

Potential Contribution of Coastal Upwelling to Carbon Sink through Interaction between Cyanobacteria and Microbial Eukaryotes




 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
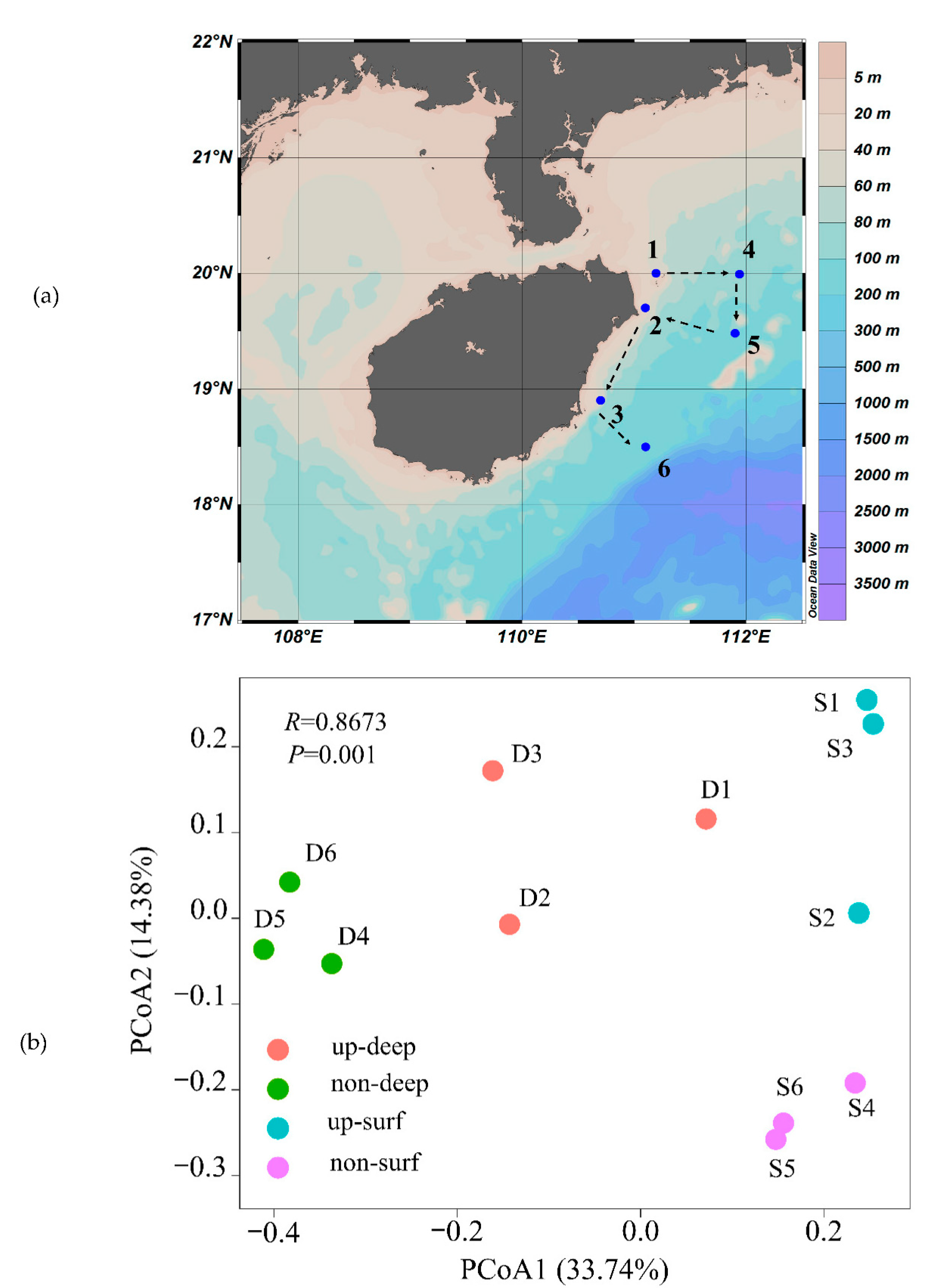
2.1. Sampling and Metadata
2.2. DNA Extraction and Amplicon Sequencing
2.3. Processing of Paired-End Sequences and Function Prediction
2.4. Statistical Analyses
3. Results
3.1. Variations of Bacterioplankton Communities
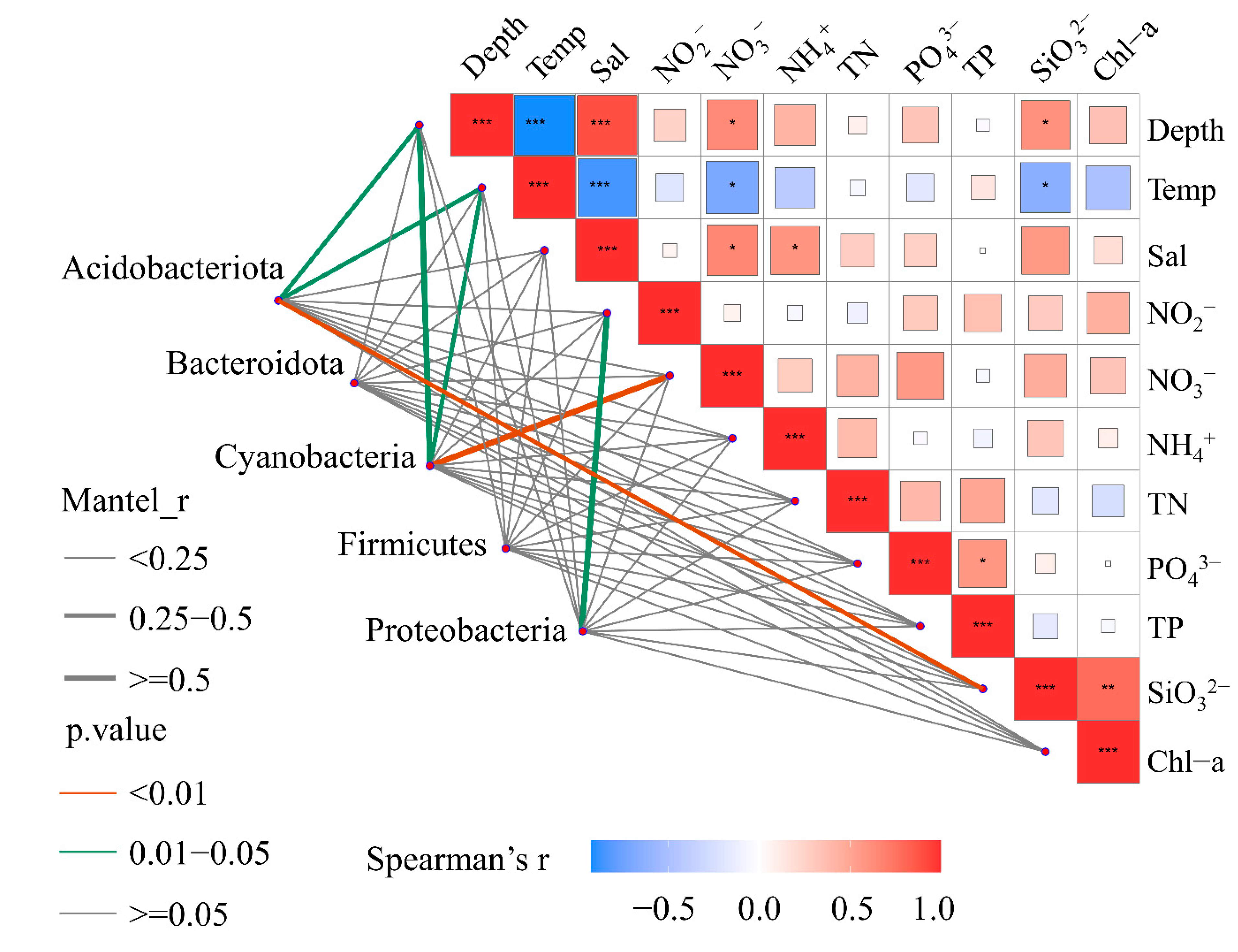
3.2. Relationship between Environmental Factors and Bacterioplankton Community Composition
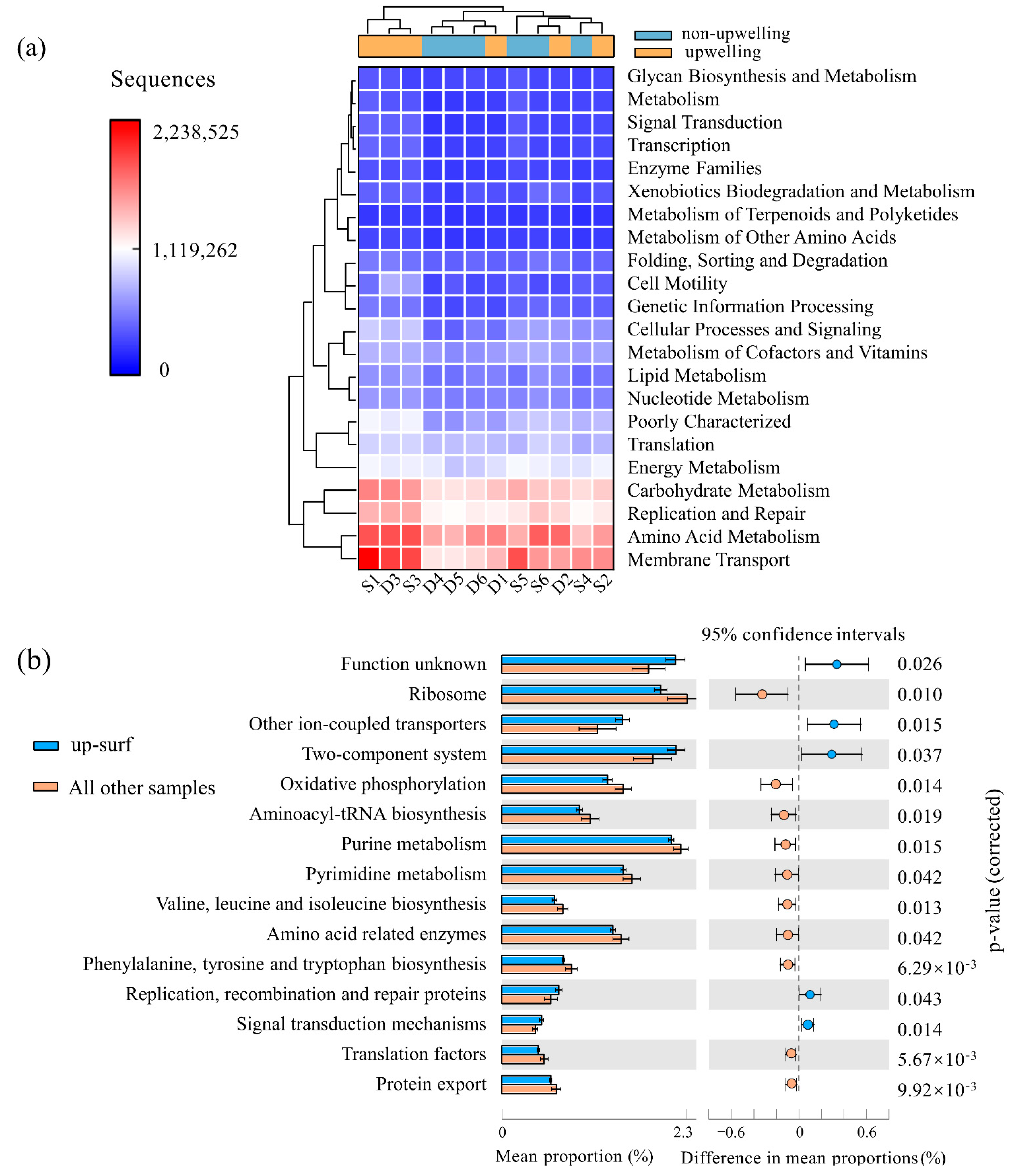
3.3. Functional Differences of the Bacterioplankton Communities
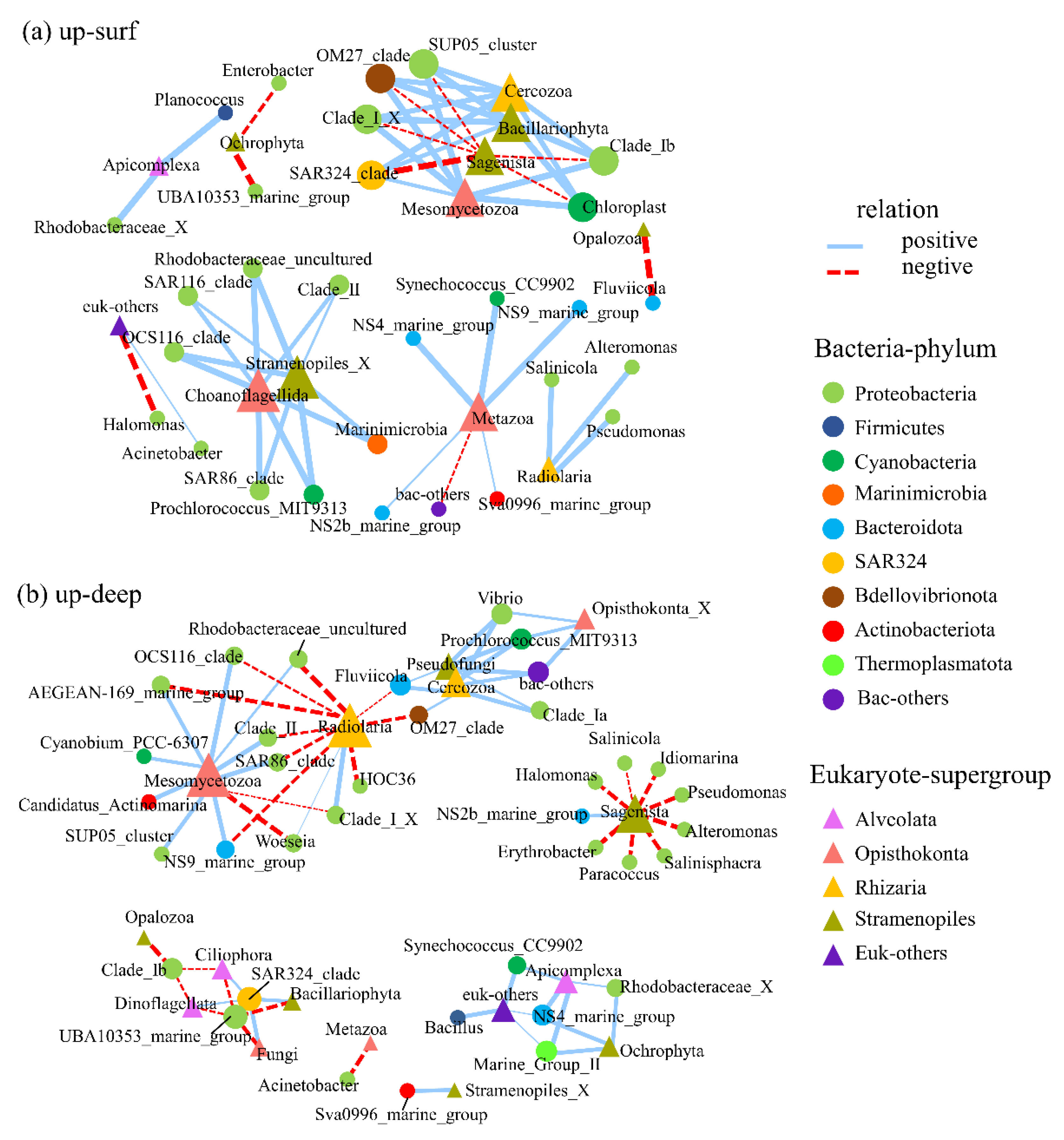
3.4. Interactions between Bacterioplankton and Microbial Eukaryotes
4. Discussion
4.1. Response of Bacterioplankton Communities to the Upwelling
4.2. Upregulated Signaling Function in Upwelling and Potential Contribution of Bacterioplankton to Carbon Sequestration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kuang, C.Y. Analysis of Green House Gases and Positive Impact of Replacing Traditional Energy with Clean Energy. In Proceedings of the 8th International Conference on Environment Pollution and Prevention (ICEPP), Electr Network, Sydney, Australia, 3–5 December 2020. [Google Scholar]
- Lawrence, M.W. Efficiency of carbon sequestration by added reactive nitrogen in ocean fertilisation. Int. J. Glob. Warm. 2014, 6, 15–33. [Google Scholar] [CrossRef]
- Rickels, W.; Lontzek, T.S. Optimal global carbon management with ocean sequestration. Oxf. Econ. Pap. -New Ser. 2012, 64, 323–349. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, L.; Li, Y. Development Status of CO2 Marine Sequestration. Geol. Sci. Techol. Inf. 2018, 37, 212–218. [Google Scholar]
- Le Quere, C.; Andrew, R.M.; Friedlingstein, P.; Sitch, S.; Hauck, J.; Pongratz, J.; Pickers, P.A.; Korsbakken, J.I.; Peters, G.P.; Canadell, J.G.; et al. Global Carbon Budget 2018. Earth Syst. Sci. Data 2018, 10, 2141–2194. [Google Scholar] [CrossRef]
- Rotko, D.; Stupar, D.; Neale, C.M.; Maltese, A. Carbon sequestering using remote sensing. In Proceedings of the Remote Sensing for Agriculture, Ecosystems, and Hydrology XXI, Strasbourg, France, 21 October 2019. [Google Scholar]
- Cao, L.; Caldeira, K. Can ocean iron fertilization mitigate ocean acidification? Clim. Change 2010, 99, 303–311. [Google Scholar] [CrossRef]
- Baumann, M.; Taucher, J.; Paul, A.J.; Heinemann, M.; Vanharanta, M.; Bach, L.T.; Spilling, K.; Ortiz, J.; Aristegui, J.; Hernandez-Hernandez, N.; et al. Effect of Intensity and Mode of Artificial Upwelling on Particle Flux and Carbon Export. Front. Mar. Sci. 2021, 8, 742142. [Google Scholar] [CrossRef]
- Pan, Y.W.; You, L.; Li, Y.F.; Fan, W.; Chen, C.T.A.; Wang, B.J.; Chen, Y. Achieving Highly Efficient Atmospheric CO2 Uptake by Artificial Upwelling. Sustainability 2018, 10, 664. [Google Scholar] [CrossRef]
- Lovelock, J.E.; Rapley, C.G. Ocean pipes could help the earth to cure itself. Nature 2007, 449, 403. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, M.; Cui, Q.; Fan, W.; Qi, J.; Chen, Y.; Zhang, Y.; Gao, K.; Fan, J.; Wang, G.; et al. Processes of coastal ecosystem carbon sequestration and approaches for increasing carbon sink. Sci. China Earth Sci. 2017, 60, 809–820. [Google Scholar] [CrossRef]
- Pan, Y.W.; Fan, W.; Huang, T.H.; Wang, S.L.; Chen, C.T.A. Evaluation of the sinks and sources of atmospheric CO2 by artificial upwelling. Sci. Total Environ. 2015, 511, 692–702. [Google Scholar] [CrossRef]
- Pan, Y.W.; Wei, F.; Zhang, D.H.; Chen, J.W.; Huang, H.C.; Liu, S.X.; Jiang, Z.P.; Di, Y.N.; Tong, M.M.; Chen, Y. Research progress in artificial upwelling and its potential environmental effects. Sci. China-Earth Sci. 2016, 59, 236–248. [Google Scholar] [CrossRef]
- Yang, G.; Nian-peng, H.; Ya-feng, W. Characteristics of Carbon Sequestration by Ecosystem and Progress in Its Research. J. Nat. Resour. 2013, 28, 1264–1274. [Google Scholar]
- Beaulieu, S.E. Accumulation and fate of phytodetritus on the sea floor. In Oceanography and Marine Biology; Gibson, R.N., Barnes, M., Atkinson, R.J.A., Eds.; Taylor & Francis: Abingdon, UK, 2002; Volume 40, pp. 171–232. [Google Scholar]
- O’Brien, M.C.; Melling, H.; Pedersen, T.F.; Macdonald, R.W. The role of eddies on particle flux in the Canada Basin of the Arctic Ocean. Deep. -Sea Res. Part I-Oceanogr. Res. Pap. 2013, 71, 1–20. [Google Scholar] [CrossRef]
- Oschlies, A. Can eddies make ocean deserts bloom? Glob. Biogeochem. Cycles 2002, 16, 53-1–53-11. [Google Scholar] [CrossRef]
- Chen, F.Z.; Cai, W.J.; Benitez-Nelson, C.; Wang, Y.C. Sea surface pCO2-SST relationships across a cold-core cyclonic eddy: Implications for understanding regional variability and air-sea gas exchange. Geophys. Res. Lett. 2007, 34, L10603. [Google Scholar] [CrossRef]
- Jiao, N.; Zhang, Y.; Zhou, K.; Li, Q.; Dai, M.; Liu, J.; Guo, J.; Huang, B. Revisiting the CO2 “source” problem in upwelling areas—A comparative study on eddy upwellings in the South China Sea. Biogeosciences 2014, 11, 2465–2475. [Google Scholar] [CrossRef]
- Anas, A.; Sheeba, V.A.; Jasmin, C.; Gireeshkumar, T.R.; Mathew, D.; Krishna, K.; Nair, S.; Muraleedharan, K.R.; Jayalakshmy, K.V. Upwelling induced changes in the abundance and community structure of archaea and bacteria in a recurring mud bank along the southwest coast of India. Reg. Stud. Mar. Sci. 2018, 18, 113–121. [Google Scholar] [CrossRef]
- Sun, F.; Wu, M.; Wang, Y.; Sun, C.; Xu, Z. Diversity and potential function of bacterial communities in different upwelling systems. Estuar. Coast. Shelf Sci. 2020, 237, 106698. [Google Scholar] [CrossRef]
- Bergen, B.; Herlemann, D.P.; Jurgens, K. Zonation of bacterioplankton communities along aging upwelled water in the northern Benguela upwelling. Front. Microbiol. 2015, 6, 621. [Google Scholar] [CrossRef]
- Vijayan, J.; Ammini, P.; Nathan, V.K. Diversity pattern of marine culturable heterotrophic bacteria in a region with coexisting upwelling and mud banks in the southeastern Arabian Sea. Environ. Sci. Pollut. Res. 2021, 29, 3967–3982. [Google Scholar] [CrossRef]
- Liu, X.; Xie, N.; Bai, M.; Li, J.; Wang, G. Composition change and decreased diversity of microbial eukaryotes in the coastal upwelling waters of South China Sea. Sci. Total Environ. 2021, 795, 148892. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sen, B.; Zhou, S.; Xie, N.; Zhang, Y.; Zhang, J.; Wang, G. Distinct Seasonal Patterns of Bacterioplankton Abundance and Dominance of Phyla alpha-Proteobacteria and Cyanobacteria in Qinhuangdao Coastal Waters Off the Bohai Sea. Front. Microbiol. 2017, 8, 1579. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.B.; Stedmon, C.A.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindstrom, E.S.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Verbaendert, I.; Boon, N.; De Vos, P.; Heylen, K. Denitrification is a common feature among members of the genus Bacillus . Syst. Appl. Microbiol. 2011, 34, 385–391. [Google Scholar] [CrossRef]
- Tsai, A.-Y.; Gong, G.-C.; Sanders, R.W.; Chao, C.-F.; Chiang, K.-P. Microbial Dynamics in an Oligotrophic Bay of the Western Subtropical Pacific: Impact of Short-Term Heavy Freshwater Runoff and Upwelling. J. Oceanogr. 2010, 66, 873–883. [Google Scholar] [CrossRef]
- Malik, A.; Fernandes, C.E.G.; Gonsalves, M.J.B.D.; Subina, N.S.; Mamatha, S.S.; Krishna, K.; Varik, S.; Kumari, R.; Gauns, M.; Cejoice, R.P.; et al. Interactions between trophic levels in upwelling and non-upwelling regions during summer monsoon. J. Sea Res. 2015, 95, 56–69. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.X.; Mann, A.J.; Waldmann, J.; et al. Substrate-Controlled Succession of Marine Bacterioplankton Populations Induced by a Phytoplankton Bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef]
- Pinhassi, J.; Sala, M.M.; Havskum, H.; Peters, F.; Guadayol, O.; Malits, A.; Marrase, C. Changes in bacterioplankton composition under different phytoplankton regimens. Appl. Environ. Microbiol. 2004, 70, 6753–6766. [Google Scholar] [CrossRef]
- Grossart, H.P.; Levold, F.; Allgaier, M.; Simon, M.; Brinkhoff, T. Marine diatom species harbour distinct bacterial communities. Environ. Microbiol. 2005, 7, 860–873. [Google Scholar] [CrossRef]
- Irwin, A.J.; Finkel, Z.V.; Schofield, O.M.E.; Falkowski, P.G. Scaling-up from nutrient physiology to the size-structure of phytoplankton communities. J. Plankton Res. 2006, 28, 459–471. [Google Scholar] [CrossRef]
- Liu, H.B.; Suzukil, K.; Minami, C.; Saino, T.; Watanabe, M. Picoplankton community structure in the subarctic Pacific Ocean and the Bering Sea during summer 1999. Mar. Ecol. Prog. Ser. 2002, 237, 1–14. [Google Scholar] [CrossRef][Green Version]
- DuRand, M.D.; Olson, R.J.; Chisholm, S.W. Phytoplankton population dynamics at the Bermuda Atlantic Time-series station in the Sargasso Sea. Deep. Sea Res. II 2001, 48, 1983–2003. [Google Scholar] [CrossRef]
- Gomes, J.; Khandeparker, R.; Naik, H.; Shenoy, D.; Meena, R.M.; Ramaiah, N. Denitrification rates of culturable bacteria from a coastal location turning temporally hypoxic. J. Mar. Syst. 2020, 209, 103089. [Google Scholar] [CrossRef]
- Sun, Q.; Song, J.; Li, X.; Yuan, H.; Ma, J.; Wang, Q. Bacterial vertical and horizontal variability around a deep seamount in the Tropical Western Pacific Ocean. Mar. Pollut. Bull. 2020, 158, 111419. [Google Scholar] [CrossRef] [PubMed]
- Divya, B.; Parvathi, A.; Loka Bharathi, P.A.; Nair, S. 16S rRNA-based bacterial diversity in the organic-rich sediments underlying oxygen-deficient waters of the eastern Arabian Sea. World J. Microbiol. Biotechnol. 2011, 27, 2821–2833. [Google Scholar] [CrossRef]
- Velmurugan, N.; Kalpana, D.; Cho, J.-Y.; Lee, G.-H.; Park, S.-H.; Lee, Y.-S. Phylogenetic analysis of culturable marine bacteria in sediments from South Korean Yellow Sea. Microbiology 2011, 80, 261–272. [Google Scholar] [CrossRef]
- Tran, V.; Geraci, K.; Midili, G.; Satterwhite, W.; Wright, R.; Bonilla, C.Y. Resilience to oxidative and nitrosative stress is mediated by the stressosome, RsbP and SigB in Bacillus subtilis. J. Basic Microbiol. 2019, 59, 834–845. [Google Scholar] [CrossRef]
- Aldhafiri, S.; Mahmoud, H.; Al-Sarawi, M.; Ismail, W.A. Natural Attenuation Potential of Polychlorinated Biphenyl-Polluted Marine Sediments. Pol. J. Microbiol. 2018, 67, 37–48. [Google Scholar] [CrossRef]
- Guldimann, C.; Boor, K.J.; Wiedmann, M.; Guariglia-Oropeza, V. Resilience in the Face of Uncertainty: Sigma Factor B Fine-Tunes Gene Expression To Support Homeostasis in Gram-Positive Bacteria. Appl. Environ. Microbiol. 2016, 82, 4456–4469. [Google Scholar] [CrossRef]
- Chollett, I.; Mumby, P.J.; Cortés, J. Upwelling areas do not guarantee refuge for coral reefs in a warming ocean. Mar. Ecol. Prog. Ser. 2010, 416, 47–56. [Google Scholar] [CrossRef]
- Kozak, E.R.; Franco-Gordo, C.; Suarez-Morales, E.; Palomares-Garcia, R. Seasonal and interannual variability of the calanoid copepod community structure in shelf waters of the Eastern Tropical Pacific. Mar. Ecol. Prog. Ser. 2014, 507, 95–110. [Google Scholar] [CrossRef]
- Hirota, Y.; Itoh, H.; MoriMoto, H.; Ichikawa., T.; Horikawa., H. Comparison of copepod communities during upwelling and non-upwelling in the summers from 1992 to 2010 in Tosa Bay, western Japan. Plankton Benthos Res. 2017, 12, 201–211. [Google Scholar] [CrossRef][Green Version]
- Shivaji, S.; Prakash, J.S. How do bacteria sense and respond to low temperature? Arch. Microbiol. 2010, 192, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sun, D.; Zhu, J.; Liu, W. Two-Component Signal Transduction Systems: A Major Strategy for Connecting Input Stimuli to Biofilm Formation. Front. Microbiol. 2018, 9, 3279. [Google Scholar] [CrossRef] [PubMed]
- Los, D.A.; Suzuki, I.; Zinchenko, V.V.; Murata, N. Stress Responses in Synechocystis: Regulated Genes and Regulatory Systems. In the Cyanobacteria: Molecular Biology, Genomics and Evolution; Caister Academic Press: Norfolk, UK, 2008. [Google Scholar]
- Aguilar, P.S.; Hernandez-Arriaga, A.M.; Cybulski, L.E.; Erazo, A.C.; de Mendoza, D. Molecular basis of thermosensing: A two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 2001, 20, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Mironov, K.S.; Sidorov, R.A.; Trofimova, M.S.; Bedbenov, V.S.; Tsydendambaev, V.D.; Allakhverdiev, S.I.; Los, D.A. Light-dependent cold-induced fatty acid unsaturation, changes in membrane fluidity, and alterations in gene expression in Synechocystis. Biochim. Et Biophys. Acta-Bioenerg. 2012, 1817, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, I.; Kanesaki, Y.; Mikam, K.; Kanehisa, M.; Murata, N. Cold-regulated genes under control of the cold sensor Hik33 in Synechocystis. Mol. Microbiol. 2001, 40, 235–244. [Google Scholar] [CrossRef]
- Liu, Z.X.; Li, H.C.; Wei, Y.P.; Chu, W.Y.; Chong, Y.L.; Long, X.H.; Liu, Z.P.; Qin, S.; Shao, H.B. Signal transduction pathways in Synechocystis sp. PCC 6803 and biotechnological implications under abiotic stress. Crit. Rev. Biotechnol. 2015, 35, 269–280. [Google Scholar] [CrossRef]
- Shapiro, R.S.; Cowen, L.E. Thermal control of microbial development and virulence: Molecular mechanisms of microbial temperature sensing. mBio 2012, 3, e00238-12. [Google Scholar] [CrossRef]
- Skerker, J.M.; Prasol, M.S.; Perchuk, B.S.; Biondi, E.G.; Laub, M.T. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: A system-level analysis. PLoS Biol. 2005, 3, e334. [Google Scholar] [CrossRef]
- Harshey, R.M.; Kawagishi, I.; Maddock, J.; Kenney, L.J. Function, Diversity, and Evolution of Signal Transduction in Prokaryotes. Dev. Cell 2003, 4, 459–465. [Google Scholar] [CrossRef]
- Dudler, R.; Eberl, L. Interactions between bacteria and eukaryotes via small molecules. Curr. Opin. Biotechnol. 2006, 17, 268–273. [Google Scholar] [CrossRef]
- Wu, L.C.; Estrada, O.; Zaborina, O.; Bains, M.; Shen, L.; Kohler, J.E.; Patel, N.; Musch, M.W.; Chang, E.B.; Fu, Y.X.; et al. Recognition of host immune activation by Pseudomonas aeruginosa. Science 2005, 309, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Kamennaya, N.A.; Zemla, M.; Mahoney, L.; Chen, L.; Holman, E.; Holman, H.Y.; Auer, M.; Ajo-Franklin, C.M.; Jansson, C. High pCO2-induced exopolysaccharide-rich ballasted aggregates of planktonic cyanobacteria could explain Paleoproterozoic carbon burial. Nat. Commun. 2018, 9, 2116. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Monks, L.; Neuer, S. Effects of clay minerals on the aggregation and subsequent settling of marine Synechococcus. Limnol. Oceanogr. 2015, 60, 805–816. [Google Scholar] [CrossRef]
- Richardson, T.L. Mechanisms and Pathways of Small-Phytoplankton Export from the Surface Ocean. Annu. Rev. Mar. Sci. 2019, 11, 57–74. [Google Scholar] [CrossRef]
- Hamamoto, Y.; Honda, D. Nutritional intake of Aplanochytrium (Labyrinthulea, Stramenopiles) from living diatoms revealed by culture experiments suggesting the new prey-predator interactions in the grazing food web of the marine ecosystem. PLoS ONE 2019, 14, e0208941. [Google Scholar] [CrossRef]
- Bai, M.; Xie, N.; He, Y.; Li, J.; Collier, J.L.; Hunt, D.E.; Johnson, Z.I.; Jiao, N.; Wang, G. Vertical community patterns of Labyrinthulomycetes protists reveal their potential importance in the oceanic biological pump. Environ. Microbiol. 2021, 24, 1703–1713. [Google Scholar] [CrossRef]
- Lundgreen, R.B.C.; Jaspers, C.; Traving, S.J.; Ayala, D.J.; Lombard, F.; Grossart, H.P.; Nielsen, T.G.; Munk, P.; Riemann, L. Eukaryotic and cyanobacterial communities associated with marine snow particles in the oligotrophic Sargasso Sea. Sci. Rep. 2019, 9, 8891. [Google Scholar] [CrossRef]
- Thiele, S.; Fuchs, B.M.; Amann, R.; Iversen, M.H. Colonization in the photic zone and subsequent changes during sinking determine bacterial community composition in marine snow. Appl. Environ. Microbiol. 2015, 81, 1463–1471. [Google Scholar] [CrossRef]
- Sano, M.; Maki, K.; Nishibe, Y.; Nagata, T.; Nishida, S. Feeding habits of mesopelagic copepods in Sagami Bay: Insights from integrative analysis. Prog. Oceanogr. 2013, 110, 11–26. [Google Scholar] [CrossRef]
- Vojvoda, J.; Lamy, D.; Sintes, E.; Garcia, J.A.L.; Turk, V.; Herndl, G.J. Seasonal variation in marine-snow-associated and ambient-water prokaryotic communities in the northern Adriatic Sea. Aquat. Microb. Ecol. 2014, 73, 211–224. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Xie, N.; Li, J.; Bai, M.; Sen, B.; Wang, G. Potential Contribution of Coastal Upwelling to Carbon Sink through Interaction between Cyanobacteria and Microbial Eukaryotes. Water 2022, 14, 3097. https://doi.org/10.3390/w14193097
Liu X, Xie N, Li J, Bai M, Sen B, Wang G. Potential Contribution of Coastal Upwelling to Carbon Sink through Interaction between Cyanobacteria and Microbial Eukaryotes. Water. 2022; 14(19):3097. https://doi.org/10.3390/w14193097
Chicago/Turabian StyleLiu, Xiuping, Ningdong Xie, Jiaqian Li, Mohan Bai, Biswarup Sen, and Guangyi Wang. 2022. "Potential Contribution of Coastal Upwelling to Carbon Sink through Interaction between Cyanobacteria and Microbial Eukaryotes" Water 14, no. 19: 3097. https://doi.org/10.3390/w14193097
APA StyleLiu, X., Xie, N., Li, J., Bai, M., Sen, B., & Wang, G. (2022). Potential Contribution of Coastal Upwelling to Carbon Sink through Interaction between Cyanobacteria and Microbial Eukaryotes. Water, 14(19), 3097. https://doi.org/10.3390/w14193097