
Decomposition Kinetics of Non-Volatile Alkanes on Urban Aerosol


Abstract
:1. Introduction
2. Experiments
2.1. PM2.5 Collection, Exposure in Reactors, and Ancillary Measurements
2.2. Materials, Extraction, and Analyses
3. Results
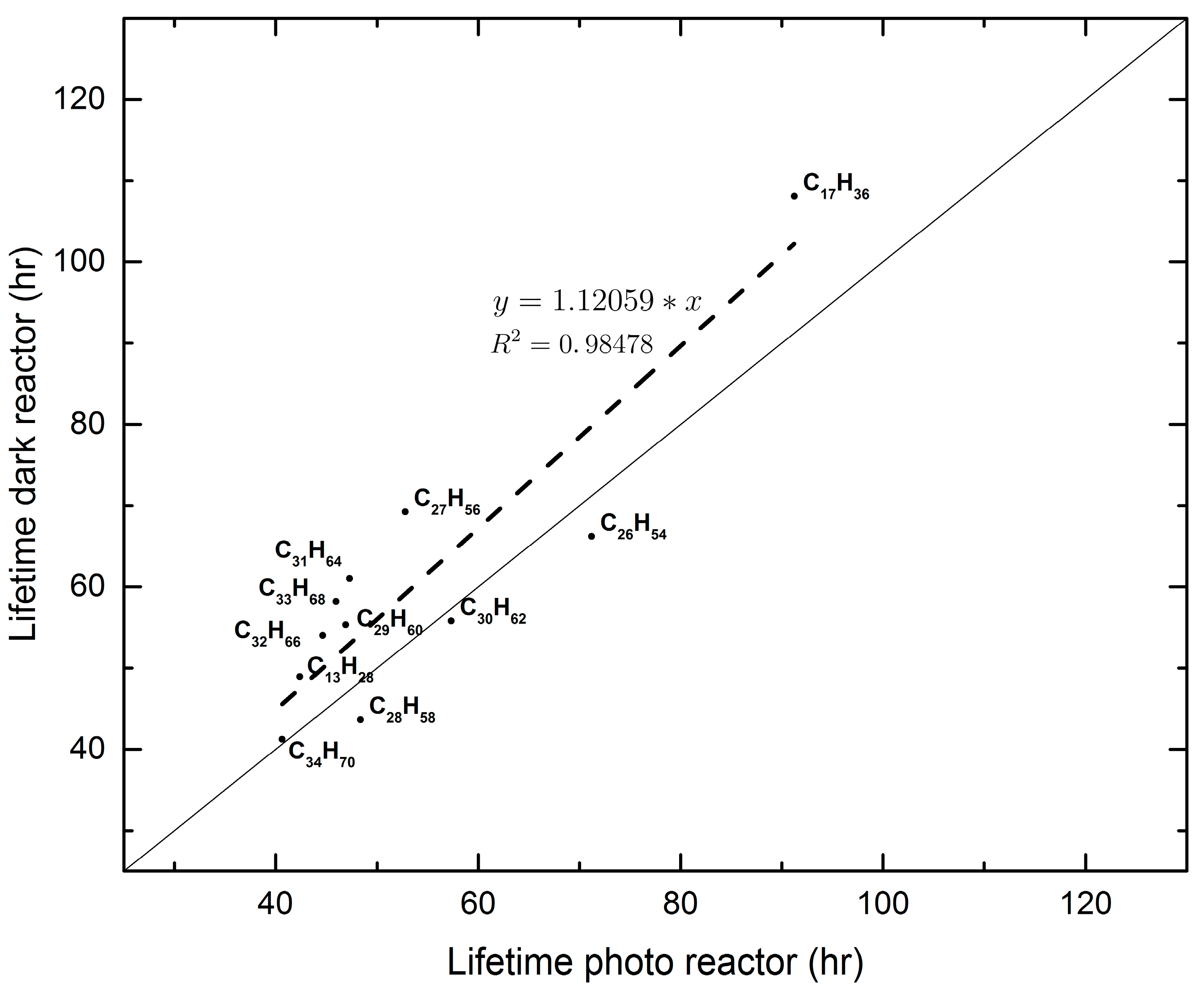
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pope, C.A., III; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Lewtas, J. Air pollution combustion emissions: Characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat. Res. Rev. Mutat. Res. 2007, 636, 95–133. [Google Scholar] [CrossRef] [PubMed]
- Cubison, M.J.; Ervens, B.; Feingold, G.; Docherty, K.S.; Ulbrich, I.M.; Shields, L.; Prather, K.; Hering, S.; Jimenez, J.L. The influence of chemical composition and mixing state of Los Angeles urban aerosol on CCN number and cloud properties. Atmos. Chem. Phys. 2008, 8, 5649–5667. [Google Scholar] [CrossRef]
- Fuzzi, S.; Andreae, M.O.; Huebert, B.J.; Kulmala, M.; Bond, T.C.; Boy, M.; Doherty, S.J.; Guenther, A.; Kanakidou, M.; Kawamura, K.; et al. Critical assessment of the current state of scientific knowledge, terminology, and research needs concerning the role of organic aerosol in the atmosphere, climate, and global change. Atmos. Chem. Phys. 2006, 6, 2017–2038. [Google Scholar] [CrossRef]
- Andreou, G.; Alexiou, S.D.; Loupa, G.; Rapsomanikis, S. Identification, Abundance and Origin of Aliphatic Hydrocarbons in the Fine Atmospheric Particulate Matter of Athens, Greece. Water Air Soil Pollut. Focus 2008, 8, 99–106. [Google Scholar] [CrossRef]
- Andreou, G.; Rapsomanikis, S. Origins of n-alkanes, carbonyl compounds and molecular biomarkers in atmospheric fine and coarse particles of Athens, Greece. Sci. Total Environ. 2009, 407, 5750–5760. [Google Scholar] [CrossRef] [PubMed]
- Behnke, W.; Holländer, W.; Koch, W.; Nolting, F.; Zetzsch, C. A smog chamber for studies of the photochemical degradation of chemicals in the presence of aerosol. Atmos. Environ. 1988, 22, 1113–1120. [Google Scholar] [CrossRef]
- Ma, J.; Liu, Y.; Ma, Q.; Liu, C.; He, H. Heterogeneous photochemical reaction of ozone with anthracene adsorbed on mineral dust. Atmos. Environ. 2013, 72, 165–170. [Google Scholar] [CrossRef]
- Ruehl, C.R.; Nah, T.; Isaacman, G.; Worton, D.R.; Chan, A.W.H.; Kolesar, K.R.; Cappa, C.D.; Goldstein, A.H.; Wilson, K.R. The Influence of Molecular Structure and Aerosol Phase on the Heterogeneous Oxidation of Normal and Branched Alkanes by OH. J. Phys. Chem. A 2013, 117, 3990–4000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Worton, D.R.; Shen, S.; Nah, T.; Isaacman-VanWertz, G.; Wilson, K.R.; Goldstein, A.H. Fundamental Time Scales Governing Organic Aerosol Multiphase Partitioning and Oxidative Aging. Environ. Sci. Technol. 2015, 49, 9768–9777. [Google Scholar] [CrossRef] [PubMed]
- Derwent, R.G.; Jenkin, M.E.; Saunders, S.M.; Pilling, M.J. Characterization of the Reactivities of Volatile Organic Compounds Using a Master Chemical Mechanism. J. Air Waste Manag. Assoc. 2001, 51, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Derwent, R.G.; Jenkin, M.E.; Saunders, S.M.; Pilling, M.J.; Passant, N.R. Multi-day ozone formation for alkenes and carbonyls investigated with a master chemical mechanism under European conditions. Atmos. Environ. 2005, 39, 627–635. [Google Scholar] [CrossRef]
- Saunders, S.M.; Jenkin, M.E.; Derwent, R.G.; Pilling, M.J. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): Tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. [Google Scholar] [CrossRef]
- Růžička, K.; Majer, V. Simultaneous Treatment of Vapor Pressures and Related Thermal Data between the Triple and Normal Boiling Temperatures for n-Alkanes C5–C20. J. Phys. Chem. Reference Data 1994, 23, 1–39. [Google Scholar] [CrossRef]
- Cickos, J. Sublimation Vapor Pressures as Evaluated by Correlation-Gas Chromatography. J. Chem. Eng. Data 2010, 55, 1558–1563. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications; Elsevier Science: San Diego, CA, USA, 2000. [Google Scholar]
- Atkinson, R.; Carter, W.P.L. Kinetics and mechanisms of the gas-phase reactions of ozone with organic compounds under atmospheric conditions. Chem. Rev. 1984, 84, 437–470. [Google Scholar] [CrossRef]
- Atkinson, R. Gas-phase tropospheric chemistry of organic compounds: A review. Atmos. Environ. 2007, 41, 200–240. [Google Scholar] [CrossRef]
- Calvert, J.G.; Derwent, R.G.; Orlando, J.J.; Tyndall, G.S.; Wallington, T.J. Mechanisms of Atmospheric Oxidation of the Alkanes; Oxford University Press: New York, NY, USA, 2008. [Google Scholar]
- Shiraiwa, M.; Garland, R.M.; Pöschl, U. Kinetic double-layer model of aerosol surface chemistry and gas-particle interactions (K2-SURF): Degradation of polycyclic aromatic hydrocarbons exposed to O3, NO2, H2O, OH and NO3. Atmos. Chem. Phys. 2009, 9, 9571–9586. [Google Scholar] [CrossRef]
- Gehling, W.; Khachatryan, L.; Dellinger, B. Hydroxyl Radical Generation from Environmentally Persistent Free Radicals (EPFRs) in PM2.5. Environ. Sci. Technol. 2014, 48, 4266–4272. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Date | Latitude & Longitude | Zenith Angle (°) | Total Incoming Radiation (W·m−2) | Mean O3 (ppb) | Mean NO2 (ppb) |
|---|---|---|---|---|---|---|
| 1 | 24–28 September 2014 | 41.146458 24.919022 | 41.74 | 1,501,047.498 | 67.645 | 3.618 |
| 2 | 7–11 October 2014 | 46.71 | 853,626.555 | 74.704 | 1.136 | |
| 3 | 6–10 November 2014 | 57.14 | 348,921.870 | 55.322 | 11.645 |
| Compounds | Formula | Boiling Point (°C at 760 mmHg) | Vapor Pressure (mmHg at 35 °C) | |
|---|---|---|---|---|
| Tridecane | C13H28 | 234.5 ± 3.0 | 4.26 × 10−2 | Natural |
| Tetradecane | C14H30 | 253.9 ± 3.0 | 1.35 × 10−2 | Natural |
| Hexadecane | C16H34 | 286.6 ± 3.0 | 1.43 × 10−3 | Natural |
| Heptadecane | C17H36 | 301.8 ± 5.0 | 4.61 × 10−4 | Natural |
| Tetracosane | C24H50 | 391.1 ± 5.0 | 2.25 × 10−7 | Natural |
| Hexacosane | C26H54 | 412.2 ± 8.0 | 1.27 × 10−8 | Natural |
| Heptacosane | C27H56 | 422.1 ± 8.0 | 7.05 × 10−9 | Anthropogenic |
| Octacosane | C28H58 | 431.7 ± 8.0 | 8.25 × 10−10 | Natural |
| Nonacosane | C29H60 | 440.9 ± 8.0 | 2.40 × 10−10 | Anthropogenic |
| Triacontane | C30H62 | 449.8 ± 8.0 | 5.70 × 10−11 | Natural |
| Hentriacontane | C31H64 | 458.4 ± 8.0 | 1.20 × 10−11 | Anthropogenic |
| Dotriacontane | C32H66 | 466.7 ± 8.0 | 4.30 × 10−12 | Natural |
| Tritriacontane | C33H68 | 474.7 ± 8.0 | 9.00 × 10−13 | Anthropogenic |
| Tetratriacontane | C34H70 | 482.4 ± 8.0 | 1.88 × 10−13 | Natural |
| Pentatriacontane | C35H72 | 489.9 ± 8.0 | 5.03 × 10−14 | Anthropogenic |
| Hexatriacontane | C36H74 | 497.1 ± 8.0 | na | Natural |
| Tetracontane | C40H82 | 523.9 ± 13.0 | na | Natural |
| Tritetracontane | C43H88 | 541.9 ± 13.0 | na | Anthropogenic |
| Compounds | Pseudo First Order k1 [s−1] | Life Time τ [h] | Half-Lives t1/2 [h] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | Sample | Sample | |||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| C13H28 | 6.55 × 10−6 | 42.40 | 29.39 | ||||||
| C14H30 | 7.37 × 10−7 | 376.99 | 261.31 | ||||||
| C16H34 | 9.10 × 10−6 | 3.12 × 10−6 | 30.54 | 88.91 | 21.17 | 61.63 | |||
| C17H36 | 3.04 × 10−6 | 6.86 × 10−7 | 91.24 | 404.86 | 63.25 | 280.63 | |||
| C24H50 | 3.84 × 10−6 | 2.71 × 10−6 | 72.39 | 102.67 | 50.18 | 71.17 | |||
| C26H54 | 3.90 × 10−6 | 2.88 × 10−6 | 2.14 × 10−6 | 71.19 | 96.53 | 129.63 | 49.34 | 66.91 | 89.85 |
| C27H56 | 5.26 × 10−6 | 2.69 × 10−6 | 1.89 × 10−6 | 52.77 | 103.23 | 146.69 | 36.58 | 71.55 | 101.68 |
| C28H58 | 5.75 × 10−6 | 2.05 × 10−6 | 2.00 × 10−6 | 48.34 | 135.45 | 138.73 | 33.51 | 93.88 | 96.16 |
| C29H60 | 5.92 × 10−6 | 3.84 × 10−6 | 1.85 × 10−6 | 46.89 | 72.35 | 150.54 | 32.50 | 50.15 | 104.34 |
| C30H62 | 4.85 × 10−6 | 4.38 × 10−6 | 2.31 × 10−6 | 57.31 | 63.42 | 120.46 | 39.73 | 43.96 | 83.50 |
| C31H64 | 5.88 × 10−6 | 4.39 × 10−6 | 2.02 × 10−6 | 47.28 | 63.33 | 137.65 | 32.77 | 43.90 | 95.41 |
| C32H66 | 6.23 × 10−6 | 4.77 × 10−6 | 1.64 × 10−6 | 44.62 | 58.21 | 169.50 | 30.93 | 40.35 | 117.49 |
| C33H68 | 6.05 × 10−6 | 4.94 × 10−6 | 2.18 × 10−6 | 45.93 | 56.21 | 127.38 | 31.84 | 38.96 | 88.29 |
| C34H70 | 6.83 × 10−6 | 5.93 × 10−6 | 2.28 × 10−6 | 40.65 | 46.85 | 121.98 | 28.18 | 32.47 | 84.55 |
| C35H72 | 2.34 × 10−6 | 118.89 | 82.41 | ||||||
| C36H74 | 5.07 × 10−6 | 2.07 × 10−6 | 54.83 | 134.40 | 38.01 | 93.16 | |||
| C40H82 | 1.93 × 10−6 | 143.63 | 99.56 | ||||||
| C43H88 | 2.87 × 10−6 | 96.83 | 67.12 | ||||||
| Compounds | Pseudo First Order k1 [s−1] | Life Time τ [h] | Half-Lives t1/2 [h] | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | Sample | Sample | |||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| C13H28 | 5.68 × 10−6 | 3.78 × 10−6 | 48.92 | 73.45 | 33.91 | 50.91 | |||
| C14H30 | 2.70 × 10−6 | 103.08 | 71.45 | ||||||
| C16H34 | 2.22 × 10−6 | 2.01 × 10−6 | 3.36 × 10−6 | 125.04 | 137.91 | 82.70 | 86.67 | 95.59 | 57.33 |
| C17H36 | 2.57 × 10−6 | 1.91× 10−6 | 108.10 | 145.67 | 74.93 | 100.97 | |||
| C24H50 | 8.27 × 10−6 | 5.15 × 10−6 | 2.55 × 10−6 | 33.57 | 53.99 | 108.79 | 23.27 | 37.42 | 75.41 |
| C26H54 | 4.20 × 10−6 | 3.99 × 10− | 1.82 × 10−6 | 66.22 | 69.68 | 152.57 | 45.90 | 48.30 | 105.75 |
| C27H56 | 4.01 × 10−6 | 2.43 × 10−6 | 1.55 × 10−6 | 69.24 | 114.26 | 179.70 | 47.99 | 79.20 | 124.56 |
| C28H58 | 6.36 × 10−6 | 3.66 × 10−6 | 2.03 × 10−6 | 43.65 | 75.97 | 136.62 | 30.26 | 52.66 | 94.69 |
| C29H60 | 5.02 × 10−6 | 4.48 × 10−6 | 2.33 × 10−6 | 55.35 | 62.06 | 119.25 | 38.36 | 43.02 | 82.66 |
| C30H62 | 4.98 × 10−6 | 5.62× 10−6 | 2.48 × 10−6 | 55.83 | 49.45 | 111.81 | 38.70 | 34.27 | 77.50 |
| C31H64 | 4.55 × 10−6 | 5.00 × 10−6 | 2.22 × 10−6 | 61.02 | 55.60 | 125.39 | 42.30 | 38.54 | 86.92 |
| C32H66 | 5.14 × 10−6 | 6.88 × 10−6 | 2.92 × 10−6 | 54.05 | 40.41 | 95.14 | 37.47 | 28.01 | 65.94 |
| C33H68 | 4.77 × 10−6 | 5.87 × 10−6 | 2.53 × 10−6 | 58.20 | 47.34 | 109.76 | 40.34 | 32.81 | 76.08 |
| C34H70 | 6.73 × 10−6 | 5.01 × 10−6 | 3.23 × 10−6 | 41.26 | 55.40 | 86.07 | 28.60 | 38.40 | 59.66 |
| C35H72 | 2.57 × 10−6 | 108.15 | 74.97 | ||||||
| C36H74 | 4.89 × 10−6 | 3.08 × 10−6 | 56.82 | 90.17 | 39.39 | 62.50 | |||
| C40H82 | 2.06 × 10−6 | 134.80 | 93.44 | ||||||
| C43H88 | 2.76 × 10−6 | 100.49 | 69.65 | ||||||
| Compounds | ln(k1) 1st Order | RSQ | Slope = −[Ea·R−1] | Ea [kJ·mol−1] | ||
|---|---|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | ||||
| C13H28 | NSD | NSD | NSD | NSD | NSD | NSD |
| C14H30 | NSD | NSD | NSD | NSD | NSD | NSD |
| C16H34 | NSD | NSD | NSD | NSD | NSD | NSD |
| C17H36 | NSD | NSD | NSD | NSD | NSD | NSD |
| C24H50 | NSD | NSD | NSD | NSD | NSD | NSD |
| C26H54 | NSD | NSD | NSD | NSD | NSD | NSD |
| C27H56 | NSD | NSD | NSD | NSD | NSD | NSD |
| C28H58 | NSD | NSD | NSD | NSD | NSD | NSD |
| C29H60 | NSD | NSD | NSD | NSD | NSD | NSD |
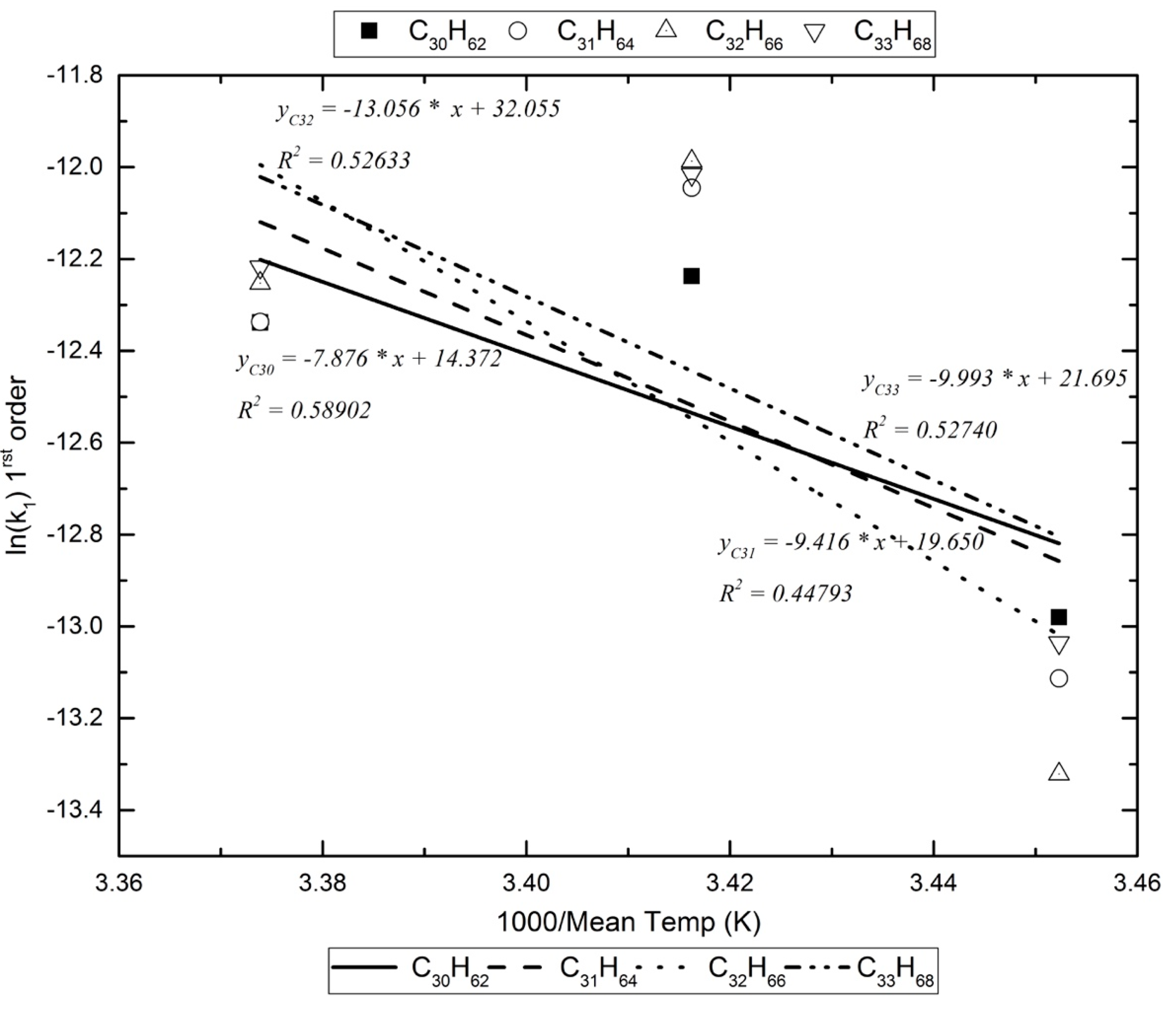
| C30H62 | −12.237 | −12.338 | −12.980 | 0.589 | −7876.237 | 65.487 |
| C31H64 | NSD | NSD | NSD | NSD | NSD | NSD |
| C32H66 | −11.987 | −12.253 | −13.322 | 0.526 | −13,056.140 | 108.555 |
| C33H68 | −12.016 | −12.218 | −13.036 | 0.527 | −9993.166 | 83.088 |
| C34H70 | −11.894 | −12.036 | −12.993 | 0.596 | −11,753.232 | 97.722 |
| C35H72 | NSD | NSD | NSD | NSD | NSD | NSD |
| C36H74 | NSD | NSD | NSD | NSD | NSD | NSD |
| C40H82 | NSD | NSD | NSD | NSD | NSD | NSD |
| C43H88 | NSD | NSD | NSD | NSD | NSD | NSD |
| Compounds | ln(k1) 1st Order | RSQ | Slope = −[Ea·R−1] | Ea [kJ·mol−1] | ||
|---|---|---|---|---|---|---|
| Sample 1 | Sample 2 | Sample 3 | ||||
| C13H28 | NSD | NSD | NSD | NSD | NSD | NSD |
| C14H30 | NSD | NSD | NSD | NSD | NSD | NSD |
| C16H34 | NSD | NSD | NSD | NSD | NSD | NSD |
| C17H36 | NSD | NSD | NSD | NSD | NSD | NSD |
| C24H50 | NSD | NSD | NSD | NSD | NSD | NSD |
| C26H54 | −12.382 | −12.433 | −13.216 | 0.614 | −12,124.477 | 100.809 |
| C27H56 | NSD | NSD | NSD | NSD | NSD | NSD |
| C28H58 | NSD | NSD | NSD | NSD | NSD | NSD |
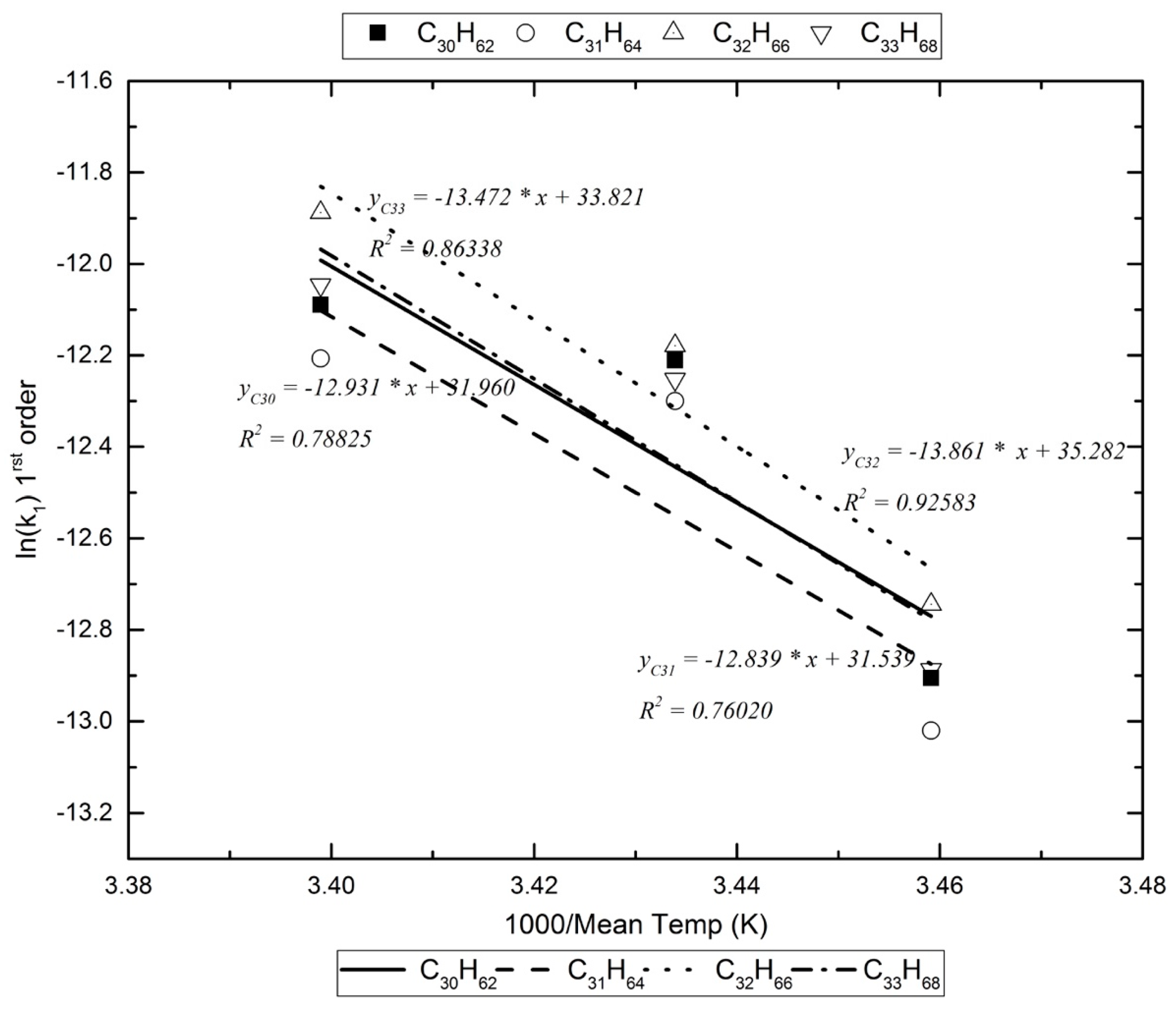
| C29H60 | −12.202 | −12.317 | −12.970 | 0.530 | −9974.793 | 82.935 |
| C30H62 | −12.211 | −12.090 | −12.905 | 0.788 | −12,930.895 | 107.514 |
| C31H64 | −12.300 | −12.207 | −13.020 | 0.760 | −12,839.333 | 106.753 |
| C32H66 | −12.179 | −11.888 | −12.744 | 0.926 | −13,860.788 | 115.246 |
| C33H68 | −12.253 | −12.046 | −12.887 | 0.863 | −13,471.609 | 112.010 |
| C34H70 | NSD | NSD | NSD | NSD | NSD | NSD |
| C35H72 | NSD | NSD | NSD | NSD | NSD | NSD |
| C36H74 | NSD | NSD | NSD | NSD | NSD | NSD |
| C40H82 | NSD | NSD | NSD | NSD | NSD | NSD |
| C43H88 | NSD | NSD | NSD | NSD | NSD | NSD |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karali, D.; Rapsomanikis, S.; Christoforidis, A. Decomposition Kinetics of Non-Volatile Alkanes on Urban Aerosol. Atmosphere 2017, 8, 89. https://doi.org/10.3390/atmos8050089
Karali D, Rapsomanikis S, Christoforidis A. Decomposition Kinetics of Non-Volatile Alkanes on Urban Aerosol. Atmosphere. 2017; 8(5):89. https://doi.org/10.3390/atmos8050089
Chicago/Turabian StyleKarali, Dimitra, Spyridon Rapsomanikis, and Achilleas Christoforidis. 2017. "Decomposition Kinetics of Non-Volatile Alkanes on Urban Aerosol" Atmosphere 8, no. 5: 89. https://doi.org/10.3390/atmos8050089
APA StyleKarali, D., Rapsomanikis, S., & Christoforidis, A. (2017). Decomposition Kinetics of Non-Volatile Alkanes on Urban Aerosol. Atmosphere, 8(5), 89. https://doi.org/10.3390/atmos8050089