Abstract
The spread of fecal pollutants and antibiotic resistance in the aquatic environment represents a major public health concern and is predicted to increase in light of climate change consequences and the increasing human population pressure on the lagoon and coastal areas. The city of Venice (Italy) is affected by diverse microbial pollution sources, including domestic wastewaters that, due to the lack of modern sewage treatment infrastructure in the historical city center, are released into canals. The outflowing jets of its tidal inlets thus represent a source of contamination for the nearby beaches on the barrier island separating the lagoon from the sea. Metagenomic analyses of DNA extracted from sediment samples from six sites in the canals of the city’s historic center were undertaken to characterize the microbial community composition, the presence of fecal microbes as well as other non-enteric pathogens, and the content of genes related to antibiotic (AB) and heavy metal (HM) resistance, and virulence. The six sites hosted similar prokaryotic communities, although variations in community composition likely related to oxygen availability were observed. All sites displayed relatively high levels of fecal contamination, including the presence of Fecal Indicator Bacteria, sewage- and alternative feces-associated bacteria. Relatively high levels of other potential pathogens were also found. About 1 in 500 genes identified at these sites are related to AB and HM resistance; conversely, genes related to virulence were rare. Our data suggest the existence of widespread sediment microbial pollution in the canals of Venice, coupled with the prevalence of ARGs to antibiotics frequently used in humans as well as of HMRGs to toxic metals that still persists in the lagoon. All of this evidence raises concerns about the consequences on the water quality of the lagoon and adjacent marine areas and the potential risks for humans, deserving further studies.
1. Introduction
Transitional and coastal aquatic ecosystems may experience low to severe levels of anthropogenic impacts, which may include microbial contamination as a consequence of human (e.g., tourism, sewage, boat and ship traffic), industrial (i.e., chemicals), and agricultural activities (e.g., fertilization with zootechnical wastewaters and runoff of livestock manure from open-field farming) [1,2,3]. Microbiological pollution in such ecosystems can therefore represent a threat to aquatic life as well as to human health [4,5,6]. Indeed, it may likely include potential pathogenic organisms and impair water quality and food safety, human and ecosystem health [7], with important consequences from socio-economic, sanitary and environmental perspectives [8], including the impairment of bathing water quality [9].
Fecal Indicator Bacteria (FIB; Escherichia [E.] coli and intestinal enterococci) [9,10,11,12,13,14] are used worldwide to assess bathing water quality and as a proxy for the potential health risks posed by microbial contaminants [15,16]. However, some populations of E. coli have been shown to be environmentally adapted and able to persist and grow in aquatic environments, thus questioning their reliability as indicators of (fecal) microbial pollution [10,17]. Novel microbial taxa, such as Lachnospiraceae or Arcobacter, have been thus identified and successfully used as alternative fecal- and sewage-associated pollution indicators [8,18,19,20,21,22,23,24]. At the same time, further potential and/or emerging pathogenic, non-enteric bacteria are being explored for their potential in depicting the level of microbial pollution by microorganisms of public health concern in coastal and transitional environments [8,23,24,25].
Microbial pollution can also introduce additional risks, given the possibility of releasing antibiotic-resistant bacteria (ARBs) and their antibiotic resistance genes (ARGs) [26]. Antibiotic (AB) resistance is recognized today as one of the most significant threats to global public health [27], also in relation to the possibility of Horizontal Gene Transfer (HGT) events of ARGs between and among microorganisms [28], and it is considered ubiquitous in the seawater environment, especially in coastal areas [26]. Anthropogenic activities may represent a significant driver for the presence of ARGs in human-impacted areas [26]. Recent findings report that the presence of resistance genes can largely be explained by fecal pollution [29]. For these reasons, scientific efforts are dedicated to understanding the occurrence, threats, and potential ecological effects of ABs and ARGs in various aquatic environments worldwide [27]. In addition, heavy metals (HM) resistance genes (HMRGs) have also been suggested to co-occur with AB resistance in the environment [30], thus prompting the need to target these genes when assessing the quality of coastal waters.
Several approaches have been identified worldwide to assess microbial pollution in aquatic environments, most of them targeting Fecal Indicator Bacteria (FIB) by applying cultivation-based techniques. An alternative to such traditional approaches, High-Throughput Sequencing (HTS) approaches, such as shotgun metagenomics, offer a powerful chance to catch the microbial diversity (e.g., by analyzing 16S rRNA genes) and potential functioning (e.g., by analyzing genes involved in pathogenesis, AB and HM resistance, virulence) of microbial communities in the environment [9,22,23,24,31,32]. Given this, HTS represents a powerful means toward a safer and more sustainable management of aquatic ecosystems, including the assessment of bathing water quality.
In this work, based on shotgun metagenomic analyses, we report the diversity and community composition of prokaryotic assemblages in sediment samples collected from several locations in the historic center of the city of Venice, with the further aim of understanding the occurrence and spatial distribution of microbial pollutants, as well as genes for AB and HM resistance and virulence. Our study illustrates the significance of microbial pollution in the city of Venice. The results can be helpful in the evaluation of the role/importance of the city center as a pollution source of nearby lagoon areas and, through the tidal exchanges on the quality of bathing waters on the barrier islands.
2. Materials and Methods
2.1. Study Site
The Venice Lagoon (Northern Adriatic Sea, Italy), one of the largest lagoons in the Mediterranean Sea [33], is connected to the sea by three inlets, which guarantee tidal exchanges of water with the adjacent open sea [13,14,34,35]. Because of the presence of a city that is visited by millions of tourists yearly and of a heavily populated and industrialized mainland, the lagoon of Venice is affected by a variety of pollution sources [36]. These include human and animal wastewaters, the weathering of building materials, the inputs from agriculture in the drainage basin and industrial activities both in the city and the nearby mainland, massive tourism and intensive boat and ship traffic [24,37]. Besides the external inputs of contaminants from the drainage basin and point sources located at the lagoon–mainland interface, domestic wastewaters are also released in the historical city center and in some minor islands through a dense canal network that has a centuries-old history of management and that often appears to lack adequate sewage treatment infrastructures [38,39,40]. Despite the flushing effect of the semidiurnal tidal flow that helps reduce the levels of fecal pollution [13], sediment accumulates in the canals of the historic center at a rate that now requires periodical dredging, typically every few decades to remove excess sludge [41]. The city canal system is also partly affected by the inputs from the mainland and is drained by natural tidal channels of the lagoon that exchange with the Adriatic Sea through three tidal inlets (Lido, Malamocco, and Chioggia). The lagoon waters may, therefore, potentially impact the quality of adjacent bathing coastal waters of the barrier islands enclosing the lagoon [42].
2.2. Sampling Activities
Surface sediment samples were collected in six sites located in the canals of the historic center of Venice (Figure 1). Sampling was performed in the six sites indicated in Figure 1 (S1 to S6) using a Van Veen grab sampler (capacity 5 L). S1 was located in a small canal that has relatively low boat traffic; S5 was close to St. Mark’s basin, which is characterized by intense boat traffic; the other stations were in intermediate conditions. Once recovered, the uppermost 0–2 cm layer of each sediment sample was collected, put in sterile glass bottles, and stored at −20 °C until processing.
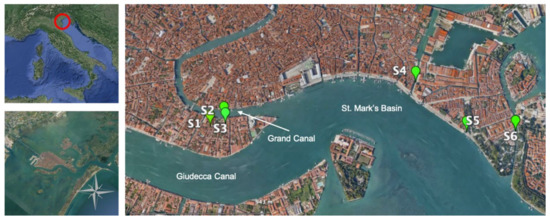
Figure 1.
Map showing the study area and the location of the six sampling stations in the canals of the city of Venice.
At all stations, water temperature (°C), salinity (PSU), dissolved oxygen (DO, mg/L), and redox potential (ORP) were measured in surface and bottom seawater layers using a Hydrolab DS5 multiparameter probe. Measured values are reported in Supplementary Table S1.
2.3. DNA Extraction and Shotgun Metagenomic Sequencing
DNA was extracted from sediment samples (1 g each) using the DNeasy PowerSoil kit (Qiagen, Germantown, MD, USA) following the manufacturer’s instructions with a few modifications to increase DNA yield and quality, as previously reported [43,44]; these included two additional vortexing steps (following the one recommended by the manufacturer) for 2 min at the maximum speed, each one preceded by incubation at 70 °C for 5 min, and one more washing step with Solution C5, as an additional removal step for contaminants. DNAs were stored at −20 °C until sequencing.
Shotgun metagenomic sequencing (approximately 100 million per sample) was performed on an Illumina MiSeq at the Wake Forest University Cancer Genomics Center (USA), which returned data as 75-nucleotide, paired-end reads. Raw sequences are available at the Github site (https://github.com/Curran-WFU, accessed on 7 March 2022).
2.4. Data Analysis
Raw paired-end sequence reads were uploaded to the MG-RAST (version 4.0.3) [45] analytical platform for gene identification and annotation using subsystems technology [46]. To facilitate file management, data for each metagenome are provided in four large datasets, each of which includes files of annotated sequences distributed among prokaryotic functional subsystems. Previous work [47] showed that all four of the datasets created by MG-RAST are essentially identical; for this reason, for our analysis, we chose one of the four datasets at random and focused on two subsystems: taxonomic identifications based on 16S rRNA gene fragments, and genes mapped to the “virulence, disease and defense” functional subsystem. Genus frequency accumulation curves analysis was performed to assess the comparability of the analyzed metagenomes (Supplementary Figure S1). All analyses were performed using programs written in Python, version 3.7. The datasets and programs are available at https://github.com/Curran-WFU (accessed on 1 February 2022).
MG-RAST provides taxonomic identifications by sequence alignment of the 16S rRNA gene fragments in the metagenomes to several large rRNA databases (see the MG-RAST documentation, https://help.mg-rast.org/user_manual.html, accessed on 1 February 2022). We focused on the genus-level identification, the deepest level that MG-RAST provides, because many metabolic characteristics are associated with genera. For the analysis of alpha diversity, we calculated the Inverse Simpson diversity index [48]. The minimum numbers of genera that contain 50% (G50) or 95% (G95) of sequences were calculated based on rarefaction plots prepared as described in Chapter 4 of [48].
To analyze beta-diversity, we performed Non-metric Multidimensional Scaling (NMDS) based on a Bray–Curtis dissimilarity matrix. NMDS was performed with the scikit-learn MDS module (https://scikit-learn.org/stable/modules/manifold.html#multi-dimensional-scaling-mds, accessed on 1 February 2022). In addition, genus correlation analyses were performed using the scikit-learn correlation module (https://pandas.pydata.org/docs/reference/api/pandas.DataFrame.corr.html, accessed on 1 February 2022).
From the metagenome-derived 16S rDNA gene dataset, we looked for bacterial taxa previously identified as potential microbial pollutants, as previously reported in several studies [9,21,22,24] and including: (i) traditional fecal bacteria (i.e., E. coli and Enterococcus), (ii) alternative feces-associated bacteria from human and non-human sources (Bacteroidaceae, Clostridiaceae, Lachnospiraceae, Porphyromonadaceae, Prevotellaceae, Rikenellaceae and Ruminococcaceae families), (iii) sewage-associated bacteria (Acinetobacter, Arcobacter and Trichococcus genera), and (iv) other common and emerging potential human pathogens (Achromobacter, Aeromonas, Bacillus, Bordetella, Brucella, Campylobacter, Citrobacter, Clostridium, Comamonas, Corynebacterium, Cronobacter, Deinococcus, Dermatophilus, Gardnerella, Halomonas, Klebsiella, Kocuria, Legionella, Leptospira, Listeria, Micrococcus, Mycobacterium, Neisseria, Photobacterium, Propionibacterium, Pseudomonas, Roseomonas, Rothia, Salmonella, Shewanella, Shigella, Staphylococcus, Stenotrophomonas, Streptococcus, Vibrio, Yersinia) [23,24].
We characterized genes associated with antibiotic resistance within the “virulence, disease and defense” (VDD) functional subsystem, as identified by MG-RAST. To identify antibiotic-resistance genes within these datasets, we searched the annotations for the list of terms (entire words or word fragments) associated with resistance. The complete set of searched terms is reported in Supplementary Table S2. The MG-RAST annotations rarely reference specific antibiotics, such as “penicillin”, but instead almost exclusively refer to antibody classes, such as “beta-lactamase”, which would include the natural and semisynthetic forms of penicillin, as well as the carbapenems and cephalosporins. Additionally, in some cases, annotations referred to both a specific antibiotic and its class, which creates the risk of double counting such sequences. To resolve these issues, annotations that listed antibiotic resistance were grouped by antibiotic class. Topoisomerase can be inhibited by members of two classes, quinolones and aminocoumarins, that were not distinguished in the VDD database. Therefore, we simply classified resistance genes as “Topoisomerase inhibitors”. The classes beta-lactamase, macrolide, acriflavine, aminoglycosides, topoisomerase inhibitor, and multidrug efflux were abundant and are reported in Results and Discussion. Classes for which few or no representative genes were detected in the canals are listed in Supplementary Table S2.
To detect genes related to resistance to toxic and heavy metals, we also searched the VDD annotations for keywords related to resistance to arsenic cadmium, chromium, cobalt, copper, lead, mercury and zinc. The keywords used are listed in Supplementary Table S3. Finally, to detect genes related to bacterial virulence, we searched for keywords related to exotoxin, hemolysin, leucocidin, collagenase, type III secretion, hyaluronic acid, and fibronectin-binding proteins.
3. Results and Discussion
3.1. Prokaryotic Diversity and Community Composition
We first analyzed the overall prokaryotic composition at the genus level at the six sample sites. The sites contained diverse populations (avg. richness 780 ± 44; avg. inverse Simpson 129 ± 36) (Table 1), as one might expect for similar environments characterized by organic enrichment and the presence of human and animal wastes. Each sample contained at least one DNA sequence from approximately 800 identifiable genera (Table 1). Interestingly, richness and Inverse Simpson index values did not differ significantly among samples (t-test, p > 0.5); however, lower Inverse Simpson values were displayed at S1, which displayed higher frequencies of the genus Beggiatoa. The numbers of genera, including at least 50% and 95% of the sequences (G50, G95, respectively), were also similar among sites (Table 1, Supplementary Figure S1). All sites also contained a substantial number of unclassified Gammaproteobacteria (between 4.4 to 10% per site) and Deltaproteobacteria (now phylum Desulfobacterota, according to SILVA database, https://www.arb-silva.de/, accessed on 1 February 2022) (1 to 3.3% per site). Because these high-level taxa are so metabolically diverse, we excluded them from further investigation.

Table 1.
Measures of alpha diversity. Values of richness (i.e., number of genus-level identifications), Inverse Simpson, G50, and G95 (i.e., number of taxa that contained 50% and 95%, respectively, of the sequence reads) per sampling site are reported.
Non-metric Multidimensional Scaling (NMDS, a two-dimensional representation of the relative similarities among sites) analysis (Supplementary Figure S2) based on Bray–Curtis dissimilarity (Supplementary Table S4) showed that Venice samples clustered in two groups, one including S1 and the second including S2 to S6 (Supplementary Figure S2).
We then compared relative genus frequencies of the top taxa within and among the datasets from these sites (Supplementary Figure S3). We found that the prokaryotic community composition at sites S2 and S3 and at S4 and S6 appeared to be relatively similar to each other, whereas sites S1 and S5 displayed a slightly different composition from the others and from each other, corroborating what was previously observed in the NMDS analysis. This was also confirmed when analyzing the percentage of shared and specific taxa across the six metagenomes. This analysis showed that more than 53% of taxa were shared among all samples and that, conversely, only 1.2 to 7.3% of taxa were specific to a single sample. Overall, analyses of community composition showed comparable frequencies of Nitrosopumilus at stations S2, S3, S4, and S6 (0.01–0.019, with a substantially higher value recorded at S5 (0.054) and a lower value (0.0035) at S1. At the same time, S1 exhibited the highest Beggiatoa (0.098) and Desulfobacterium (0.034) frequencies. Similar frequencies were found among samples for the other most abundant taxa (Supplementary Figure S3). Genus Beggiatoa mainly includes sulfur-oxidizing bacteria, growing at the oxic–anoxic interface on reduced, organic, or hydrocarbon-rich porous sediments [49,50] and, typically, in organic-rich, coastal marine sediments [51,52], suggesting a higher organic enrichment at S1. Desulfobacterium is a sulfur-reducing bacterium; interestingly, the species Desulfobacterium autotrophicum within this genus was originally isolated from anoxic sediments of Rio Marin, a canal in Venice’s historic center [53]. Finally, Nitrosopumilus, a key ammonia-oxidizer archeon [54], has been shown to be substantially enriched by co-culture with sulfur-oxidizing bacteria (SOB) in marine sediments [55], suggesting that at stations S2 to S6, the presence of SOB might couple with the higher frequency of genus Nitrosopumilus, as previously reported [56].
To determine whether taxa systematically varied among sites, we calculated correlation coefficients between pairs of taxa, with a focus on the top ten taxa, which showed notable covariant relationships (Supplementary Figure S4, panel A). Nitrosopumilus, Maribacter, and Planctomyces were found to positively covary with each other, with all of them being negatively covaried with the other taxa. Nitrosopumilus and Planctomyces share a role in nitrogen metabolism, which occurs extensively within these groups [57], while no information on this aspect is available for Maribacter. The remaining taxa (including Beggiatoa, Desulfobacterium, Bacteroides, Desulfatibacillum, Desulfococcus, and Geobacter), all known anaerobes and linked to sulfate metabolism [58], were all negatively covariant with those above but positively correlated with each other. Because the correlation matrix in Supplementary Figure S4 (panel A) tends to separate taxa based on aerobiosis, we compared the correlation coefficients among taxa known to be strict aerobes or anaerobes. In Supplementary Figure S4 (panel B), the anaerobic genera Bacteroides, Syntrophus, Anaerolinea, and Clostridium are clearly distinguishable from the aerobes Mycobacterium, Pseudomonas, Shewanella, and Maribacter.
Altogether, the correlation analyses strongly suggested that oxygen availability played a role in the observed differences in taxon composition among samples, with the highest proportions of strict anaerobes at S1 and lowest at S5. The highest proportions of Beggiatoa (3- to 20-fold higher than other sites) also characterized S1. Since members of this genus are able to oxidize sulfides using nitrate as an electron acceptor [59], we hypothesize that S1 had greater availability of nitrate than the other sites. Sites S1 and S5 were also outliers in the Bray–Curtis-based NMDS plots, and we hypothesize that, since S1 is located in a small canal that has relatively low boat traffic, it, therefore, may have contained a relatively high proportion of stabilized, low-oxygen sediments. In contrast, S5 was close to the always busy St. Mark’s basin and therefore likely experienced a greater degree of mixing due to heavy boat traffic.
3.2. Microbial Pollutants
The lagoon of Venice is known to host a number of microbial pollutants originating from a variety of sources, which are also related to the discharge of untreated or partially treated wastewaters from the city center [13,14,24,60]. Several studies have demonstrated that sediments and beach sands can be heavily colonized by fecal bacteria and that settling and resuspending colonized particles may significantly influence the distribution of microbial pollutants in the overlying water column [61,62]. However, measurements of fecal bacteria in sediments are rarely incorporated into monitoring programs, and geographic surveys of their distribution are still rare. This also holds true for the city of Venice, where studies have so far focused on assessing their distribution in water, while the contamination levels in sediments within the city canals have rarely been studied [60].
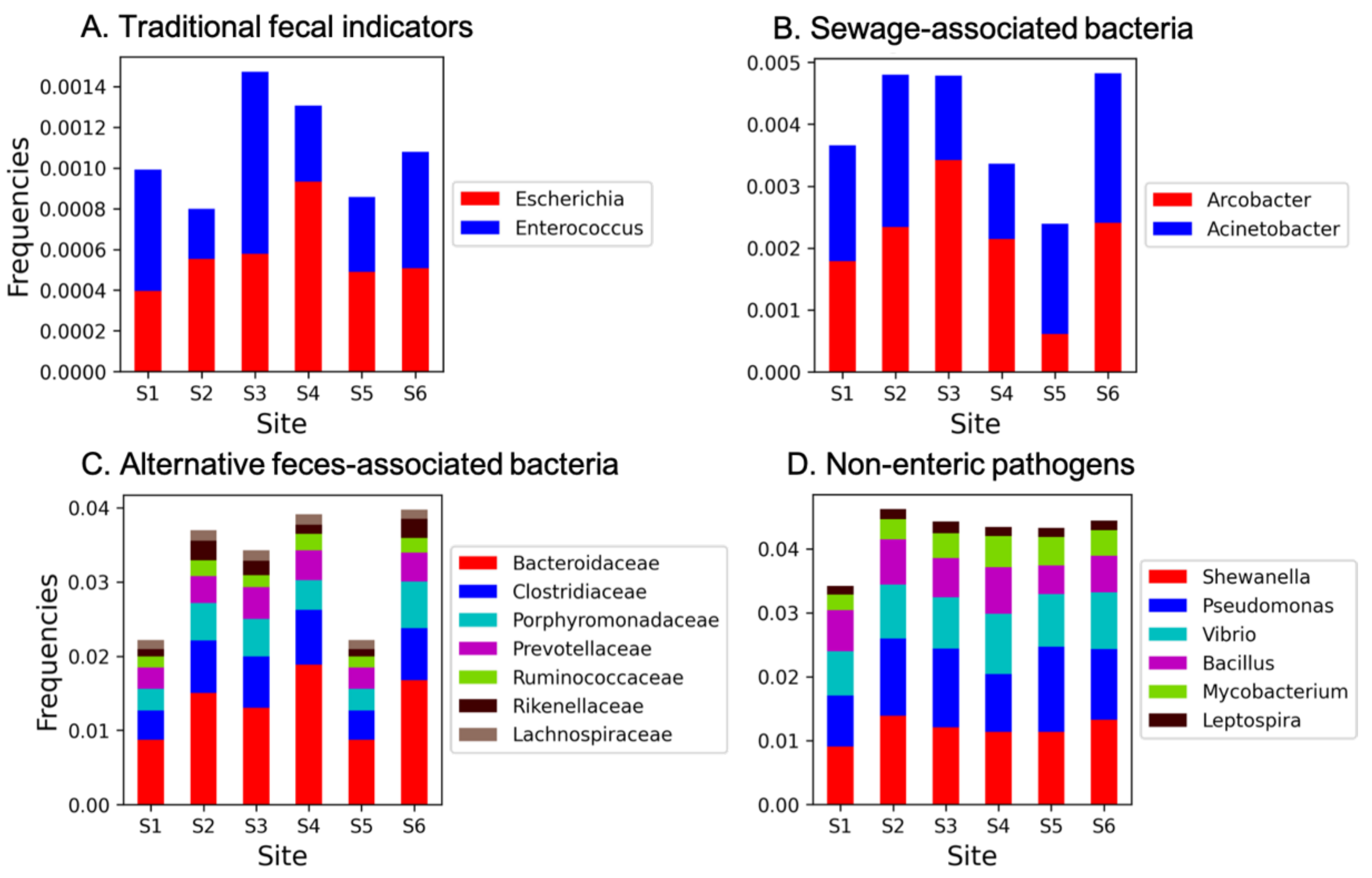
In our dataset, all the sites analyzed displayed a relatively high level of cumulative microbial fecal contamination, with lower values registered at S5 (about 8% of the total 16S rRNA gene data, respectively) and higher values at S1 (about 9.8% of the total 16S rRNA gene data extracted from metagenomes). When analyzing the different contributions to the total potential pathogenic community, we found that traditional FIB frequencies (i.e., Escherichia plus Enterococcus genera) (Figure 2A) were relatively low and ranged from ~0.07% at S2 to 0.13% at S3. Sewage-associated bacterial frequencies (i.e., Acinetobacter plus Arcobacter) ranged from ~0.2% to 0.42% at S5 and S6, respectively (Figure 2B), with Arcobacter tending to vary the most among sites. Alternative feces-associated bacteria were dominated by the families Bacteroidaceae (mainly represented by the Bacteroides genus) and Clostridiaceae and showed a similar trend as FIB, with the lowest frequencies recorded at S5 (1.8%) and highest at S6 (4.4%) (Figure 2C) and an average percentage 3.3 ± 0.83% among the six sites. Finally, relatively high levels of other potential pathogens were displayed by all sites (avg. percentage of 3.5 ± 0.28%), with a relatively lower frequency at S1 (3.2%), and with the genera Pseudomonas, Shewanella, and Vibrio dominating this fraction of the potential pathogenic community overall (Figure 2D). The total potential pathogenic signature at each sampling site is reported in Supplementary Figure S5.
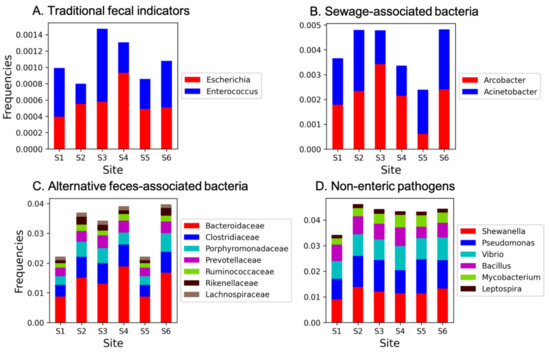
Figure 2.
(A) Frequencies of traditional, (B) sewage-associated, (C) alternative feces-associated, and (D) other non-enteric potential pathogens at each sampling station. All taxa reported in the Materials and Methods section and not reported in Figure legend were not found in our dataset. Frequencies were determined by dividing the number of sequences whose annotations contained the selected keyword(s) by the total number of sequences in that Subsystems database (e.g., VDD).
Overall, our results corroborate previous findings that report high levels of microbial pollution and, in particular, fecal contaminants in this study area [13,14,24]. Our data also suggest that sediment microbial pollution of fecal origins is ubiquitous in the sampled stations of Venetian canals. However, we hypothesize that different distances of each sampling site from the likely sources of pollution might have contributed to the different contamination levels observed. In fact, flocculation of particles and associated bacteria is predicted to be quite rapid near the sewers as the transition between freshwater and seawater is almost immediate, thus causing flocculation and accelerating particle settling that results in a patchy fecal signature in the sediments. In addition, water exchange in the different sites and the consequent different degrees of tidal flushing might also explain such differences. Moreover, it has been recently reported that microbial pollutants were almost exclusively associated with the particle-attached fraction in the Venice lagoon [24], thus supporting the need for prioritizing management plans aimed at reducing particulate matter inputs to the lagoon to limit microbial pollution.
The cumulative frequencies of other non-enteric potential (human and aquatic animals) pathogens (Figure 2D) displayed a similar distribution and abundance at all stations except for S1, which suggests a systematic and even release of these potential pathogens in the canals of the city center. Members of the Pseudomonas genus are known etiological agents of disease in humans and animals, who have the ability to become a potential pathogen in aquatic environments, also for issues related to antibiotic resistance [63] and reference therein. Shewanella is widely distributed in aquatic environments [64], but members of this genus are considered an emerging cause of human infections; multidrug resistance has increasingly been reported in this genus, thus making it a potential reservoir and vehicle for the transmission of ARGs [65,66,67,68]. However, the ability of different Shewanella spp. to convert heavy metals and toxic substances, such as chromates, into less toxic products by using them as electron acceptors has also been reported [69]. Finally, despite being indigenous to marine and brackish water, the genus Vibrio includes some pathogenic species (i.e., Vibrio cholerae, V. vulnificus, V. parahaemolyticus, V. alginolyticus).
Although there is still much to be deciphered about the true pathogenic potential of these taxa, their presence in the lagoon waters poses additional concerns on the risks for humans in this lagoon and nearby bathing waters, deserving further studies. Moreover, these results highlight the potential risks posed by the recent modifications of the inlet morphology to protect Venice from storm surges and the increased potential interaction between the tidal jets with coastal waters by redirecting part of the outflowing currents towards the adjacent beaches.
3.3. Antibiotic- and Heavy Metal-Resistance Genes
Anthropogenic impact, including wastewaters and industrial wastes, may introduce antibiotic residues (ABs) and heavy metals (HMs) as well as antibiotic and heavy metal resistance genes (ARGs and HMRGs, respectively) in the aquatic environment [70,71]. Due to their selective pressure, ABs and HMs may promote the acquisition and/or transfer of resistance genes by Horizontal Gene Transfer (HGT) to other bacteria living or released in the environment [72]; moreover, HMs pose selective pressure on ARGs [71], and HMRGs often co-occur with ARGs [70,72]. Here, aimed at exploring the contamination of Venice canals by these emerging pollutants, we looked for genes coding for AB and HM resistance in our metagenomic dataset.
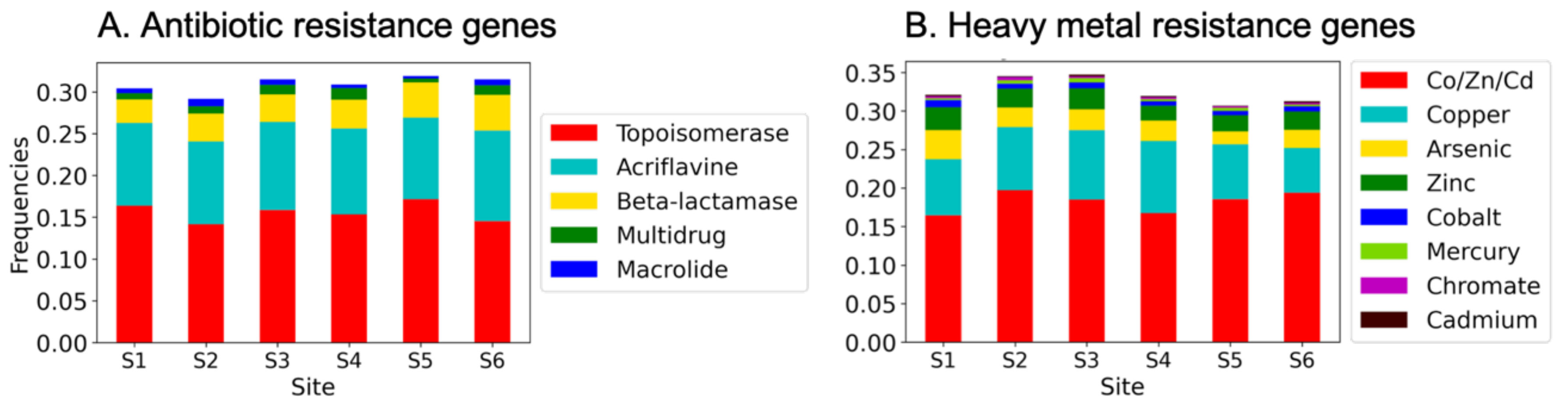
MG-RAST returned genes by functional class in multiple databases (COG, NOG, KO, and Subsystems). Subsystem genes were distributed into the greatest number of categories, including those related to “virulence, disease and defense” (VDD). The VDD subsystem comprises approximately 3% of the sequences from each site, and the genes were distributed among 470 gene functions such as “beta-lactamase”. HMR and ARG genes are each represented by about 1/3 of the VDD genes (HMR frequencies of 0.33 ± 0.016) and ARG frequencies of 0.31 ± 0.01) (Figure 3). The remainders of the genes are related primarily to stress responses and to virulence factors. To determine their approximate relative abundances of specific ARG and HMR genes in the canal populations, we searched the VDD gene annotations for words or word fragments specific to ARG or HMR names or descriptions, and genes were grouped by antibiotic class, as detailed in the Material and Method section. The relative frequencies of ARGs and HMRGs found in our dataset within the VDD database are shown in Figure 3.
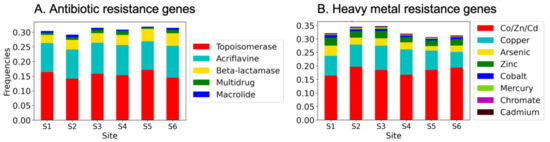
Figure 3.
(A) Antibiotic and (B) heavy metal resistance gene frequencies in the canal samples. Keywords for ARG and HMR resistance genes are in the legends in the corresponding figure panel. Shown are frequencies in genes identified by MG-RAST as related to “virulence, disease and defense”. Frequencies were determined by dividing the number of identified genes by the total number of VDD genes at each site. The total numbers of genes in the VDD category at each site: S1 = 4056; S2 = 2088; S3 = 2463; S4 = 1283; S5 = 2003; S6 = 2057.
We found that ARGs and HMRs were common in the canal samples and that, similarly to what was observed for the community composition, the sites contained comparable repertoires of these genes (Figure 3A). Notably, genes conferring resistance to the “Topoisomerase inhibitor” antibiotic group were the most abundant (mean of 16 ± 1.1% of the VDD genes and 0.47 ± 0.03% of all genes among the sites). These were detected using searches for the generic term “topoisomerase” and, more specifically within this class, “gyrase” (commonly associated with novobiocin resistance [73] and quinolone resistance [74]). It must be pointed out, however, that the presence of topoisomerase and gyrase genes is not conclusive that resistance variants are present but only that the potential for resistance exists. In contrast, the genes identified in the VDD database for the other ARG categories are specific to antibiotic resistance. These include substantial numbers of genes of resistance to other antibiotic resistance-related genes were found in the analyzed metagenomes, which included “acriflavine” and “macrolide”, as well as “beta-lactamases”, “multidrug efflux” pumps, and “macrolide” as indicated in Figure 3A. Genes related to the aminoglycoside and vancomycin classes were present at some sites, and genes conferring resistance to other classes of antibiotic (e.g., tetracycline, folate acid synthesis inhibitors) were not found. The complete list of searched terms is included in Supplementary Table S3.
Overall, the wide range of genes at relatively high frequencies and related to antibiotic resistance in our dataset confirms previous findings that sediments in human-impacted areas can act as a reservoir of antibiotic-resistant bacteria and their genes [75]. The genes found belong to antibiotic resistances previously reported as contaminants in several studies on environmental, aquatic settings [76,77,78], including other anthropically impacted lagoons [79,80]. Overall, our results suggest that the canals of Venice may represent a source of potential ARB possibly transferable to humans by food chain or by recreational activities [81]. It is noteworthy that beta-lactamases and fluoroquinolones (the latter falling in the broader category of “topoisomerases”) have been reported among the most used antibiotics in Italy, according to a recent document published by the Italian National Institute of Health (ISS) (https://www.epicentro.iss.it/antibiotico-resistenza/pdf/Rapporto_Antibiotici_2019.pdf, accessed on 1 February 2022). According to the same report, polymyxins, a last-resort type of AB [82], are still poorly used in Italy; nevertheless, the finding of genes potentially involved in the resistance to these compounds highlights possible emerging threats related to the use of such ABs. Similarly, trimethoprim (TMP) is still scarcely used at the national level, likely due to limited efficacy given the rapidity with which TMP resistance develops [83]. Taken together, our results underline the need for further study to clarify the overall ARGs contamination in this area.
Given the long history of industrial activity in Venice, we also looked for genes in the canals related to metal contaminants. After the 1960s, large industrial activities progressively left the historic center; however, activities by some remaining craftsmen produce a residual pollutant discharge. These point sources combine with diffuse inputs of a typical urban system, which, in the case of Venice, includes very high boat traffic, leading to the release of chemical contaminants (e.g., PAHs and copper from anti-fouling paints). Moreover, many modern building materials such as rooftops and gutters contain metallic components, which may be toxic to marine life and, potentially, human populations [84]. A synthesis of the results of chemical contamination assessment in the system was published by [85]. The comparative study of the recent changes in pollution levels by [86] shows that, despite a considerable reduction in heavy metal inputs after the last maintenance dredging (1995–2005), the concentration of elements of toxic concern (Cd, Cr, Cu, Hg, Pb, Zn) are still higher than international sediment quality guidelines limits. We therefore searched the VDD Subsystem datasets for genes related to resistance to HMs. Such genes were found in abundance at all sites (Figure 3B). Genes related to the multimetal resistance to cobalt, zinc and cadmium (Co/Zn/Cd) as well as to copper alone were the most common, whereas genes related to the resistance towards other metals, including mercury, instead, were rare. Mercury contamination has been problematic in the western regions of the lagoon; however, it is currently less significant than other metals, at least in the canals of the historical city center. Nevertheless, the most represented type of genes in this analysis was made up by multiple metals resistance genes (Figure 3B); although a deeper insight on this data was not possible due to the resolution of our data, previous findings report that multiple resistance genes, such as, for instance, cadmium-zinc-cobalt resistance, were found to be correlated with ARGs [30], and thus possibly involved in phenomena of genetic co-selection between heavy metals and antibiotic resistances.
Finally, we searched for virulence-related genes. In our dataset, such genes were found to be rare. For example, all sites except S4 had from one to four “fibronectin-binding protein” genes, and sites S1, S2, S3, and S5 each had up to three “hemolysin” genes. No site had a gene identified as an exotoxin, hyaluronidase, leukocidin, or type III secretion system component. It is possible that virulence factor genes do not persist long in the canal sediments. Alternatively, it is possible that the representative virulence-related genes in the VDD database, which have been characterized in a restricted set of medically relevant organisms, do not contain the sequence diversity required to detect distantly related genes from these environmental communities.
4. Conclusions
The Venetian canal system has a long history of management and may currently be in a phase of transition. For example, after rising in the latter half of the 20th century, sediment nitrogen and phosphate, along with the general level of industrial contamination, have been considerably reduced [87]. However, the lagoon and, particularly, the city center still receive high levels of domestic waste, which can potentially threaten the quality of the nearby recreational beaches. Moreover, the recent modifications of the inlet morphology to protect Venice from tides, in fact, increased the potential interaction of the tidal jets with coastal waters by redirecting part of the outflowing currents towards the adjacent beaches. Our analyses highlighted that microbial pollution is ubiquitous in the studied sites, thus raising concerns about the effects on water quality on the nearby lagoon areas and bathing waters of the adjacent beaches. This work provides useful knowledge of current levels of microbial contamination, including the presence and distribution of fecal and other potentially pathogenic contaminants as well as AB and HM resistance genes. For this reason, we expect that our study will be useful for future monitoring actions in the area. In addition, given the fluctuations in tourism, boat traffic, and tides, seasonal variations should also be considered, prompting the need to widen this kind of study to address the dynamics of microbial pollutants over time.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/w14071161/s1, Figure S1: Genus frequency accumulation curves; Figure S2: NMDS plots based on taxa frequencies; Figure S3: Frequencies of the top ten taxa from each site; Figure S4: Correlation Matrices; Figure S5: Total pathogenic signature; Table S1: Environmental parameters measured within this study; Table S2: Keywords used to search for antibiotic resistance genes in our dataset; Table S3: Keywords used to search for heavy metal resistance genes in our dataset; Table S4: Bray-Curtis dissimilarity matrix.
Author Contributions
Conceptualization, G.M.Q. and J.F.C.; methodology, G.M.Q. and J.F.C.; formal analysis, J.F.C.; investigation, J.F.C. and L.Z.; resources, J.F.C.; writing—original draft preparation, G.M.Q. and J.F.C.; writing—review and editing, G.M.Q., J.F.C. and L.Z.; visualization, J.F.C.; funding acquisition, J.F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was performed within the scope of a microbiology course (Bio 332), with funding provided by the operating budgets of the Department of Biology and the Center for Global Programs and Studies at Wake Forest University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw sequences, metadata, and programs described in this study are available at the Github site https://github.com/Curran-WFU (accessed on 1 February 2022).
Acknowledgments
We thank the fall of the 2019 Wake Forest University Microbiology Class for sample collection and DNA extraction. We also thank Isabelle Ricke, who assisted in the analysis, for her contribution and Gian Marco Luna for his constructive comments on the manuscript. We are especially grateful to the Department of Biology of Padua University for the access to the laboratory facilities “Fiore di Botta” and to Lorenzo Zane for his logistic support and for comments that have improved this manuscript. The authors also wish to acknowledge the support of the Wake Forest Baptist Comprehensive Cancer Center Cancer Genomics Shared Resource, supported by the National Cancer Institute’s Cancer Center Support Grant award number P30CA012197. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fries, J.S.; Characklis, G.W.; Noble, R.T. Attachment of fecal indicator bacteria to particles in the Neuse River Estuary, NC. J. Environ. Eng. 2006, 132, 1338–1345. [Google Scholar] [CrossRef]
- Vörösmarty, C.J.; McIntyre, P.B.; Gessner, M.O.; Dudgeon, D.; Prusevich, A.; Green, P.; Glidden, S.; Bunn, S.E.; Sullivan, C.A.; Liermann, C.R.; et al. Global threats to human water security and river biodiversity. Nature 2010, 67, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.E.; Elliott, K.H. Tracking marine pollution. Science 2013, 340, 556–558. [Google Scholar] [CrossRef]
- Bernhard, A.E.; Goyard, T.; Simonich, M.T.; Field, K.G. Application of a rapid method for identifying fecal pollution sources in a multi-use estuary. Water Res. 2003, 37, 909–913. [Google Scholar] [CrossRef]
- Lipp, E.K.; Kurz, R.; Vincent, R.; Rodriguez-Palacios, C.; Farrah, S.R.; Rose, J.B. The effects of seasonal variability and weather on microbial fecal pollution and enteric pathogens in a subtropical estuary. Estuaries 2001, 24, 266–276. [Google Scholar] [CrossRef]
- Mallin, M.A.; Williams, K.E.; Esham, E.C.; Lowe, R.P. Effect of human development on bacteriological water quality in coastal watersheds. Ecol. Appl. 2000, 10, 1047–1056. [Google Scholar] [CrossRef]
- Milledge, D.G.; Gurjar, S.K.; Bunce, J.T.; Tare, V.; Sinha, R.; Carbonneau, P.E. Population density controls on microbial pollution across the Ganga catchment. Water Res. 2018, 128, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.R.; Gast, R.J.; Fujioka, R.S.; Solo-Gabriele, H.M.; Meschke, J.S.; Amaral-Zettler, L.A.; Del Castillo, E.; Polz, M.F.; Collier, T.K.; Strom, M.S.; et al. The coastal environment and human health: Microbial indicators, pathogens, sentinels and reservoirs. Environ. Health 2008, 7, S3. [Google Scholar] [CrossRef] [Green Version]
- Luna, G.M.; Quero, G.M.; Perini, L. Next generation sequencing reveals distinct fecal pollution signatures in aquatic sediments across gradients of anthropogenic influence. Adv. Oceanogr. Limnol. 2016, 7, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Byappanahalli, M.N.; Nevers, M.B.; Korajkic, A.; Staley, Z.R.; Harwood, V.J. Enterococci in the environment. Microbiol. Mol. Biol. Rev. 2012, 76, 685–706. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Goh, S.G.; Vergara, G.G.R.V.; Fang, H.M.; Rezaeinejad, S.; Chang, S.Y.; Bayen, S.; Lee, W.A.; Sobsey, M.D.; Rose, J.B.; et al. Alternative fecal indicators and their empirical relationships with enteric viruses, Salmonella enterica, and Pseudomonas aeruginosa in surface waters of a tropical urban catchment. Appl. Environ. Microbiol. 2015, 81, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Luna, G.M.; Manini, E.; Turk, V.; Tinta, T.; D’Errico, G.; Baldrighi, E.; Baljak, V.; Buda, D.; Cabrini, M.; Campanelli, A.; et al. Status of faecal pollution in ports: A basin-wide investigation in the Adriatic Sea. Mar. Pollut. Bull. 2019, 147, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Perini, L.; Quero, G.M.; Serrano García, E.; Luna, G.M. Distribution of Escherichia coli in a coastal lagoon (Venice, Italy): Temporal patterns, genetic diversity and the role of tidal forcing. Water Res. 2015, 87, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Quero, G.M.; Fasolato, L.; Vignaroli, C.; Luna, G.M. Understanding the association of Escherichia coli with diverse macroalgae in the lagoon of Venice. Sci. Rep. 2015, 5, 10969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, K.G.; Samadpour, M. Fecal source tracking, the indicator paradigm, and managing water quality. Water Res. 2007, 41, 3517–3538. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Solo-Gabriele, H.M.; Fleming, L.E.; Elmir, S. Monitoring marine recreational water quality using multiple microbial indicators in an urban tropical environment. Water Res. 2004, 38, 3119–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Walk, S.T.; Gordon, D.M.; Feldgarden, M.; Tiedje, J.M.; Konstantinidis, K.T. Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. USA 2011, 108, 7200–7205. [Google Scholar] [CrossRef] [Green Version]
- McLellan, S.L.; Newton, R.J.; Vandewalle, J.L.; Shanks, O.C.; Huse, S.M.; Eren, A.M.; Sogin, M.L. Sewage reflects the distribution of human faecal Lachnospiraceae. Environ. Microbiol. 2013, 15, 2213–2227. [Google Scholar] [CrossRef] [Green Version]
- McLellan, S.L.; Eren, A.M. Discovering new indicators of fecal pollution. Trends Microbiol. 2014, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; VandeWalle, J.L.; Borchardt, M.A.; Gorelick, M.H.; McLellan, S.L. Lachnospiraceae and Bacteroidales alternative fecal indicators reveal chronic human sewage contamination in an Urban harbor. Appl. Environ. Microbiol. 2011, 77, 6972–6981. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Bootsma, M.J.; Morrison, H.G.; Sogin, M.L.; McLellan, S.L. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of lake Michigan. Microb. Ecol. 2013, 65, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Buccheri, M.A.; Salvo, E.; Coci, M.; Quero, G.M.; Zoccarato, L.; Privitera, V.; Rappazzo, G. Investigating microbial indicators of anthropogenic marine pollution by 16S and 18S High-Throughput Sequencing (HTS) library analysis. FEMS Microbiol. Lett. 2019, 366, fnz179. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Campanelli, A.; Frapiccini, E.; Luna, G.M.; Quero, G.M. Occurrence and distribution of microbial pollutants in coastal areas of the Adriatic Sea influenced by river discharge. Environ. Pollut. 2021, 285, 117672. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Techtmann, S.M.; Zaggia, L.; Luna, G.M.; Quero, G.M. Partitioning and sources of microbial pollution in the Venice Lagoon. Sci. Total Environ. 2021, 818, 151755. [Google Scholar] [CrossRef]
- Ming, H.; Ma, Y.; Gu, Y.; Su, J.; Guo, J.; Li, J.; Li, X.; Jin, Y.; Fan, J. Enterococci may not present the pollution of most enteric pathogenic bacteria in recreational seawaters of Xinghai bathing Beach, China. Ecol. Indicat. 2020, 110, 105938. [Google Scholar] [CrossRef]
- Li, W.; Su, H.; Cao, Y.; Wang, L.; Hu, X.; Xu, W.; Xu, Y.; Li, Z.; Wen, G. Antibiotic resistance genes and bacterial community dynamics in the seawater environment of Dapeng Cove, South China. Sci. Total Environ. 2020, 723, 138027. [Google Scholar] [CrossRef]
- Shao, S.; Hu, Y.; Cheng, J.; Chen, Y. Research progress on distribution, migration, transformation of antibiotics and antibiotic resistance genes (ARGs) in aquatic environment. Crit. Rev. Biotechnol. 2018, 38, 1195–1208. [Google Scholar] [CrossRef]
- Calero-Cáceres, W.; Balcázar, J.L. Antibiotic resistance genes in bacteriophages from diverse marine habitats. Sci. Total Environ. 2019, 654, 452–455. [Google Scholar] [CrossRef]
- Karkman, A.; Pärnänen, K.; Larsson, D.J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 2019, 10, 80. [Google Scholar] [CrossRef]
- Di Cesare, A.; Eckert, E.M.; D’Urso, S.; Bertoni, R.; Gillan, D.C.; Wattiez, R.; Corno, G. Co-occurrence of integrase 1, antibiotic and heavy metal resistance genes in municipal wastewater treatment plants. Water Res. 2016, 94, 208–214. [Google Scholar] [CrossRef]
- Vierheilig, J.; Savio, D.; Farnleitner, A.H.; Reischer, G.H.; Ley, R.E.; Mach, R.L.; Farnleitner, A.H.; Reischer, G.H. Potential applications of next generation DNA sequencing of 16S rRNA gene amplicons in microbial water quality monitoring. Water Sci. Technol. 2015, 72, 1962–1972. [Google Scholar] [CrossRef] [Green Version]
- Rotter, A.; Barbier, M.; Bertoni, F.; Bones, A.M.; Cancela, M.L.; Carlsson, J.; Carvalho, M.F.; Cegłowska, M.; Chirivella-Martorell, J.; Dalay, M.C.; et al. The essentials of marine biotechnology. Front. Mar. Sci. 2021, 8, 158. [Google Scholar] [CrossRef]
- Umgiesser, G.; Canu, D.M.; Cucco, A.; Solidoro, C. A finite element model for the Venice Lagoon. Development, set up, calibration and validation. J. Mar. Syst. 2004, 51, 123–145. [Google Scholar] [CrossRef]
- Cucco, A.; Umgiesser, G. Modelling the Venice lagoon residence time. Ecol. Model. 2006, 193, 34–51. [Google Scholar] [CrossRef]
- Lagi, F.; Corti, G.; Meli, M.; Pinto, A.; Bartoloni, A. Leptospirosis acquired by tourists in Venice, Italy. J. Travel Med. 2013, 20, 128–130. [Google Scholar] [CrossRef] [PubMed]
- Bonello, V.; Faraone, C.; Gambarotto, F.; Nicoletto, L.; Pedrini, G. Clusters in formation in a deindustrialized area: Urban regeneration and structural change in Porto Marghera (Venice). Compet. Rev. 2020, 30, 417–436. [Google Scholar] [CrossRef]
- Scarpa, G.M.; Zaggia, L.; Manfè, G.; Lorenzetti, G.; Parnell, K.; Soomere, T.; Rapaglia, J.; Molinaroli, E. The effects of ship wakes in the Venice Lagoon and implications for the sustainability of shipping in coastal waters. Sci. Rep. 2019, 9, 19014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaggia, L.; Rosso, J.; Zonta, R. Sulphate reduction in the sediment of the Venice canals (Italy). Mar. Pollut. Bull. 2007, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Sfriso, A.; Facca, C. Annual growth and environmental relationships of the invasive species Sargassum muticum and Undaria pinnatifida in the lagoon of Venice. Estuar. Coast. Shelf Sci. 2013, 129, 162–172. [Google Scholar] [CrossRef]
- Coraci, E.; Umgiesser, G.; Zonta, R. Hydrodynamic and sediment transport 752 modelling in the canals of Venice (Italy). Estuar. Coast. Shelf Sci. 2007, 75, 250–260. [Google Scholar] [CrossRef]
- Madricardo, F.; Foglini, F.; Campiani, E.; Grande, V.; Catenacci, E.; Petrizzo, A.; Kruss, A.; Toso, C.; Trincardi, F. Assessing the human footprint on the sea-floor of coastal systems: The case of the Venice Lagoon, Italy. Sci. Rep. 2019, 9, 6615. [Google Scholar] [CrossRef] [PubMed]
- Ostoich, M.; Ghezzo, M.; Umgiesser, G.; Zambon, M.; Tomiato, L.; Ingegneri, F.; Mezzadri, G. Modelling as decision support for the localisation of submarine urban wastewater outfall: Venice lagoon (Italy) as a case study. Environ. Sci. Pollut. Res. 2018, 25, 34306–34318. [Google Scholar] [CrossRef] [PubMed]
- Basili, M.; Quero, G.M.; Giovannelli, D.; Manini, E.; Vignaroli, C.; Avio, C.G.; De Marco, R.; Luna, G.M. Major role of surrounding environment in shaping biofilm community composition on marine plastic debris. Front. Mar. Sci. 2020, 7, 262. [Google Scholar] [CrossRef]
- Quero, G.M.; Perini, L.; Pesole, G.; Manzari, C.; Lionetti, C.; Bastianini, M.; Marini, M.; Luna, G.M. Seasonal rather than spatial variability drives planktonic and benthic bacterial diversity in a microtidal lagoon and the adjacent open sea. Mol. Ecol. 2017, 26, 5961–5973. [Google Scholar] [CrossRef]
- Meyer, F.; Bagchi, S.; Chaterji, S.; Gerlach, W.; Grama, A.; Harrison, T.; Paczian, T.; Trimble, W.L.; Wilke, A. MG-RAST version 4—lessons learned from a decade of low-budget ultra-high-throughput metagenome analysis. Brief. Bioinform. 2019, 20, 1151–1159. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Rosso, G.E.; Muday, J.A.; Curran, J.F. Tools for metagenomic analysis at wastewater treatment plants: Application to a foaming episode. Water Environ. Res. 2018, 90, 258–268. [Google Scholar] [CrossRef]
- Magurran, A.E.; McGill, B.J. Biological Diversity: Frontiers in Measurement and Assessment; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Jean, M.R.; Gonzalez-Rizzo, S.; Gauffre-Autelin, P.; Lengger, S.K.; Schouten, S.; Gros, O. Two new Beggiatoa species inhabiting marine mangrove sediments in the Caribbean. PLoS ONE 2015, 10, e0117832. [Google Scholar] [CrossRef] [Green Version]
- Teske, A.; Nelson, D.C. The genera Beggiatoa and Thioploca. In The Prokaryotes, 4th ed.; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; Volume 6, pp. 784–810. [Google Scholar]
- Jørgensen, B.B. Distribution of colorless sulfur bacteria (Beggiatoa spp.) in a coastal marine sediment. Mar. Biol. 1977, 41, 19–28. [Google Scholar] [CrossRef]
- Mussmann, M.; Schulz, H.N.; Strotmann, B.; Kjaer, T.; Nielsen, L.P.; Rossello-Mora, R.A.; Aman, R.I.; Jørgensen, B.B. Phylogeny and distribution of nitrate-storing Beggiatoa spp. in coastal marine sediments. Environ. Microbiol. 2003, 5, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Brysch, K.; Schneider, C.; Fuchs, G.; Widdel, F. Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch. Microbiol. 1987, 148, 264–274. [Google Scholar] [CrossRef]
- Yu, S.; Yao, P.; Liu, J.; Zhao, B.; Zhang, G.; Zhao, M.; Yu, Z.; Zhang, X.H. Diversity, abundance, and niche differentiation of ammonia-oxidizing prokaryotes in mud deposits of the eastern China marginal seas. Front. Microbiol. 2016, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.J.; Park, S.J.; Yoon, D.N.; Schouten, S.; Sinninghe Damsté, J.S.; Rhee, S.K. Cultivation of autotrophic ammonia-oxidizing archaea from marine sediments in coculture with sulfur-oxidizing bacteria. Appl. Environ. Microbiol. 2010, 76, 7575–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Huang, X.; Zheng, T.L. Responses of bacterial and archaeal communities to nitrate stimulation after oil pollution in mangrove sediment revealed by Illumina sequencing. Mar. Pollut. Bull. 2016, 109, 281–289. [Google Scholar] [CrossRef]
- Kuypers, M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Findlay, A.J.; Pellerin, A. The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Preisler, A.; De Beer, D.; Lichtschlag, A.; Lavik, G.; Boetius, A.; Jørgensen, B.B. Biological and chemical sulfide oxidation in a Beggiatoa inhabited marine sediment. ISME J. 2007, 1, 341–353. [Google Scholar] [CrossRef]
- Fattore, E.; Benfenati, E.; Marelli, R.; Cools, E.; Fanelli, R. Sterols in sediment samples from Venice Lagoon, Italy. Chemosphere 1996, 33, 2383–2393. [Google Scholar] [CrossRef]
- O’Mullan, G.D.; Juhl, A.R.; Reichert, R.; Schneider, E.; Martinez, N. Patterns of sediment-associated fecal indicator bacteria in an urban estuary: Benthic-pelagic coupling and implications for shoreline water quality. Sci. Total Environ. 2019, 656, 1168–1177. [Google Scholar] [CrossRef]
- Luna, G.M.; Vignaroli, C.; Rinaldi, C.; Pusceddu, A.; Nicoletti, L.; Gabellini, M.; Danovaro, R.; Biavasco, F. Extraintestinal Escherichia coli carrying virulence genes in coastal marine sediments. Appl. Environ. Microbiol. 2010, 76, 5659–5668. [Google Scholar] [CrossRef] [Green Version]
- Jurelevicius, D.; Cotta, S.R.; Montezzi, L.F.; Dias, A.C.; Mason, O.U.; Picao, R.C.; Janet, K.J.; Seldin, L. Enrichment of potential pathogens in marine microbiomes with different degrees of anthropogenic activity. Environ. Poll. 2021, 268, 115757. [Google Scholar] [CrossRef] [PubMed]
- Satomi, M. The family Shewanellaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: New York, NY, USA, 2014; pp. 597–625. [Google Scholar]
- Janda, J.M. Shewanella: A marine pathogen as an emerging cause of human disease. Clin. Microbiol. Newsl. 2014, 36, 25–29. [Google Scholar] [CrossRef]
- Poirel, L.; Héritier, C.; Nordmann, P. Chromosome-encoded ambler class D β-lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob. Agents Chemother. 2004, 48, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Potron, A.; Poirel, L.; Nordmann, P. Origin of OXA-181, an emerging carbapenem-hydrolyzing oxacillinase, as a chromosomal gene in Shewanella xiamenensis. Antimicrob. Agents Chemother. 2011, 55, 4405–4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousfi, K.; Touati, A.; Lefebvre, B.; Fournier, É.; Côté, J.C.; Soualhine, H.; Walker, M.; Bougdour, D.; Tremblay, C.; Bekal, S. A novel plasmid, pSx1, harboring a new Tn1696 derivative from extensively drug-resistant Shewanella xiamenensis encoding OXA-416. Microb. Drug Resist. 2016, 23, 429–436. [Google Scholar] [CrossRef]
- Baaziz, H.; Lemaire, O.N.; Jourlin-Castelli, C.; Iobbi-Nivol, C.; Méjean, V.; Alatou, R.; Fons, M. Draft genome sequence of Shewanella algidipiscicola H1, a highly chromate-resistant strain isolated from Mediterranean marine sediments. Microbiol. Resour. Announc. 2018, 7, e00905-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.C.; Hugie, C.N.; Kile, M.L.; Navab-Daneshmand, T. Association between heavy metals and antibiotic-resistant human pathogens in environmental reservoirs: A review. Front. Environ. Sci. Eng. 2019, 13, 46. [Google Scholar] [CrossRef]
- Zheng, D.; Yin, G.; Liu, M.; Chen, C.; Jiang, Y.; Hou, L.; Zheng, Y. A systematic review of antibiotics and antibiotic resistance genes in estuarine and coastal environments. Sci. Total Environ. 2021, 777, 146009. [Google Scholar] [CrossRef]
- Vignaroli, C.; Pasquaroli, S.; Citterio, B.; Di Cesare, A.; Mangiaterra, G.; Fattorini, D.; Biavasco, F. Antibiotic and heavy metal resistance in enterococci from coastal marine sediment. Environ. Pollut. 2018, 237, 406–413. [Google Scholar] [CrossRef]
- Vickers, A.A.; Chopra, I.; O’neill, A.J. Intrinsic novobiocin resistance in Staphylococcus saprophyticus. Antimicrob. Agents Chemother. 2007, 51, 4484–4485. [Google Scholar] [CrossRef] [Green Version]
- Hooper, D.C.; Jacoby, G.A. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cesare, A.; Pasquaroli, S.; Vignaroli, C.; Paroncini, P.; Luna, G.M.; Manso, E.; Biavasco, F. The marine environment as a reservoir of enterococci carrying resistance and virulence genes strongly associated with clinical strains. Environ. Microbiol. Rep. 2014, 6, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Eckert, E.M.; Quero, G.M.; Di Cesare, A.; Manfredini, G.; Mapelli, F.; Borin, S.; Fontaneto, D.; Luna, G.M.; Corno, G. Antibiotic disturbance affects aquatic microbial community composition and food web interactions but not community resilience. Mol. Ecol. 2019, 28, 1170–1182. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Petrin, S.; Fontaneto, D.; Losasso, C.; Eckert, E.M.; Tassistro, G.; Borello, A.; Ricci, A.; Wilson, W.H.; Pruzzo, C.; et al. ddPCR applied on archived Continuous Plankton Recorder samples reveals long-term occurrence of class 1 integrons and a sulphonamide resistance gene in marine plankton communities. Environ. Microbiol. Rep. 2018, 10, 458–464. [Google Scholar] [CrossRef]
- Su, S.; Li, C.; Yang, J.; Xu, Q.; Qiu, Z.; Xue, B.; Wang, S.; Zhao, C.; Xiao, Z.; Wang, J.; et al. Distribution of antibiotic resistance genes in three different natural water bodies-a lake, river and sea. Int. J. Environ. Res. Public Health 2020, 17, 552. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Wang, H.; Chen, Q.; Su, J.Q.; Gad, M.; Li, J.; Mulla, S.I.; Yu, C.P.; Hu, A. Fecal pollution mediates the dominance of stochastic assembly of antibiotic resistome in an urban lagoon (Yundang lagoon), China. J. Hazard. Mater. 2021, 417, 126083. [Google Scholar] [CrossRef]
- Wang, H.; Hou, L.; Liu, Y.; Liu, K.; Zhang, L.; Huang, F.; Rashid, A.; Hu, A.; Yu, C. Horizontal and vertical gene transfer drive sediment antibiotic resistome in an urban lagoon system. J. Environ. Sci. 2021, 102, 11–23. [Google Scholar] [CrossRef]
- Sabatino, R.; Di Cesare, A.; Dzhembekova, N.; Fontaneto, D.; Eckert, E.M.; Corno, G.; Moncheva, S.; Bertoni, R.; Callieri, C. Spatial distribution of antibiotic and heavy metal resistance genes in the Black Sea. Mar. Pollut. Bull. 2020, 160, 111635. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [Green Version]
- Manna, M.S.; Tamer, Y.T.; Gaszek, I.; Poulides, N.; Ahmed, A.; Wang, X.; Toprak, F.C.R.; Woodard, D.R.; Koh, A.Y.; Williams, N.S.; et al. A trimethoprim derivative impedes antibiotic resistance evolution. Nat. Commun. 2021, 12, 2949. [Google Scholar] [CrossRef]
- Müller, A.; Österlund, H.; Marsalek, J.; Viklander, M. The pollution conveyed by urban runoff: A review of sources. Sci. Total Environ. 2020, 709, 136125. [Google Scholar] [CrossRef] [PubMed]
- Zonta, R.; Zaggia, L.; Collavini, F.; Costa, F.; Scattolin, M. Sediment contamination assessment of the Venice canal network (Italy). In Flooding and Environmental Challenges for Venice and Its Lagoon: State of Knowledge; Fletcher, C., Spencer, T., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 603–615. [Google Scholar]
- Zonta, R.; Cassin, D.; Pini, R.; Dominik, J. Substantial decrease in contaminant concentrations in the sediments of the Venice (Italy) canal network in the last two decades—implications for sediment management. Water 2020, 12, 1965. [Google Scholar] [CrossRef]
- Sfriso, A.; Buosi, A.; Tomio, Y.; Juhmani, A.-S.; Mistri, M.; Munari, C.; Sfriso, A.A. Trends of nitrogen and phosphorus in surface sediments of the lagoons of the Northern Adriatic Sea as a study case. Water 2021, 13, 2914. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).