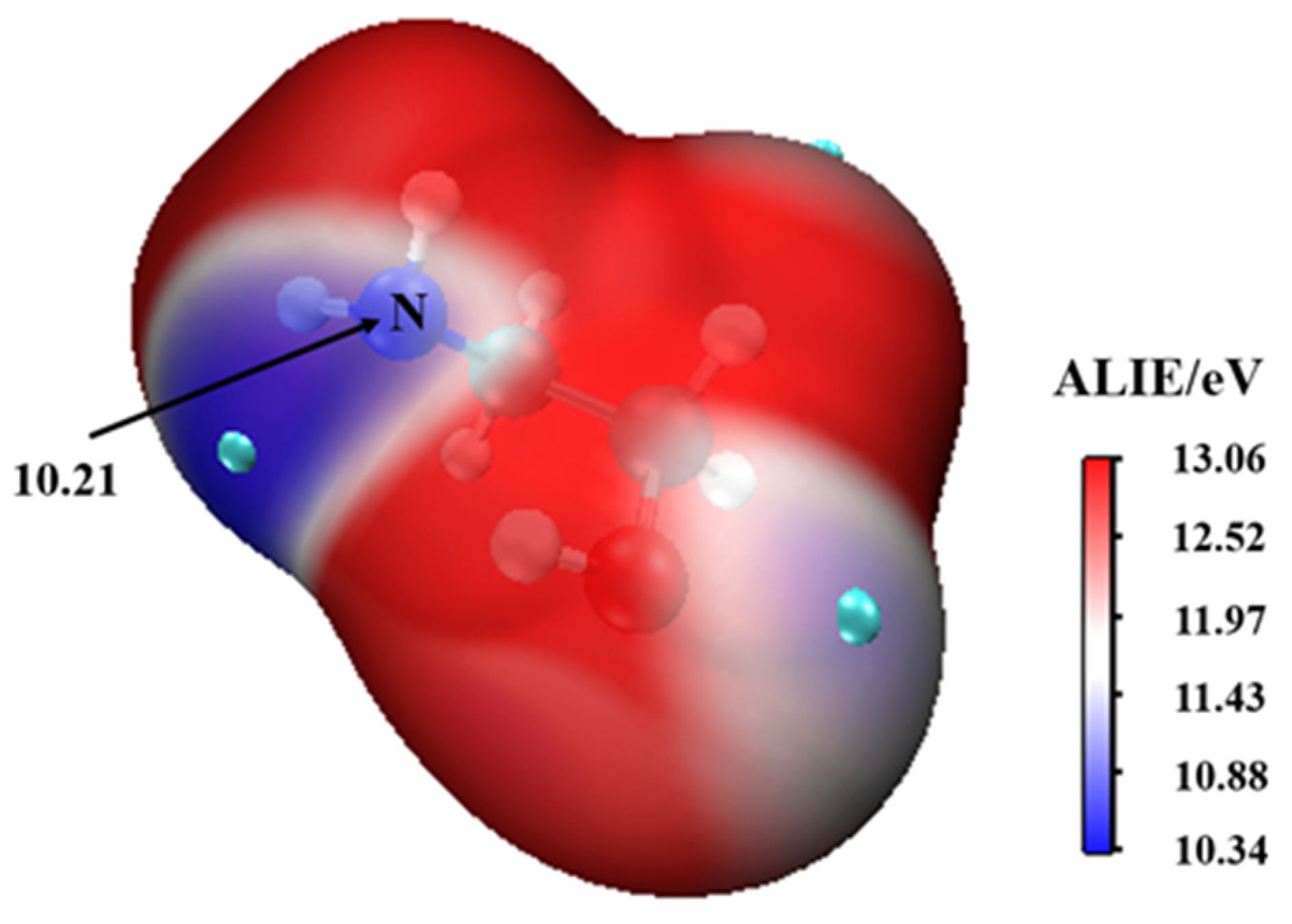
This complex rearranges through a transition state configuration located at −6.21 kcal mol−1 below separate reactants, to form •. As expected from the average local ionization energy mapped van der Waals surface shown in Figure 2, the nitrogen atom is the reactive site of MEA in this process. This is the site with the most deficient potential, which is hence the most susceptible to undergo an electrophilic addition.
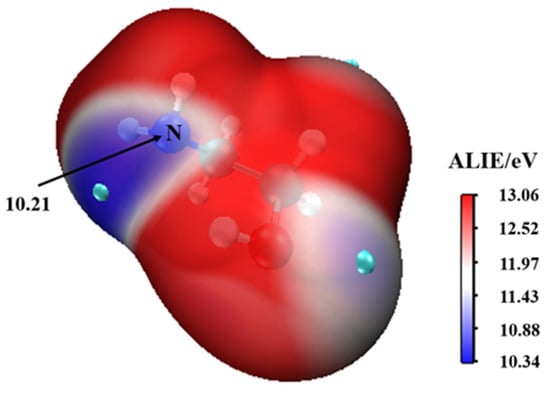
Figure 2.
The average local ionization energy (ALIE/eV) of MEA.
The electronic energy barrier height in formation is 6.83 kcal mol−1, much lower than the 33.9 kcal mol−1 energy barrier in the + → reaction, as reported in a much earlier study [48]. A previous quantum chemical study showed that ammonia can lower the barrier height of hydrolysis to ~12.0 kcal mol−1 [49]; however, it is still higher than that reported for the MEAassisted hydrolysis in the current study. Much lower barriers were determined in hydrolysis assisted by methylamine (5.80 kcal mol−1) and dimethylamine (3.17 kcal mol−1) [50]. Despite little differences that may arise as the result of different theoretical methods, the decreasing effect of ammonia, MEA, methylamine, and dimethylamine on the energy barrier in hydrolysis is in line with the order of their basicity [51]. This further confirms the stronger ability of amines, compared to ammonia, to promote hydrolysis.
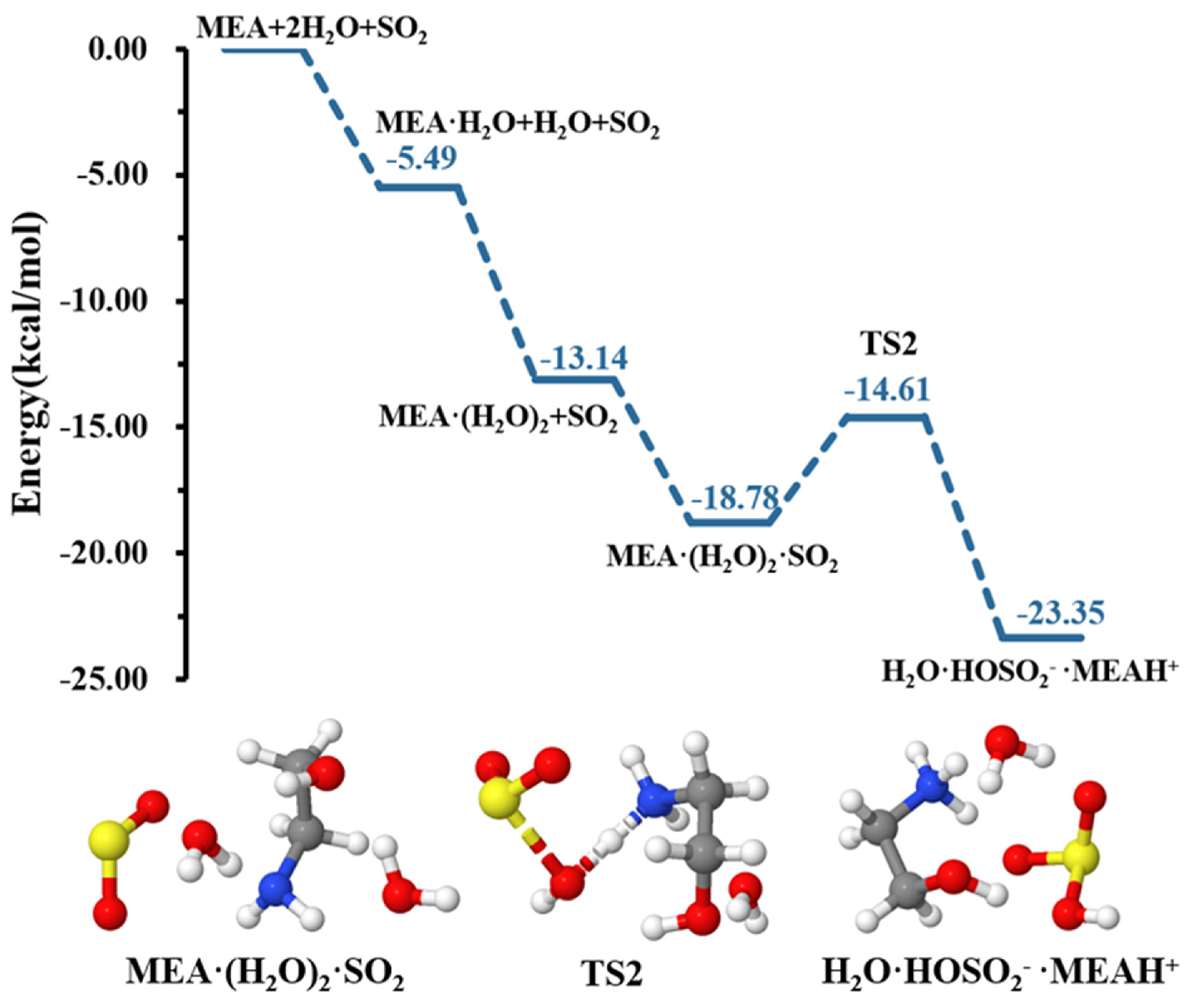
Our calculations indicate that although a second water molecule did not explicitly participate in the reaction, it contributed to further reducing the energy barrier height for formation down to 4.18 kcal mol−1, rendering the hydrolysis more favorable, as shown in the energy profile of Figure 3. Similar effects of water in decreasing energy barrier heights in chemical processes have been observed in several previous studies [49,52,53,54].
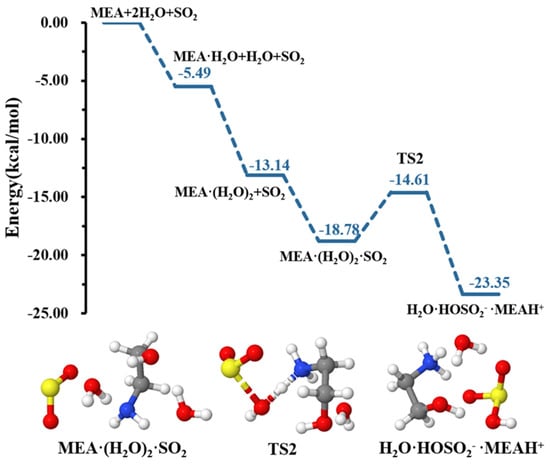
Figure 3.
Energy surface for the MEAcatalyzed hydrolysis with the presence of an additional water molecule. Color coding is yellow for sulfur atoms, red for oxygen atoms, white for hydrogen atoms, and blue for nitrogen atoms. Electronic energy values of all stationary states are indicated, and corresponding Gibbs-free energy values are presented in Table 1.
3.2. Further Reaction with
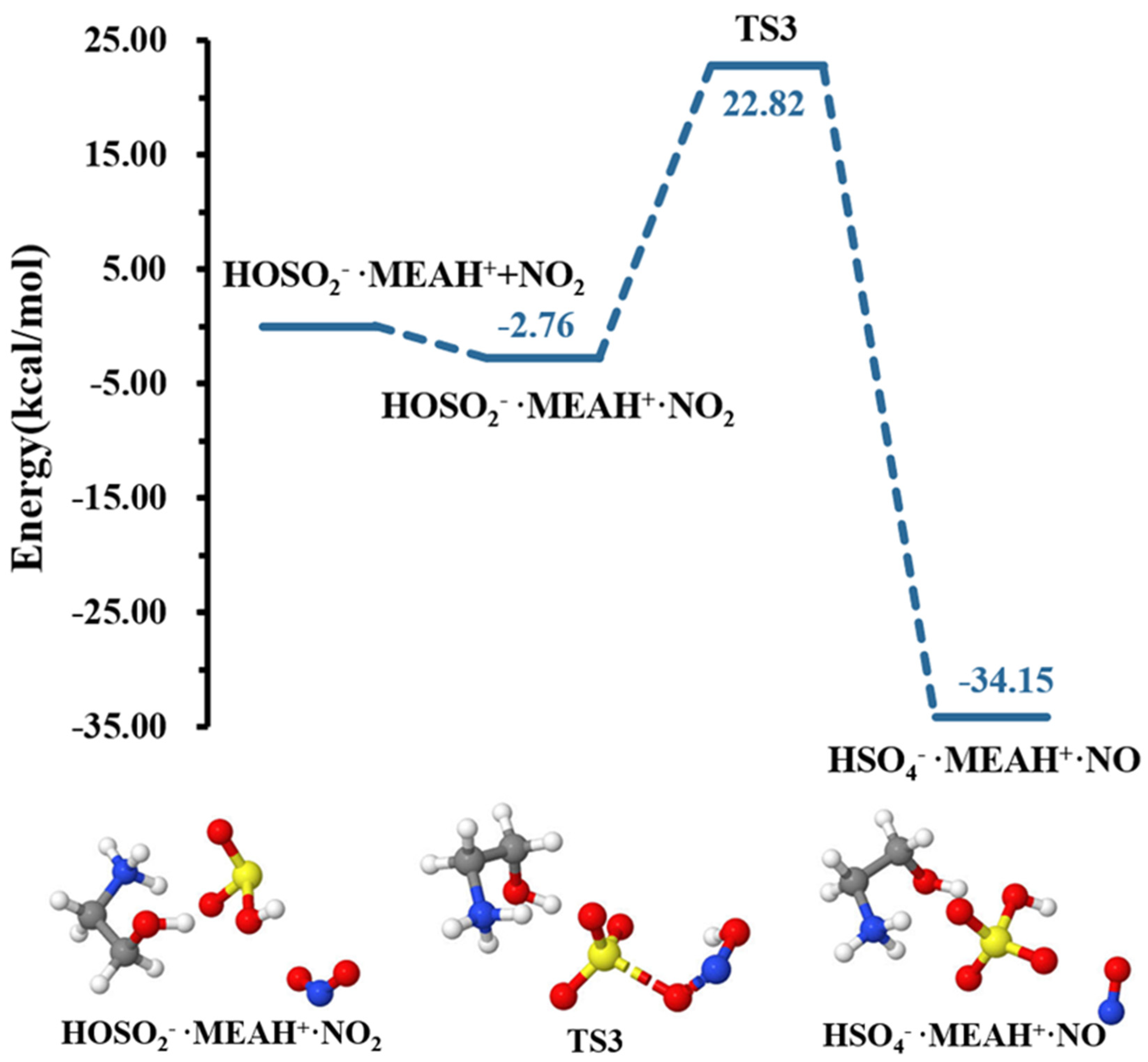
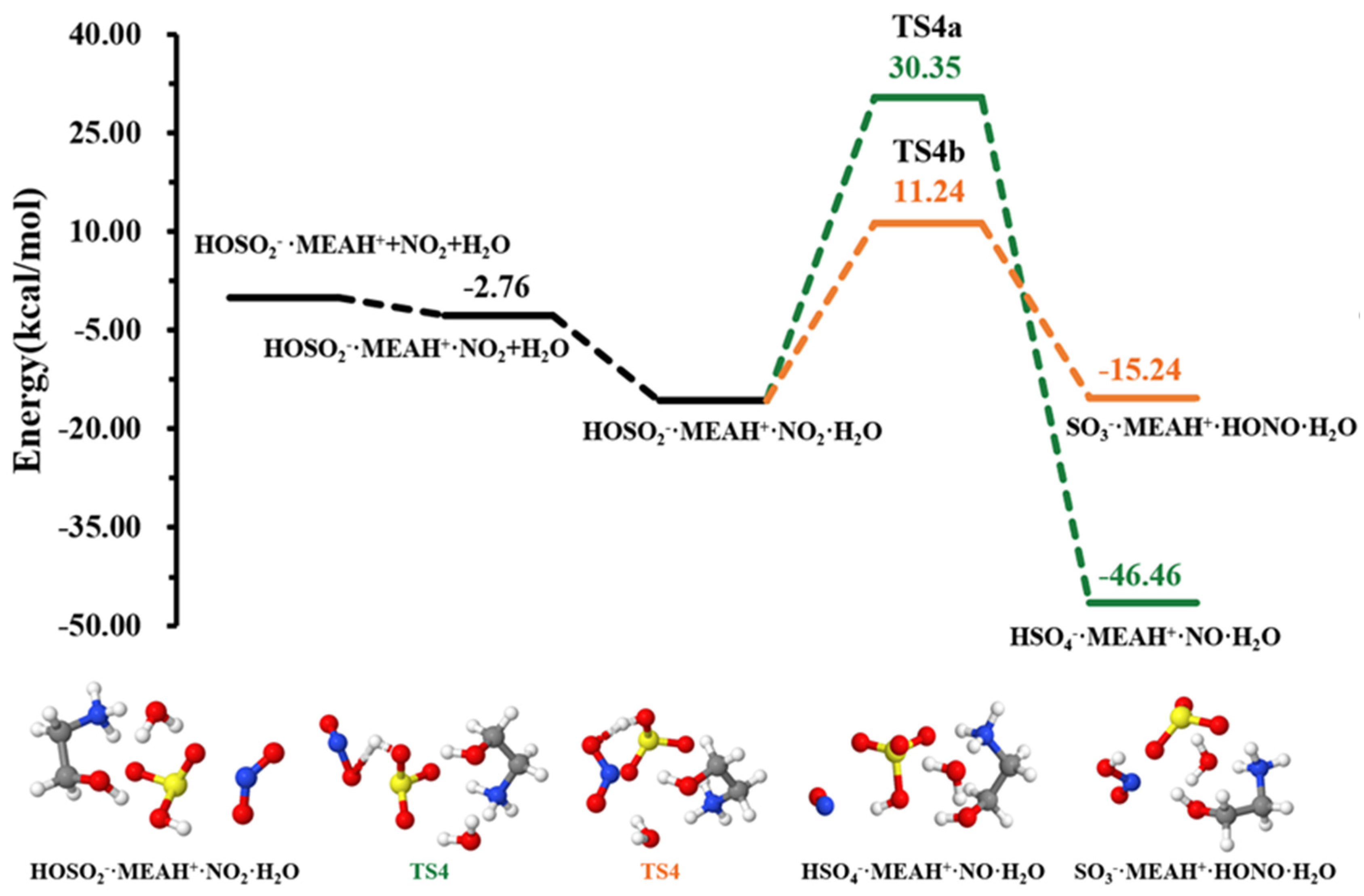
The sulfite ion in is susceptible to react with relevant atmospheric oxidants. For example, it can be oxidized by to form the (reaction (R4)). The different steps in this process are the formation of an intermediate complex, lying at −2.76 kcal mol−1, and overcoming a relatively high energy barrier prior to formation (see Figure 4). The energy barrier in this process is 25.58 kcal mol−1, which drastically increases to 46.09 kcal mol−1 in the presence of an additional water molecule (reaction (R5), Figure 5). This substantial increase in the energy barrier height is the result of a strong stability of the reactant complex due to additional water that facilitates the formation of a tighter ring structure than without water. Even though this process is highly exergonic (with −23.42 kcal mol−1 free energy change at 298.15 K and 1 atm), the high energy barrier would prevent this reaction at standard conditions. Moreover, the formation of nitrous oxide (HONO) in the product complex was observed to be a potential product in decomposition in the presence of an additional water molecule (reaction (R6)).
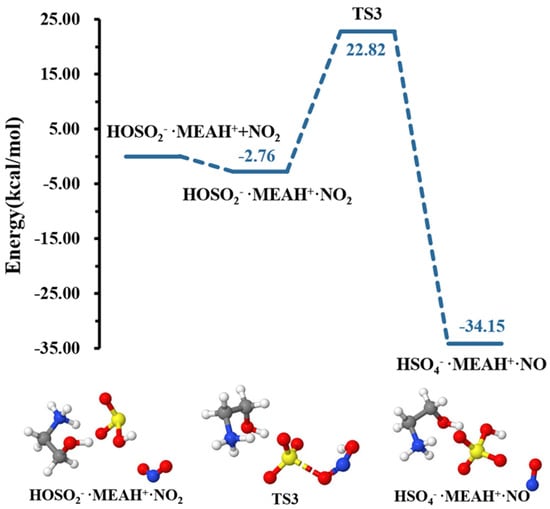
Figure 4.
Potential energy profile for the oxidation reaction of • and . Color coding is yellow for sulfur atoms, red for oxygen atoms, white for hydrogen atoms, and blue for nitrogen atoms. Electronic energy values of all stationary states are indicated, and corresponding Gibbs-free energy values are presented in Table 1.
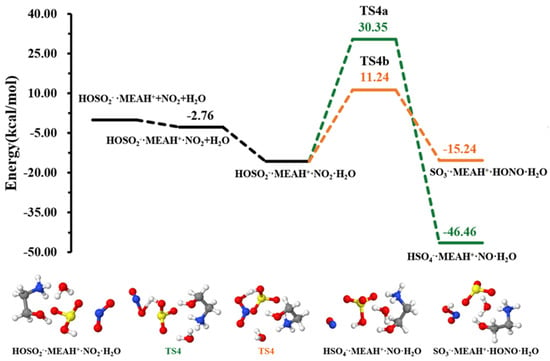
Figure 5.
Potential energy profiles for the oxidation reaction for • and , with additional . Color coding is yellow for sulfur atoms, red for oxygen atoms, white for hydrogen atoms, and blue for nitrogen atoms. Electronic energy values of all stationary states are indicated, and corresponding Gibbs-free energy values are presented in Table 1.
The energy barrier in this decomposition is 26.97 kcal mol−1, being 19.11 kcal mol−1 lower than the barrier in formation. Despite the somewhat more favorable formation of HONO than from oxidation by in the presence of water, the overall rate of HONO formation would be limited given the relatively high energy barrier to its formation. A similar conclusion was observed by Wang et al. while studying the hydrolysis assisted by methylamine and dimethylamine [50]. The fate of would then depends on other oxidants, such as .
3.3. Further Reaction with
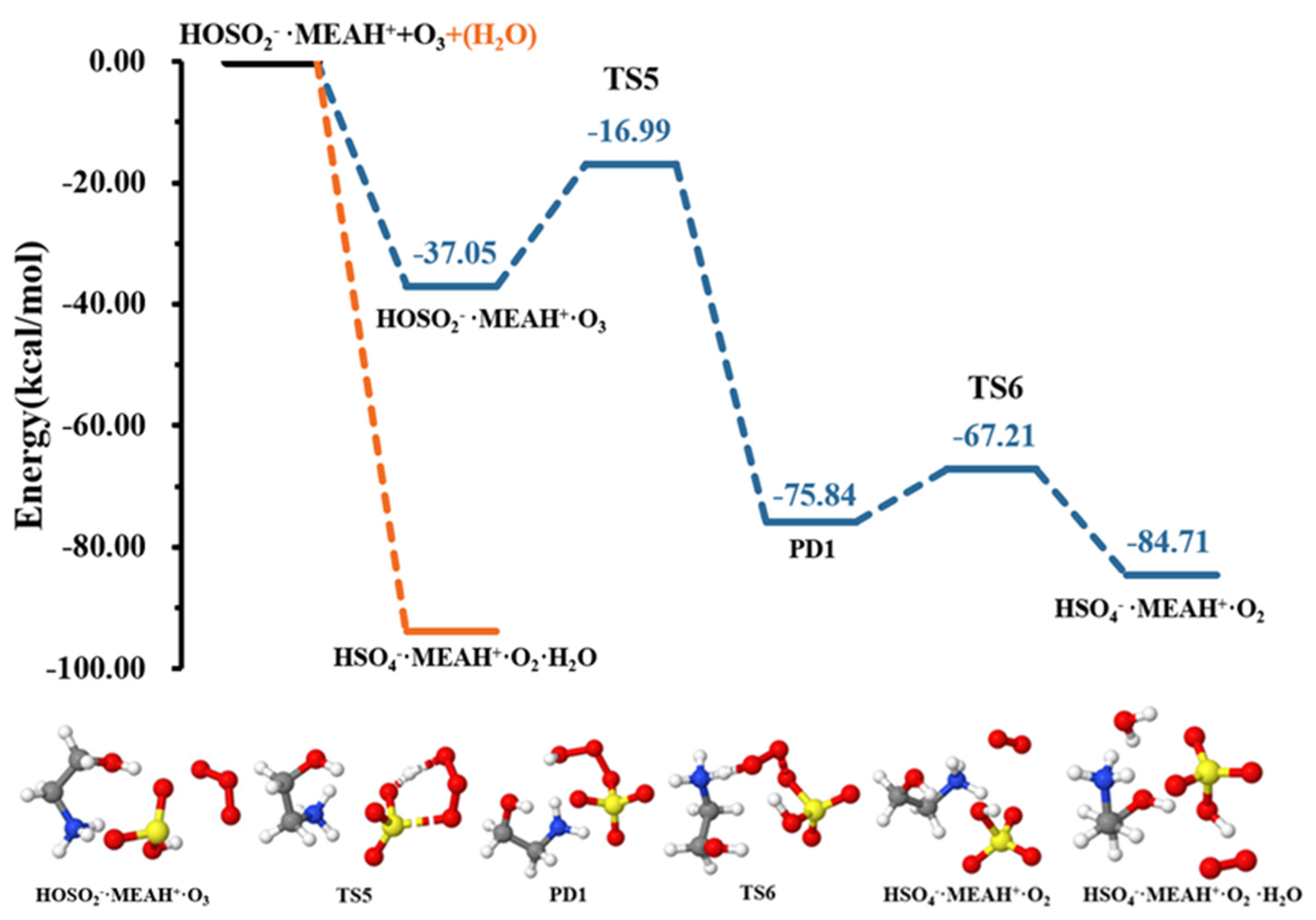
The reaction of with proceeds through the formation of the intermediate complex (reaction (R7)). Thereafter, a double intramolecular transfer occurs within the complex: O transfer from to to form and H transfer from fragment to fragment, resulting in the formation of , as shown in Figure 6. These two processes were separated by energy barriers of 20.05 and 8.63 kcal mol−1, respectively. Although the H transfer is relatively easy, i.e., characterized by a relatively low barrier, the O transfer constitutes the limiting step in formation. Compared to the reaction with , the conversion of by reaction with is kinetically and thermodynamically more favorable, especially given the relatively low energy barrier and the substantial energy gain in the later process. It is well-known that molecular oxygen can adopt singlet and triplet spin configurations. Our attempts to compute the reaction with on the triplet surface did not converge to . Hence, the calculations in this process were performed on the singlet surface, exclusively. It is then obvious that although the molecule in would initially form in the singlet state, collision with other atmospheric species will quickly convert it to the triplet state, the most stable state of molecular oxygen in the atmosphere.
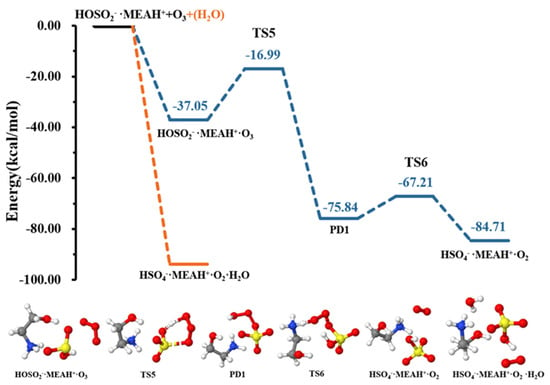
Figure 6.
Potential energy profiles for the oxidation reaction for • and (with additional ). Color coding is yellow for sulfur atoms, red for oxygen atoms, white for hydrogen atoms, and blue for nitrogen atoms. Electronic energy values of all stationary states are indicated, and corresponding Gibbs-free energy values are presented in Table 1.
Contrary to the effect of water in the + , the presence of additional water molecules significantly promotes the conversion of to by reaction with , which becomes barrierless (see Figure 6). This effect of additional water is contrary to that observed in the reaction with . The effect of water in promoting oxidation reactions was also observed in our previous studies [52]. It is speculated that different reactive behaviors of MEAassisted hydrolysis towards and compared to methylamine-assisted and dimethylamine-assisted hydrolysis [51] can be attributed to the electronic effect of the •OH group in MEA.
3.4. Kinetics of MEAAssisted Hydrolysis and Implications for Atmospheric Sulfate Formation
As introduced in Section 2.2, the rate constant of MEAassisted hydrolysis is determined by considering the formation of the pre-reactive complex from + and + MEA interactions, given the high atmospheric concentration of water relative to those of other reactants. This is further justified by the values of equilibrium constants and equilibrium concentrations (presented in Table 1) of the three possible binary complexes susceptible to form from the interactions between MEA, and . Assuming [] ~1012 molecule cm−3, [MEA] ~2.43 × 1015 molecule cm−3 and [] ~7.77 × 1017 molecule cm−3 corresponding to highly polluted conditions [29,55,56,57], has the highest equilibrium concentration among all the binary complexes. Hence, we determined the bimolecular rate constant of the MEAassisted hydrolysis based on the + interaction to be 1.90 × 10−14 cm3 molecule−1 s−1 at 298.15 K. All reaction rate constants at this temperature are presented in Table 2, while the positive temperature-dependency of the rate constant is plotted in Figure 7. Taking into account the equilibrium concentration of , which is equal to 3.63 × 109 cm3 molecule−1 as provided in Table 1, and the concentration given above, we obtain a reaction rate (calculated according to Equation (1) of 6.89 × 107 molecule cm−3 s−1 for the hydrolysis assisted by MEA at 300 K. This rate is one order of magnitude higher than the estimated rate of oxidation by •OH determined to be 1.50 × 106 molecule cm−3 s−1 at the same temperature [57], considering an average •OH concentration of ~106 molecule cm−3 during daytime and 1.30 × 10−12 cm3 molecule −1 s−1 rate constant of oxidation by •OH.
Table 2.
Unimolecular (kuni, s−1) and bimolecular (kbimol, cm3 molecule−1s−1) rate constants in the MEAassisted oxidation, all calculated at 298.15 K and 1 atm.
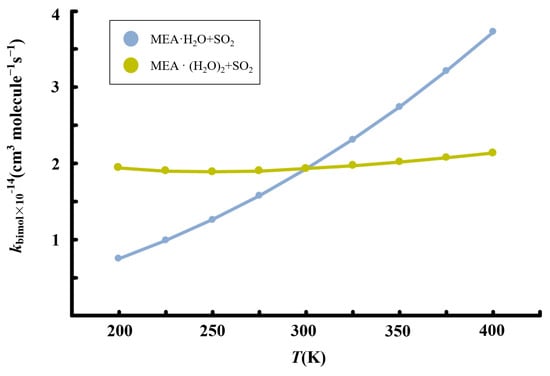
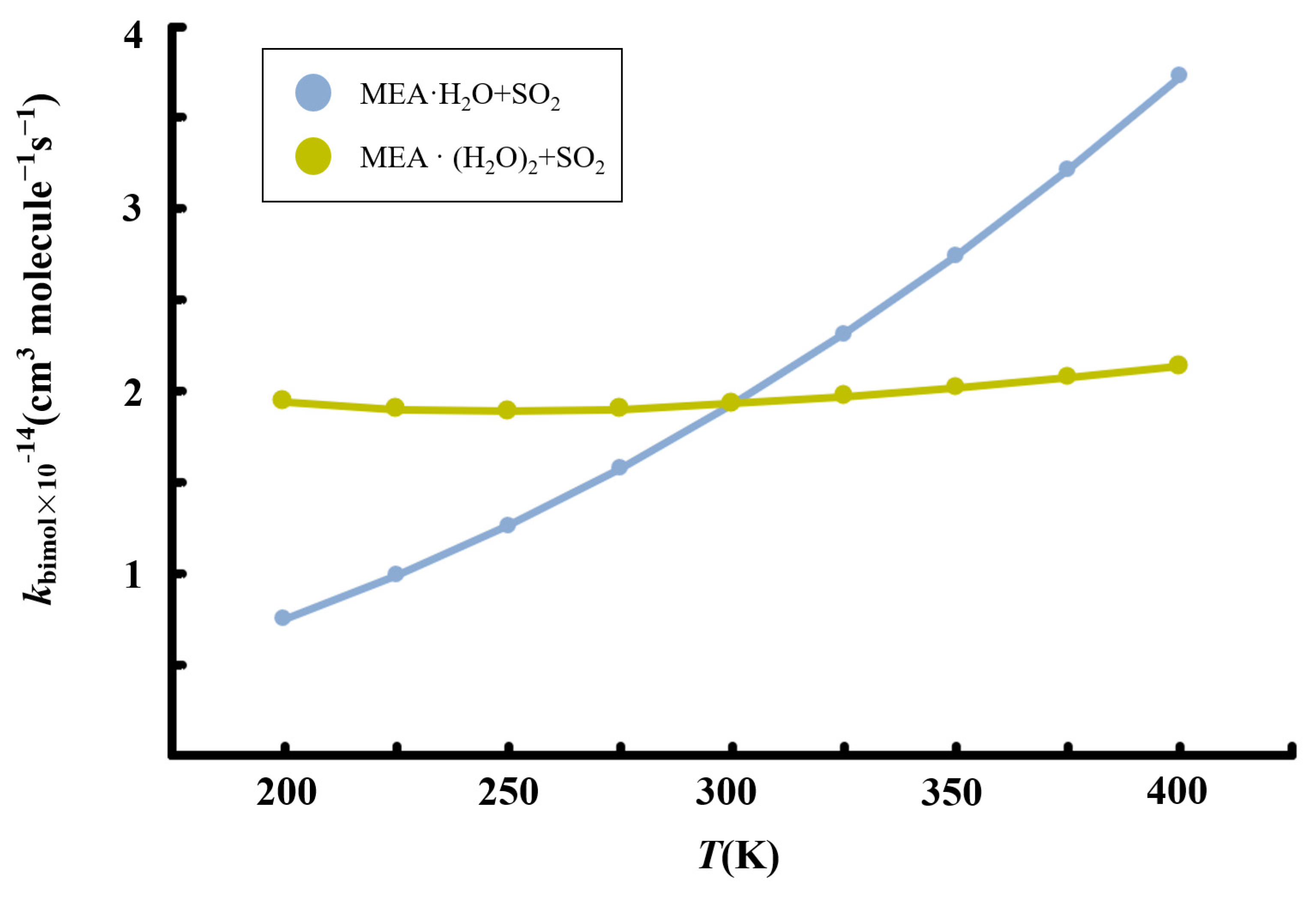
Figure 7.
Calculated temperature-dependent rate constants for the MEA + + reaction at 1 atm.
Although the presence of additional water molecules increases the unimolecular rate constant of hydrated • formation by two orders of magnitude relative to the reaction with one water molecule less, the overall effect on the bimolecular rate constant is reduced by the weaker binding between and to form the reactant complex, . For this reaction, we obtain a rate constant of 1.93 × 10−14 cm3 molecule −1 s−1 at 298.15 K. Similar to the reaction without additional water, the rate constant of the reaction in the presence of additional water exhibits a positive temperature-dependent variation in the range 200–400 K, though the effect is significantly weak. Considering [] ~4.35 × 1012 molecule cm−3 according to our calculations, the reaction rate of SO2 hydrolysis assisted by MEA with additional water was estimated to be 3.88 × 103 molecule cm−3 s−1, three order of magnitude lower than the estimated rate of oxidation by •OH reported previously [57].
Given the high rate of MEAassisted hydrolysis, it is evident that under extremely polluted conditions such as during haze events where there is insufficient •OH production due to low ultraviolet solar radiation, the investigated pathway might be a highly competitive process in atmospheric oxidation.
Further exploration of the chemistry of • shows that oxidation by and to form • occurs with unimolecular rate constants of 1.29 × 10−6 s−1 and 1.35 × 10−2 s−1, respectively. With the presence of additional water molecules, • decomposition by to form • is essentially barrierless, whereas decomposition by is further prevented by a high energy barrier. For the reaction with in the presence of water, instead, the decomposition of • to • is slightly preferred. The relevance of • can be found both in •OH production through HONO and in sulfate formation through . • is formed at a unimolecular rate constant of 2.41 × 10−7 s−1; however, this is still much lower than the rate constant of the reaction with . This indicates that the reaction with is the most likely process for • oxidation leading to sulfate formation in the gas phase.
Besides its fate in sulfate formation, the studied mechanism could also be an efficient removal pathway for MEA, which is believed to potentially represent an environmental risk [56,58]. So far, the reported sink processes for MEA include gas-phase oxidation by •OH and •Cl, and reactive uptake by sulfuric acid molecules [22,59,60]. Rate constants of 7.10 × 10−11 cm3 molecule −1 s−1 and ~10−10 cm3 molecule −1 s−1 were reported for MEA oxidation by •OH and •Cl, respectively [59,60]. With average •OH concentrations of ~106 molecule cm−3 during daytime and •Cl concentration of ~105 molecule cm−3, the estimated rates of MEA+•OH (1.73 × 1011 molecule cm−3 s−1) and MEA + •Cl (2.43 × 1010 molecule cm−3 s−1) are respectively 5 × 103 and 7 × 102 times higher than the rate of + reaction. Under highly polluted conditions with elevated concentrations of MEA and low •OH concentration induced by low solar radiation, the mechanism reported in this study may also be a competitive pathway in MEA removal processes.
4. Conclusions
Sulfate formation was investigated from MEAassisted oxidation using first-principles simulations. Results indicate that decomposition of the direct hydrolysis product, •, by is more favored than by . The presence of additional water was found to play a varied role in the further oxidation process of •, significantly facilitating the decomposition by while substantially preventing the reaction with . Besides sulfate formation mechanisms including ion-mediated and acid-catalyzed mechanisms that have already been elucidated in some previous studies, the current mechanism is an alternative pathway for sulfate formation and can be used to explain elevated sulfate formation observed under severe haze events where there is insufficient •OH production for oxidation. Moreover, the presence of water introduces an additional feature, i.e., the HONO formation, in the reaction with , though at a relatively lower rate. This latter mechanism might be a potential source for •OH under low ultraviolet solar radiation. We found that the title reaction can significantly outperform + •OH reaction under low •OH conditions, providing new pathways for sulfate formation that would prevail under conditions of heavy pollution, high humidity, and low solar radiation, such as during haze events. The MEAassisted oxidation would also potentially compete with the •OH oxidation pathway in MEA removal processes under such conditions. This study highlights the role of alkanolamines in oxidation, with implication in sulfate aerosol formation.
Author Contributions
Conceptualization, N.T.T.; methodology, N.T.T. and X.L.; formal analysis, X.L., N.T.T., S.N.T. and J.N.G.; investigation, X.L.; data curation, X.L. and N.T.T.; writing—original draft preparation, X.L.; writing—review and editing, M.L., S.N.T., J.N.G. and N.T.T.; supervision, L.D. and N.T.T.; funding acquisition, L.D. and N.T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key Research and Development Program of China (2023YFC3706203), National Natural Science Foundation of China (22376121), and Natural Science Foundation of Shandong Province (ZR2023MD041).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding authors. The data are not publicly available due to data sharing policy.
Acknowledgments
The authors acknowledge support from the Wuxi Hengding Supercomputing Center Co., Ltd.
Conflicts of Interest
The authors declare no conflicts of interests.
References
- Hu, M.; Wu, Z.; Slanina, J.; Lin, P.; Liu, S.; Zeng, L. Acidic gases, ammonia and water-soluble ions in PM2.5 at a coastal site in the Pearl River Delta, China. Atmos. Environ. 2008, 42, 6310–6320. [Google Scholar] [CrossRef]
- Lai, S.C.; Zou, S.C.; Cao, J.J.; Lee, S.C.; Ho, K.F. Characterizing ionic species in PM2.5 and PM10 in four pearl river delta cities, south china. J. Environ. Sci. 2007, 19, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zheng, G.; Wei, C.; Mu, Q.; Zheng, B.; Wang, Z.; Gao, M.; Zhang, Q.; He, K.; Carmichael, G.; et al. Reactive nitrogen chemistry in aerosol water as a source of sulfate during haze events in China. Sci. Adv. 2016, 2, e1601530. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study 2015. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chan, A.W.H.; Abbatt, J.P.D. Multiphase oxidation of sulfur dioxide in aerosol particles: Implications for sulfate formation in polluted environments. Environ. Sci. Technol. 2021, 55, 4227–4242. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, Y.; Lin, Y.; Li, J.; Cheng, H.; An, N.; Sun, Y.; Qiu, Y.; Cao, F.; Fu, P. Roles of Sulfur Oxidation Pathways in the Variability in Stable Sulfur Isotopic Composition of Sulfate Aerosols at an Urban Site in Beijing, China. Environ. Sci. Technol. Lett. 2020, 7, 883–888. [Google Scholar] [CrossRef]
- Zheng, B.; Zhang, Q.; Zhang, Y.; He, K.B.; Wang, K.; Zheng, G.J.; Duan, F.K.; Ma, Y.L.; Kimoto, T. Heterogeneous chemistry: A mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China. Atmos. Chem. Phys. 2015, 15, 2031–2049. [Google Scholar] [CrossRef]
- Li, Y.; Han, Z.; Song, Y.; Li, J.; Sun, Y.; Wang, T. Impacts of the COVID-19 lockdown on atmospheric oxidizing capacity and secondary aerosol formation over the Beijing-Tianjin-Hebei region in Winter-Spring 2020. Atmos. Environ. 2023, 295, 119540. [Google Scholar] [CrossRef]
- Wang, J.; Xing, J.; Wang, S.; Mathur, R.; Wang, J.; Zhang, Y.; Liu, C.; Pleim, J.; Ding, D.; Chang, X.; et al. The pathway of impacts of aerosol direct effects on secondary inorganic aerosol formation. Atmos. Chem. Phys. 2022, 22, 5147–5156. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Liu, H.; Lv, W.; Aikawa, M.; Liu, B.; Wang, J. Pollution sources of atmospheric fine particles and secondary aerosol characteristics in Beijing. J. Environ. Sci. 2020, 95, 91–98. [Google Scholar] [CrossRef]
- Shao, J.; Chen, Q.; Wang, Y.; Lu, X.; He, P.; Sun, Y.; Shah, V.; Martin, R.V.; Philip, S.; Song, S.; et al. Heterogeneous sulfate aerosol formation mechanisms during wintertime Chinese haze events: Air quality model assessment using observations of sulfate oxygen isotopes in Beijing. Atmos. Chem. Phys. 2019, 19, 6107–6123. [Google Scholar] [CrossRef]
- Gen, M.; Zhang, R.; Huang, D.D.; Li, Y.; Chan, C.K. Heterogeneous SO2 Oxidation in Sulfate Formation by Photolysis of Particulate Nitrate. Environ. Sci. Technol. Lett. 2019, 6, 86–91. [Google Scholar] [CrossRef]
- Xue, J.; Yu, X.; Yuan, Z.; Griffith, S.M.; Lau, A.K.H.; Seinfeld, J.H.; Yu, J.Z. Efficient control of atmospheric sulfate production based on three formation regimes. Nat. Geosci. 2019, 12, 977–982. [Google Scholar] [CrossRef]
- Itahashi, S.; Hattori, S.; Ito, A.; Sadanaga, Y.; Yoshida, N.; Matsuki, A. Role of Dust and Iron Solubility in Sulfate Formation during the Long-Range Transport in East Asia Evidenced by 17O-Excess Signatures. Environ. Sci. Technol. 2022, 56, 13634–13643. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.; Allman, D.J.; Amos, H.M.; Fairlie, T.D.; Dachs, J.; Hegg, D.A.; Sletten, R.S. Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean. J. Geophys. Res. Atmos. 2012, 117, D06304. [Google Scholar] [CrossRef]
- Chen, Q.; Sherwen, T.; Evans, M.; Alexander, B. DMS oxidation and sulfur aerosol formation in the marine troposphere: A focus on reactive halogen and multiphase chemistry. Atmos. Chem. Phys. 2018, 18, 13617–13637. [Google Scholar] [CrossRef]
- Harris, E.; Sinha, B.; van Pinxteren, D.; Tilgner, A.; Fomba, K.W.; Schneider, J.; Roth, A.; Gnauk, T.; Fahlbusch, B.; Mertes, S.; et al. Enhanced role of transition metal ion catalysis during in-cloud oxidation of SO2. Science 2013, 340, 727–730. [Google Scholar] [CrossRef]
- Zhao, D.; Song, X.; Zhu, T.; Zhang, Z.; Liu, Y.; Shang, J. Multiphase oxidation of SO2 by NO2 on CaCO3 particles. Atmos. Chem. Phys. 2018, 18, 2481–2493. [Google Scholar] [CrossRef]
- Gao, J.; Shi, G.; Zhang, Z.; Wei, Y.; Tian, X.; Feng, Y.; Russell, A.G.; Nenes, A. Targeting Atmospheric Oxidants Can Better Reduce Sulfate Aerosol in China: H2O2 Aqueous Oxidation Pathway Dominates Sulfate Formation in Haze. Environ. Sci. Technol. 2022, 56, 10608–10618. [Google Scholar] [CrossRef]
- Liu, T.; Clegg, S.L.; Abbatt, J.P.D. Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles. Proc. Natl. Acad. Sci. USA 2020, 117, 1354–1359. [Google Scholar] [CrossRef]
- Karl, M.; Wright, R.F.; Berglen, T.F.; Denby, B. Worst case scenario study to assess the environmental impact of amine emissions from a CO2 capture plant. Int. J. Greenh. Gas Control 2011, 5, 439–447. [Google Scholar] [CrossRef]
- Tian, X.; Chu, Y.; Chan, C.K. Reactive Uptake of Monoethanolamine by Sulfuric Acid Particles and Hygroscopicity of Monoethanolaminium Salts. Environ. Sci. Technol. Lett. 2022, 9, 16–21. [Google Scholar] [CrossRef]
- Xie, H.; Elm, J.; Halonen, R.; Myllys, N.; Kurtén, T.; Kulmala, M.; Vehkamäki, H. Atmospheric Fate of Monoethanolamine: Enhancing New Particle Formation of Sulfuric Acid as an Important Removal Process. Environ. Sci. Technol. 2017, 51, 8422–8431. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Han, H.; Li, W.; Ma, X.; Xu, L. Experimental study on the absorption enhancement of CO2 by MDEA-MEA based nanofluids. Can. J. Chem. Eng. 2022, 100, 3335–3344. [Google Scholar] [CrossRef]
- Zhang, T.; Yu, Y.; Zhang, Z. Effects of non-aqueous solvents on CO2 absorption in monoethanolamine: Ab initio calculations. Mol. Simul. 2018, 44, 815–825. [Google Scholar] [CrossRef]
- Feron, P.; Conway, W.; Puxty, G.; Wardhaugh, L.; Green, P.; Maher, D.; Fernandes, D.; Cousins, A.; Shiwang, G.; Lianbo, L.; et al. Amine Based Post-Combustion Capture Technology Advancement for Application in Chinese Coal Fired Power Stations. Energy Procedia 2014, 63, 1399–1406. [Google Scholar] [CrossRef]
- Reynolds, A.J.; Verheyen, T.V.; Adeloju, S.B.; Chaffee, A.L.; Meuleman, E. Primary sources and accumulation rates of inorganic anions and dissolved metals in a MEA absorbent during PCC at a brown coal-fired power station. Int. J. Greenh. Gas Control 2015, 41, 239–248. [Google Scholar] [CrossRef]
- Veltman, K.; Singh, B.; Hertwich, E.G. Human and Environmental Impact Assessment of Postcombustion CO2 Capture Focusing on Emissions from Amine-Based Scrubbing Solvents to Air. Environ. Sci. Technol. 2010, 44, 1496–1502. [Google Scholar] [CrossRef]
- Miller, D.D.; Chuang, S.S.C. Experimental and Theoretical Investigation of SO2 Adsorption over the 1,3-Phenylenediamine/SiO2 System. J. Phys. Chem. C 2015, 119, 6713–6727. [Google Scholar] [CrossRef]
- You, Y.; Kanawade, V.P.; de Gouw, J.A.; Guenther, A.B.; Madronich, S.; Sierra-Hernández, M.R.; Lawler, M.; Smith, J.N.; Takahama, S.; Ruggeri, G.; et al. Atmospheric amines and ammonia measured with a chemical ionization mass spectrometer (CIMS). Atmos. Chem. Phys. 2014, 14, 12181–12194. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, D.; Shen, Y.; Gao, Y.; Gao, H.; Yao, X. Mapping gaseous dimethylamine, trimethylamine, ammonia, and their particulate counterparts in marine atmospheres of China’s marginal seas—Part 2: Spatiotemporal heterogeneity, causes, and hypothesis. Atmos. Chem. Phys. 2022, 22, 1515–1528. [Google Scholar] [CrossRef]
- Zhong, Q.E.; Cheng, C.; Wang, Z.; Li, L.; Li, M.; Ge, D.; Wang, L.; Li, Y.; Nie, W.; Chi, X.; et al. Diverse mixing states of amine-containing single particles in Nanjing, China. Atmos. Chem. Phys. 2021, 21, 17953–17967. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Elm, J.; Jen, C.N.; Kurtén, T.; Vehkamäki, H. Strong Hydrogen Bonded Molecular Interactions between Atmospheric Diamines and Sulfuric Acid. J. Phys. Chem. A 2016, 120, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Elm, J.; Myllys, N.; Hyttinen, N.; Kurtén, T. Computational Study of the Clustering of a Cyclohexene Autoxidation Product C6H8O7 with Itself and Sulfuric Acid. J. Phys. Chem. A 2015, 119, 8414–8421. [Google Scholar] [CrossRef]
- Elm, J.; Myllys, N.; Luy, J.; Kurtén, T.; Vehkamäki, H. The effect of water and bases on the clustering of a cyclohexene autoxidation product C6H8O7 with sulfuric acid. J. Phys. Chem. A 2016, 120, 2240–2249. [Google Scholar] [CrossRef]
- Li, H.; Zhong, J.; Vehkamäki, H.; Kurtén, T.; Wang, W.; Ge, M.; Zhang, S.; Li, Z.; Zhang, X.; Francisco, J.S.; et al. Self-Catalytic Reaction of SO3 and NH3 To Produce Sulfamic Acid and Its Implication to Atmospheric Particle Formation. J. Am. Chem. Soc. 2018, 140, 11020–11028. [Google Scholar] [CrossRef]
- Myllys, N.; Olenius, T.; Kurtén, T.; Vehkamäki, H.; Riipinen, I.; Elm, J. Effect of Bisulfate, Ammonia, and Ammonium on the Clustering of Organic Acids and Sulfuric Acid. J. Phys. Chem. A 2017, 121, 4812–4824. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system.Wires. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Duchovic, R.J.; Pettigrew, J.D.; Welling, B.; Shipchandler, T. Conventional transition state theory/Rice–Ramsperger–Kassel–Marcus theory calculations of thermal termolecular rate coefficients for H(D)+O2+M. J. Chem. Phys. 1996, 105, 10367–10379. [Google Scholar] [CrossRef]
- Elm, J.; Jørgensen, S.; Bilde, M.; Mikkelsen, K.V. Ambient reaction kinetics of atmospheric oxygenated organics with the oh radical: A computational methodology study. Phys. Chem. Chem. Phys. 2013, 15, 9636–9645. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Pan, S.; Li, Y.; Wang, L. Atmospheric oxidation mechanism of toluene. J. Phys. Chem. A 2014, 118, 4533–4547. [Google Scholar] [CrossRef] [PubMed]
- Buszek, R.J.; Barker, J.R.; Francisco, J.S. Water Effect on the OH + HCl Reaction. J. Phys. Chem. A 2012, 116, 4712–4719. [Google Scholar] [CrossRef]
- Wine, P.H.; Thompson, R.J.; Ravishankara, A.R.; Semmes, D.H.; Gump, C.A.; Torabi, A.; Nicovich, J.M. Kinetics of the Reaction OH + SO2 + M→ HOSO2 + M. Temperature and Pressure Dependence in the Falloff Region. J. Chem. Phys. 1984, 88, 2095–2104. [Google Scholar] [CrossRef]
- Liu, J.; Fang, S.; Liu, W.; Wang, M.; Tao, F.; Liu, J. Mechanism of the Gaseous Hydrolysis Reaction of SO2: Effects of NH3 versus H2O. J. Phys. Chem. A 2015, 119, 102–111. [Google Scholar] [CrossRef]
- Buszek, R.J.; Torrent-Sucarrat, M.; Anglada, J.M.; Francisco, J.S. Effects of a Single Water Molecule on the OH + H2O2 Reaction. Phys. Chem. A 2012, 116, 5821–5829. [Google Scholar] [CrossRef]
- Wang, S.; Zeng, X.C.; Li, H.; Francisco, J.S. A possible unaccounted source of atmospheric sulfate formation: Amine-promoted hydrolysis and non-radical oxidation of sulfur dioxide. Chem. Sci. 2020, 11, 2093–2102. [Google Scholar] [CrossRef]
- Hall, H.K., Jr. Correlation of base strengths of amines. J. Am. Chem. Soc. 1957, 79, 5441–5444. [Google Scholar] [CrossRef]
- Franchin, A.; Ehrhart, S.; Leppä, J.; Nieminen, T.; Gagné, S.; Schobesberger, S.; Wimmer, D.; Duplissy, J.; Riccobono, F.; Dunne, E.M.; et al. Experimental investigation of ion–ion recombination under atmospheric conditions. Atmos. Chem. Phys. 2015, 15, 7203–7216. [Google Scholar] [CrossRef]
- Jara-Toro, R.A.; Hernandez, F.J.; Garavagno, M.; Taccone, R.A.; Pino, G.A. Water catalysis of the reaction between hydroxyl radicals and linear saturated alcohols (ethanol and n-propanol) at 294 k. Phys. Chem. Chem. Phys. 2018, 20, 27885–27896. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Tsona, N.T.; Tang, S.; Li, J.; Du, L. Role of (H2O)n (n = 1–2) in the Gas-Phase Reaction of Ethanol with Hydroxyl Radical: Mechanism, Kinetics, and Products. ACS Omega 2019, 4, 5805–5817. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.B.; Rubin, E.S. A Technical, Economic, and Environmental Assessment of Amine-Based CO2 Capture Technology for Power Plant Greenhouse Gas Control. Environ. Sci. Technol. 2002, 36, 4467–4475. [Google Scholar] [CrossRef]
- Sharma, S.D.; Azzi, M. A critical review of existing strategies for emission control in the monoethanolamine-based carbon capture process and some recommendations for improved strategies. Fuel 2014, 121, 178–188. [Google Scholar] [CrossRef]
- Long, B.; Bao, J.L.; Truhlar, D.G. Reaction of SO2 with OH in the atmosphere. Phys. Chem. Chem. Phys. 2017, 19, 8091–8100. [Google Scholar] [CrossRef]
- Nielsen, C.J.; Herrmannb, H.; Wellerb, C. Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS). Chem. Soc. Rev. 2012, 41, 6684–6704. [Google Scholar] [CrossRef]
- Borduas, N.; Abbatt, J.P.D.; Murphy, J.G. Gas Phase Oxidation of Monoethanolamine (MEA) with OH Radical and Ozone: Kinetics, Products, and Particles. Environ. Sci. Technol. 2013, 47, 6377–6383. [Google Scholar] [CrossRef]
- Xie, H.; Ma, F.; Wang, Y.; He, N.; Yu, Q.; Chen, J. Quantum Chemical Study on ·Cl-Initiated Atmospheric Degradation of Monoethanolamine. Environ. Sci. Technol. 2015, 49, 13246–13255. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).