
Peroxydisulfate Persistence in ISCO for Groundwater Remediation: Temperature Dependence, Batch/Column Comparison, and Sulfate Fate
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Soil and Aquifer Materials
2.3. Soil Characterization
2.4. S2O82− and SO42− Analysis
2.5. Experimental Setup
2.5.1. Batch Experiments
2.5.2. Column Experiments
3. Results and Discussions
3.1. Characterization of Soil/Aquifer Materials
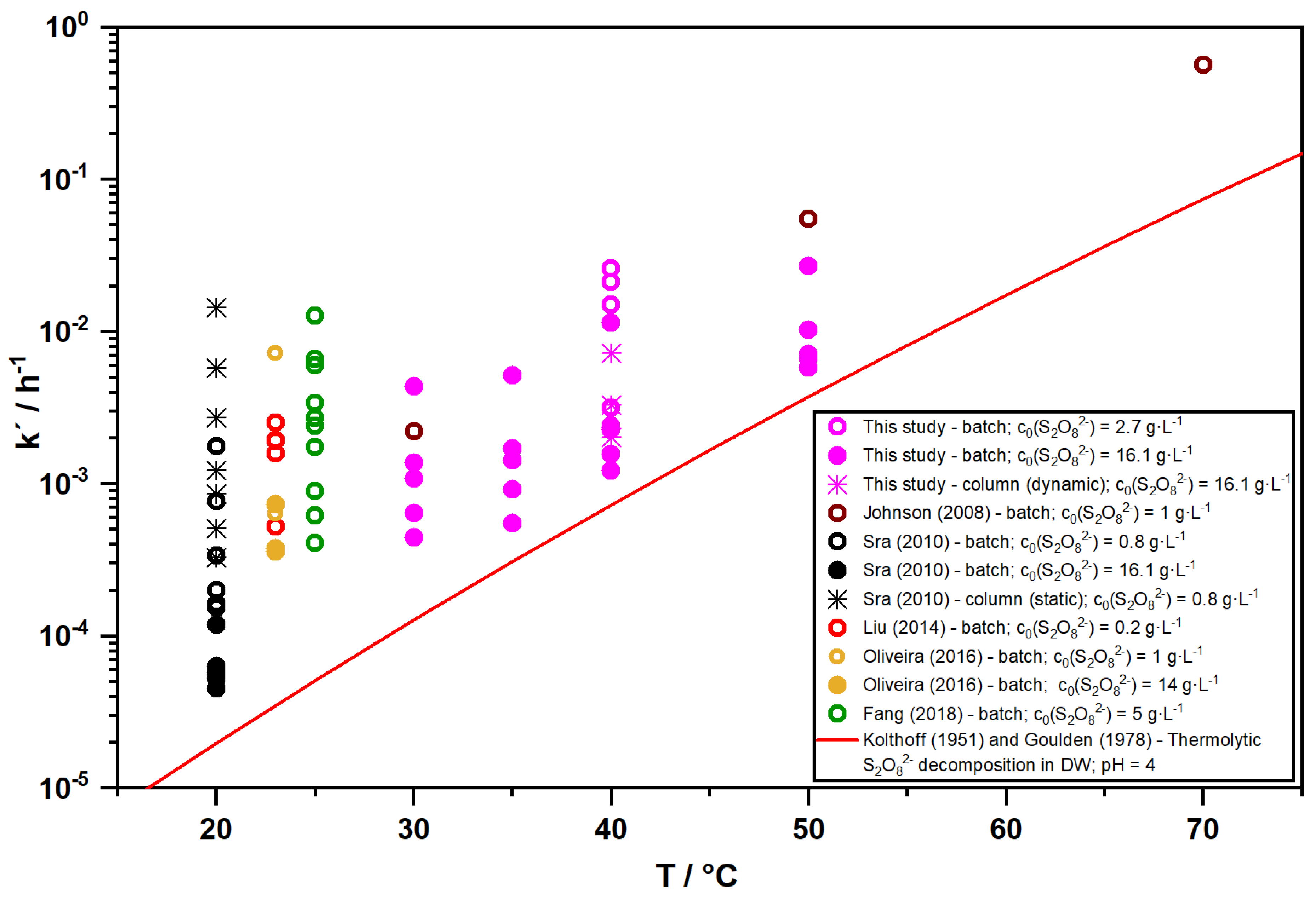
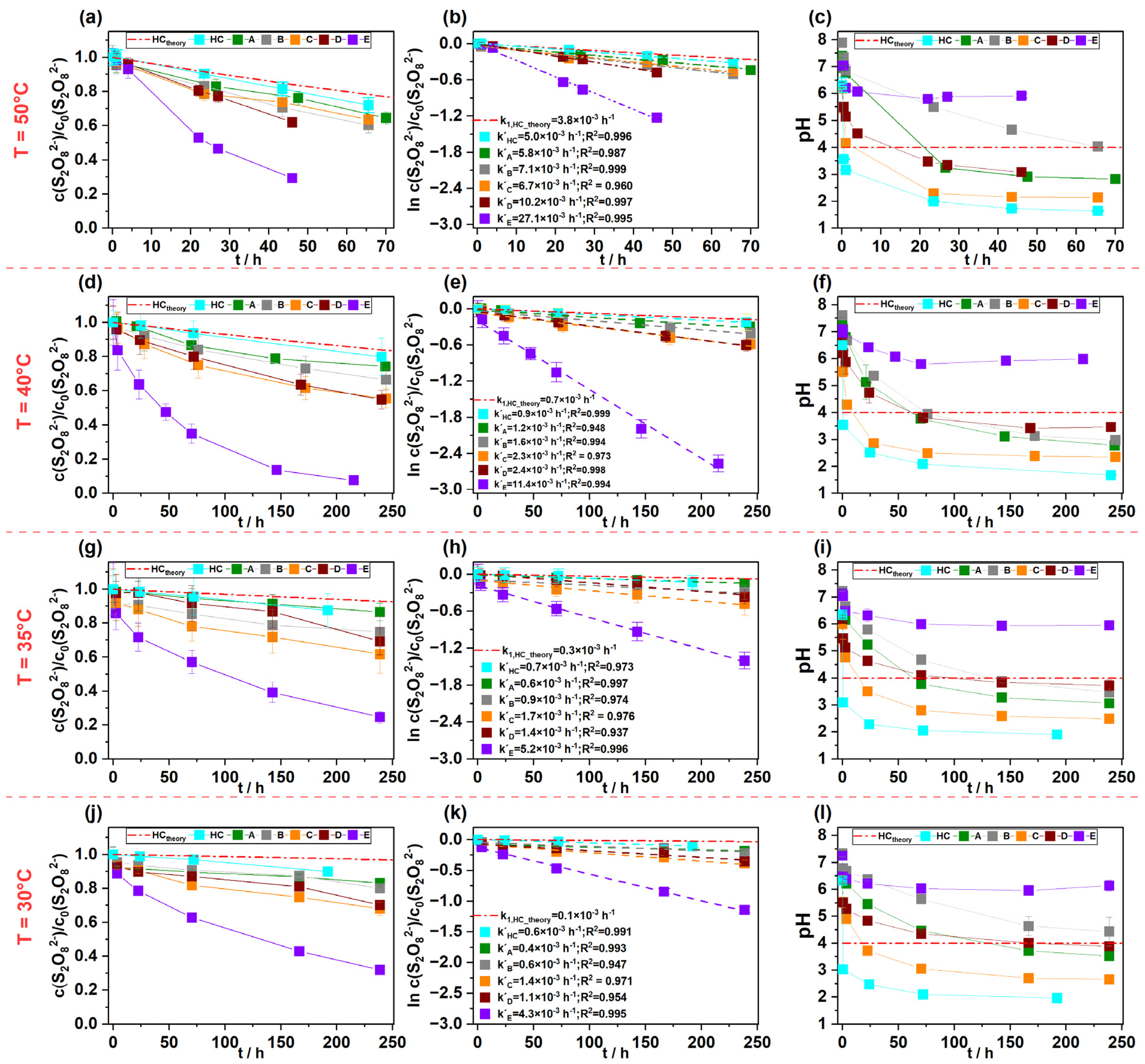
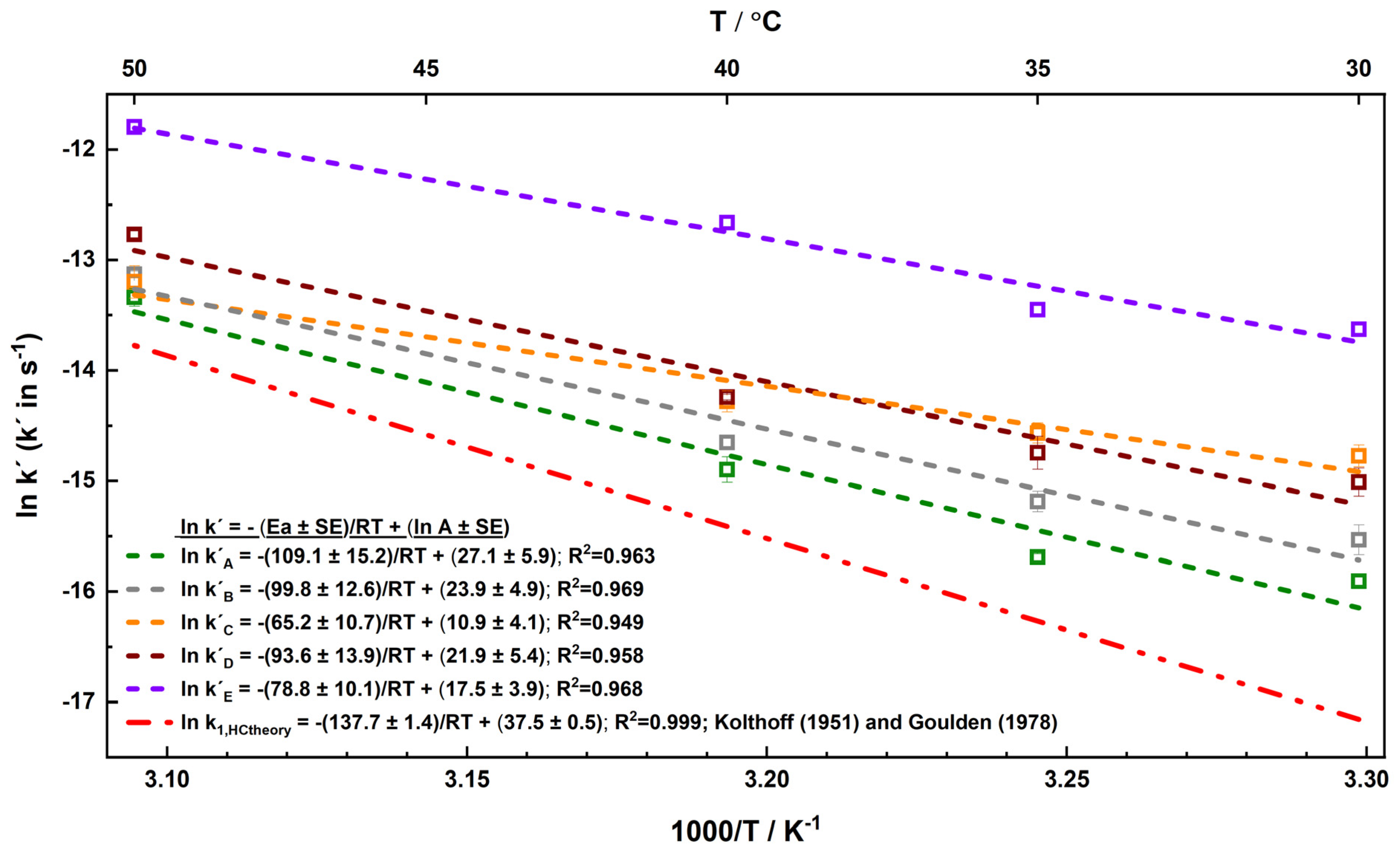
3.2. Influence of Temperature on S2O82− Persistence in Soil/Aquifer Materials
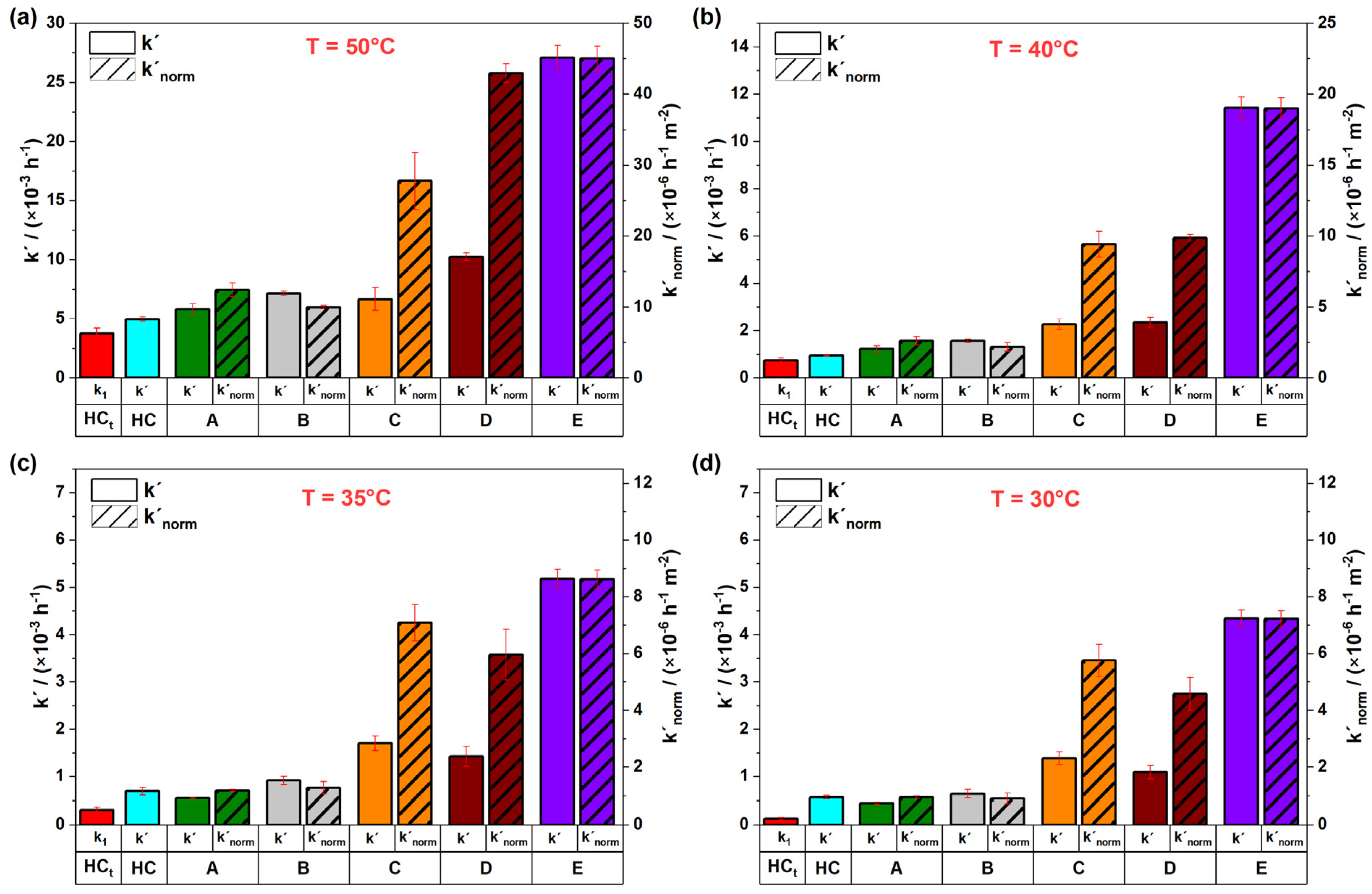
3.3. Influence of Initial S2O82− Concentration on Its Persistence in Soil Materials
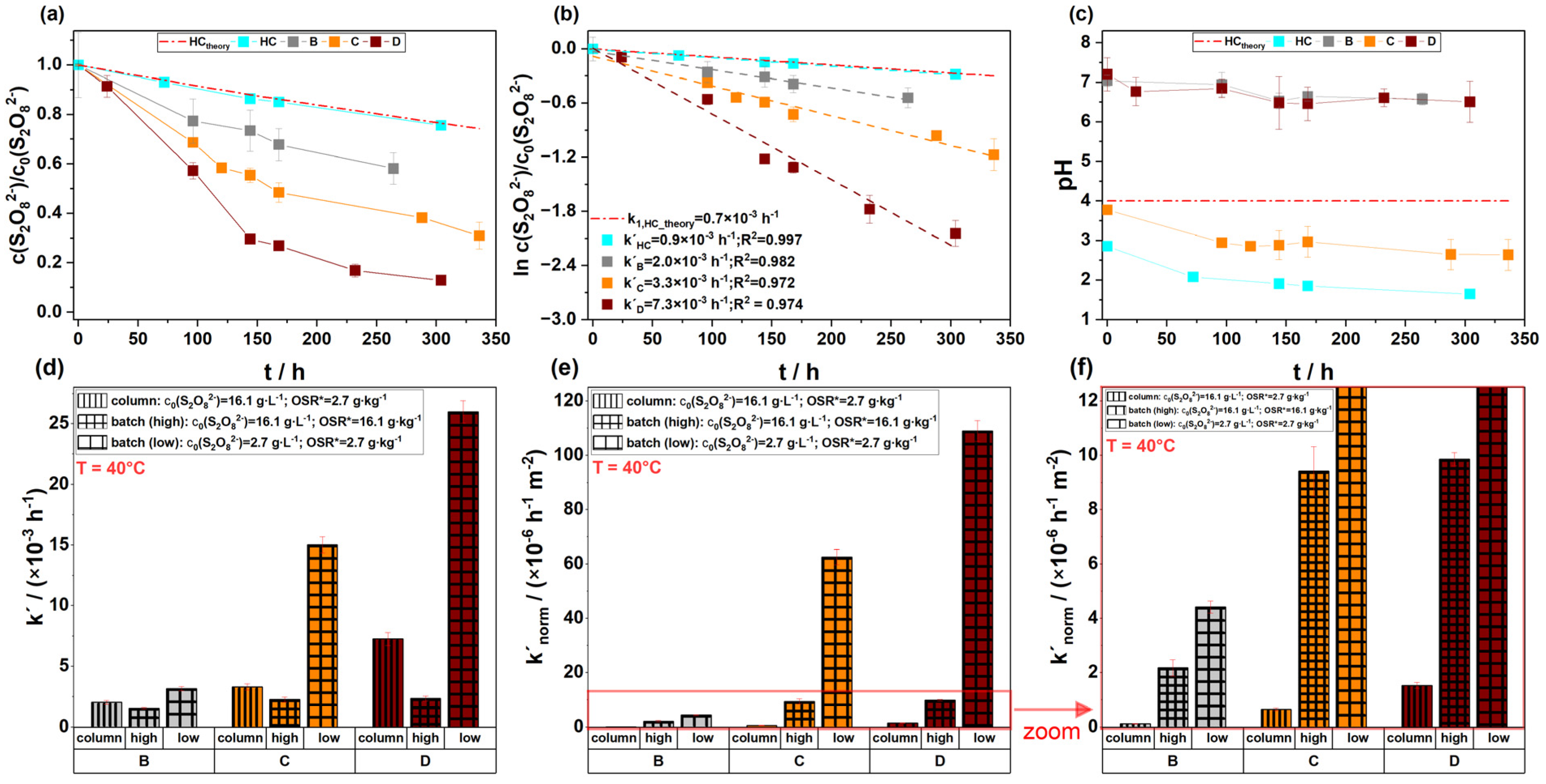
3.4. Comparison of Experimental Designs: Batch Versus Dynamic Column Tests
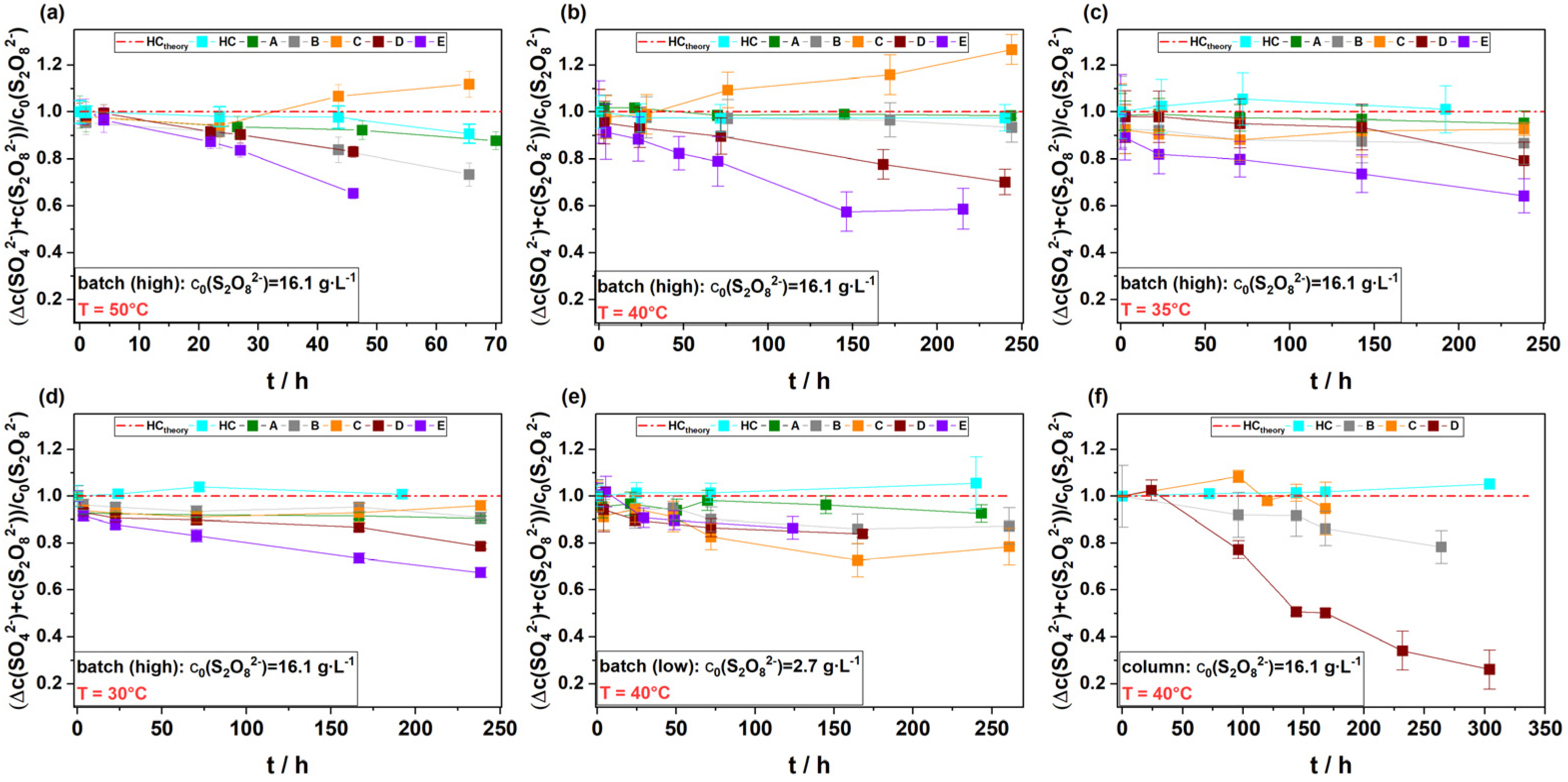
3.5. Fate of Generated SO42− in the Experimental Systems
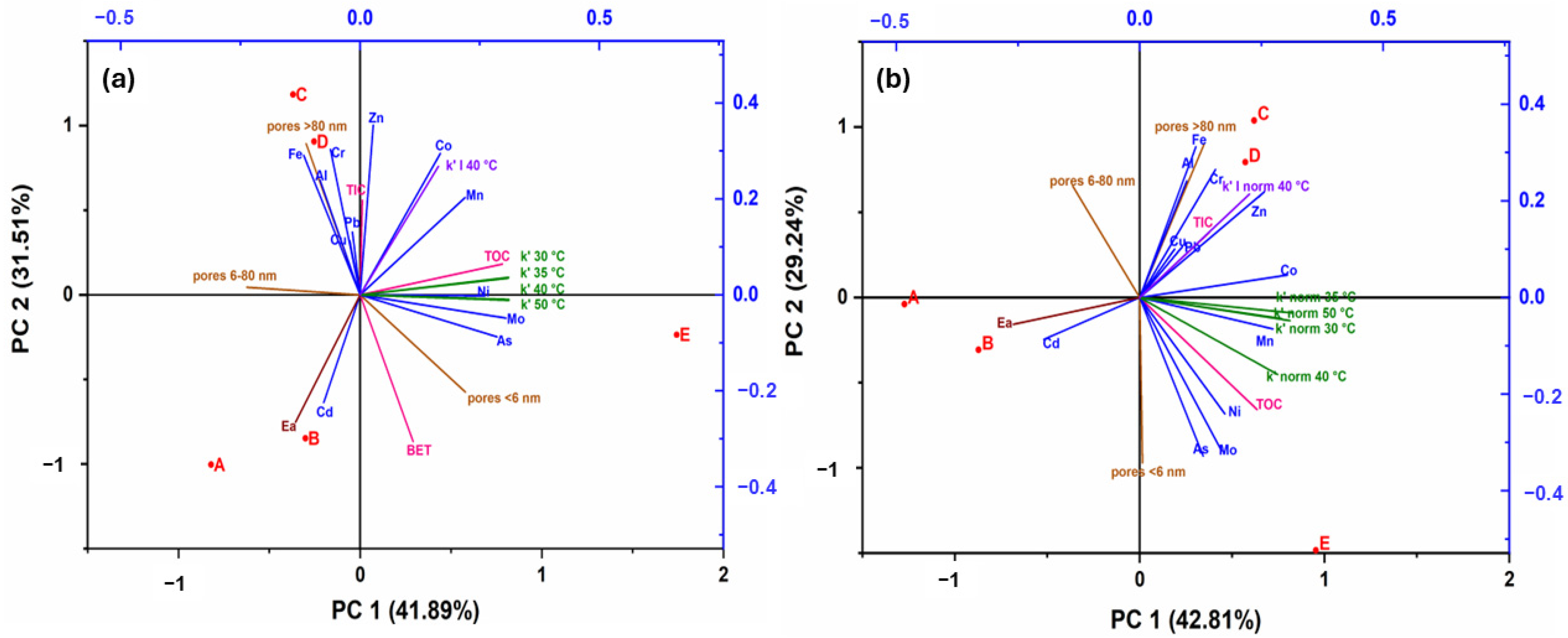
3.6. PCA Analysis
4. Implications for Environmental Applications
- Soil characteristics, especially TOC, strongly influence S2O82− decomposition rates;
- Lower initial concentrations of S2O82− increased decomposition rates across most soil systems, likely due to reduced ionic strength, thus, enhanced interaction with soil/aquifer minerals and SOM. This concentration-dependent reactivity suggests that adjusting the initial oxidant dose could optimize decomposition rates;
- Differences in Ea among the materials indicate varied thermal requirements for S2O82− activation, suggesting that materials with higher Ea may necessitate additional heat input to drive effective decomposition. This underscores the potential for temperature modulation as a control measure for S2O82− persistence in practical applications;
- While batch tests yielded higher S2O82− decomposition rates normalized to the total BET surfaces, dynamic column tests revealed that limited reactive surface accessibility and the formation of preferential flow paths may reduce the reliability of this setup in representing in situ conditions. Therefore, using batch or static stop–flow column tests may be more suitable for the evaluation of S2O82− persistence during bench-scale treatability tests;
- The main parameters affecting the decomposition rate of S2O82− were closely related to TOC, Ni, and Mo concentrations, particularly at higher concentrations (c0(S2O82−) = 16.1 g∙L⁻1) and temperatures between 30 °C and 50 °C. Additionally, at a lower concentration (c0(S2O82−) = 2.7 g∙L⁻1) and a temperature of 40 °C, the decomposition rate was more strongly associated with Co and Mn.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhan, C.; Dai, Z.; Yin, S.; Carroll, K.C.; Soltanian, M.R. Conceptualizing Future Groundwater Models through a Ternary Framework of Multisource Data, Human Expertise, and Machine Intelligence. Water Res. 2024, 257, 121679. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, R.L.; Crimi, M.; Simpkin, T.J. In Situ Chemical Oxidation for Groundwater Remediation; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011; ISBN 978-1-4419-7826-4. [Google Scholar]
- Lee, J.; Von Gunten, U.; Kim, J.-H. Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks. Environ. Sci. Technol. 2020, 54, 3064–3081. [Google Scholar] [CrossRef] [PubMed]
- McGachy, L.; Sedlak, D.L. From Theory to Practice: Leveraging Chemical Principles To Improve the Performance of Peroxydisulfate-Based In Situ Chemical Oxidation of Organic Contaminants. Environ. Sci. Technol. 2024, 58, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.-H.; Ma, J.; Xi, B.-D.; Yu, M.-D.; Cui, J.; Chen, B.-L.; Li, Y.; Gu, Q.-B.; He, X.-S. Recent Progress on In-Situ Chemical Oxidation for the Remediation of Petroleum Contaminated Soil and Groundwater. J. Hazard. Mater. 2022, 432, 128738. [Google Scholar] [CrossRef]
- Guerra-Rodríguez, S.; Rodríguez, E.; Singh, D.N.; Rodríguez-Chueca, J. Assessment of Sulfate Radical-Based Advanced Oxidation Processes for Water and Wastewater Treatment: A Review. Water 2018, 10, 1828. [Google Scholar] [CrossRef]
- Wacławek, S.; Lutze, H.V.; Grübel, K.; Padil, V.V.T.; Černík, M.; Dionysiou, D.D. Chemistry of Persulfates in Water and Wastewater Treatment: A Review. Chem. Eng. J. 2017, 330, 44–62. [Google Scholar] [CrossRef]
- Xie, J.; Yang, C.; Li, X.; Wu, S.; Lin, Y. Generation and Engineering Applications of Sulfate Radicals in Environmental Remediation. Chemosphere 2023, 339, 139659. [Google Scholar] [CrossRef]
- Li, N.; Wu, S.; Dai, H.; Cheng, Z.; Peng, W.; Yan, B.; Chen, G.; Wang, S.; Duan, X. Thermal Activation of Persulfates for Organic Wastewater Purification: Heating Modes, Mechanism and Influencing Factors. Chem. Eng. J. 2022, 450, 137976. [Google Scholar] [CrossRef]
- Wu, Z.; Gong, S.; Liu, J.; Shi, J.; Deng, H. Progress and Problems of Water Treatment Based on UV/Persulfate Oxidation Process for Degradation of Emerging Contaminants: A Review. J. Water Process. Eng. 2024, 58, 104870. [Google Scholar] [CrossRef]
- Furman, O.S.; Teel, A.L.; Watts, R.J. Mechanism of Base Activation of Persulfate. Environ. Sci. Technol. 2010, 44, 6423–6428. [Google Scholar] [CrossRef]
- Liu, B.; Huang, B.; Wang, Z.; Tang, L.; Ji, C.; Zhao, C.; Feng, L.; Feng, Y. Homogeneous/Heterogeneous Metal-Catalyzed Persulfate Oxidation Technology for Organic Pollutants Elimination: A Review. J. Environ. Chem. Eng. 2023, 11, 109586. [Google Scholar] [CrossRef]
- Lei, Z.; Song, X.; Ma, G.; Zhao, T.; Meng, K.; Zhang, M.; Ren, J.; Dai, L. A Review of Recent Studies on Nano Zero-Valent Iron Activated Persulfate Advanced Oxidation Technology for the Degradation of Organic Pollutants. New J. Chem. 2023, 47, 14585–14599. [Google Scholar] [CrossRef]
- Gujar, S.K.; Divyapriya, G.; Gogate, P.R.; Nidheesh, P.V. Environmental Applications of Ultrasound Activated Persulfate/Peroxymonosulfate Oxidation Process in Combination with Other Activating Agents. Crit. Rev. Environ. Sci. Technol. 2023, 53, 780–802. [Google Scholar] [CrossRef]
- Huang, W.; Xiao, S.; Zhong, H.; Yan, M.; Yang, X. Activation of Persulfates by Carbonaceous Materials: A Review. Chem. Eng. J. 2021, 418, 129297. [Google Scholar] [CrossRef]
- Oyekunle, D.T.; Zhou, X.; Shahzad, A.; Chen, Z. Review on Carbonaceous Materials as Persulfate Activators: Structure–Performance Relationship, Mechanism and Future Perspectives on Water Treatment. J. Mater. Chem. A 2021, 9, 8012–8050. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, J.; Wang, X.; Liu, G.; Zhou, M. A Critical Review of Persulfate-Based Electrochemical Advanced Oxidation Processes for the Degradation of Emerging Contaminants: From Mechanisms and Electrode Materials to Applications. Sci. Total Environ. 2024, 944, 173839. [Google Scholar] [CrossRef]
- Zhi, D.; Lin, Y.; Jiang, L.; Zhou, Y.; Huang, A.; Yang, J.; Luo, L. Remediation of Persistent Organic Pollutants in Aqueous Systems by Electrochemical Activation of Persulfates: A Review. J. Environ. Manag. 2020, 260, 110125. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Zhang, X.; Coulon, F.; Wang, C. Harnessing the Power of Natural Minerals: A Comprehensive Review of Their Application as Heterogeneous Catalysts in Advanced Oxidation Processes for Organic Pollutant Degradation. Chemosphere 2023, 337, 139404. [Google Scholar] [CrossRef]
- Mustapha, N.A.; Liu, H.; Ibrahim, A.O.; Huang, Y.; Liu, S. Degradation of Aniline in Groundwater by Persulfate with Natural Subsurface Sediment as the Activator. Chem. Eng. J. 2021, 417, 128078. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Y.; Zhong, S.; Zhang, L. AOPs-Based Remediation of Petroleum Hydrocarbons-Contaminated Soils: Efficiency, Influencing Factors and Environmental Impacts. Chemosphere 2020, 246, 125726. [Google Scholar] [CrossRef]
- Liu, H.; Bruton, T.A.; Doyle, F.M.; Sedlak, D.L. In Situ Chemical Oxidation of Contaminated Groundwater by Persulfate: Decomposition by Fe(III)- and Mn(IV)-Containing Oxides and Aquifer Materials. Environ. Sci. Technol. 2014, 48, 10330–10336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Dai, H.; Yuan, S. The Competition between Heterotrophic Denitrification and DNRA Pathways in Hyporheic Zone and Its Impact on the Fate of Nitrate. J. Hydrol. 2023, 626, 130175. [Google Scholar] [CrossRef]
- Ahmad, M.; Teel, A.L.; Watts, R.J. Persulfate Activation by Subsurface Minerals. J. Contam. Hydrol. 2010, 115, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Orozco, R.; Camargos, N.; Liu, H. Mechanisms on the Impacts of Alkalinity, pH, and Chloride on Persulfate-Based Groundwater Remediation. Environ. Sci. Technol. 2017, 51, 3948–3959. [Google Scholar] [CrossRef]
- Liu, H.; Bruton, T.A.; Li, W.; Buren, J.V.; Prasse, C.; Doyle, F.M.; Sedlak, D.L. Oxidation of Benzene by Persulfate in the Presence of Fe(III)- and Mn(IV)-Containing Oxides: Stoichiometric Efficiency and Transformation Products. Environ. Sci. Technol. 2016, 50, 890–898. [Google Scholar] [CrossRef]
- Peng, F.; Wang, X.; Fang, G.; Gao, Y.; Yang, X.; Gao, J.; Wang, Y.; Zhou, D. New Insights into Persulfate Decomposition by Soil Minerals: Radical and Non-Radical Pathways. Environ. Sci. Pollut. Res. Int. 2023, 30, 55922–55931. [Google Scholar] [CrossRef]
- Teel, A.L.; Ahmad, M.; Watts, R.J. Persulfate Activation by Naturally Occurring Trace Minerals. J. Hazard. Mater. 2011, 196, 153–159. [Google Scholar] [CrossRef]
- Zhu, S.; Li, X.; Kang, J.; Duan, X.; Wang, S. Persulfate Activation on Crystallographic Manganese Oxides: Mechanism of Singlet Oxygen Evolution for Nonradical Selective Degradation of Aqueous Contaminants. Environ. Sci. Technol. 2019, 53, 307–315. [Google Scholar] [CrossRef]
- Ahmad, M.; Teel, A.L.; Watts, R.J. Mechanism of Persulfate Activation by Phenols. Environ. Sci. Technol. 2013, 47, 5864–5871. [Google Scholar] [CrossRef]
- Fang, G.; Gao, J.; Dionysiou, D.; Liu, C.; Zhou, D. Activation of Persulfate by Quinones: Free Radical Reactions and Implication for the Degradation of PCBs. Environ. Sci. Technol. 2013, 47, 4605–4611. [Google Scholar] [CrossRef]
- Kim, C.; Chin, Y.-P.; Son, H.; Hwang, I. Activation of Persulfate by Humic Substances: Stoichiometry and Changes in the Optical Properties of the Humic Substances. Water Res. 2022, 212, 118107. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhong, H.; Brusseau, M.L. The Natural Activation Ability of Subsurface Media to Promote In-Situ Chemical Oxidation of 1,4-Dioxane. Water Res. 2019, 149, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Feng, R.; Jiang, Y.; Cai, T.; Jiang, C. The Impacts of Temperature, Soil-Water Ratio, and Background Multiplied Inorganic Anions on the Degradation of Organophosphorus Flame Retardants in Soil by Peroxydisulfate-Based Advanced Oxidation Processes. Process Saf. Environ. Prot. 2022, 168, 422–433. [Google Scholar] [CrossRef]
- Dong, X.; Yang, X.; Hua, S.; Wang, Z.; Cai, T.; Jiang, C. Unraveling the Mechanisms for Persulfate-Based Remediation of Triphenyl Phosphate-Contaminated Soils: Complicated Soil Constituent Effects on the Formation and Propagation of Reactive Oxygen Species. Chem. Eng. J. 2021, 426, 130662. [Google Scholar] [CrossRef]
- Ferreira, I.D.; Prieto, T.; Freitas, J.G.; Thomson, N.R.; Nantes, I.L.; Bechara, E.J.H. Natural Persulfate Activation for Anthracene Remediation in Tropical Environments. Water Air Soil Pollut. 2017, 228, 146. [Google Scholar] [CrossRef]
- Qutob, M.; Rafatullah, M.; Muhammad, S.A.; Siddiqui, M.R.; Alam, M. Advanced Oxidation of Polycyclic Aromatic Hydrocarbons in Tropical Soil: Self-Catalytic Utilization of Natural Iron Contents in an Oxygenation Reactor Supported with Persulfate. Sci. Total Environ. 2024, 926, 171843. [Google Scholar] [CrossRef]
- Liang, J.; Duan, X.; Xu, X.; Zhang, Z.; Zhang, J.; Zhao, L.; Qiu, H.; Cao, X. Critical Functions of Soil Components for In Situ Persulfate Oxidation of Sulfamethoxazole: Inherent Fe(II) Minerals-Coordinated Nonradical Pathway. Environ. Sci. Technol. 2024, 58, 915–924. [Google Scholar] [CrossRef]
- Bolourani, G.; Ioannidis, M.A.; Craig, J.R.; Thomson, N.R. Persulfate-Based ISCO for Field-Scale Remediation of NAPL-Contaminated Soil: Column Experiments and Modeling. J. Hazard. Mater. 2023, 449, 131000. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chen, T.Y.; Tsai, Y.P.; Chen, K.F. Remediation of Trichloroethene (TCE)-Contaminated Groundwater by Persulfate Oxidation: A Field-Scale Study. RSC Adv. 2018, 8, 2433–2440. [Google Scholar] [CrossRef]
- Ding, X.; Wei, C.; Wei, Y.; Liu, P.; Wang, D.; Wang, Q.; Chen, X.; Song, X. Field Test of Thermally Activated Persulfate for Remediation of PFASs Co-Contaminated with Chlorinated Aliphatic Hydrocarbons in Groundwater. Water Res. 2023, 249, 120993. [Google Scholar] [CrossRef]
- Dahmani, M.A.; Huang, K.; Hoag, G.E. Sodium Persulfate Oxidation for the Remediation of Chlorinated Solvents (USEPA Superfund Innovative Technology Evaluation Program). Water Air Soil Pollut. Focus 2006, 6, 127–141. [Google Scholar] [CrossRef]
- Liang, C.; Chien, Y.-C.; Lin, Y.-L. Impacts of ISCO Persulfate, Peroxide and Permanganate Oxidants on Soils: Soil Oxidant Demand and Soil Properties. Soil Sediment Contam. Int. J. 2012, 21, 701–719. [Google Scholar] [CrossRef]
- Ma, J.; Wen, J.; Yang, X.; Liu, Y.; Zhai, G.; Liu, S.; Liu, H.; Yue, G.; Dong, H.; Zhang, P. Persistence of Two Common Chemical Oxidants for Soil and Groundwater Remediation: Impacts of Water Chemistry and Subsurface Minerals. Water Air Soil Pollut. 2022, 233, 326. [Google Scholar] [CrossRef]
- Fang, G.; Chen, X.; Wu, W.; Liu, C.; Dionysiou, D.D.; Fan, T.; Wang, Y.; Zhu, C.; Zhou, D. Mechanisms of Interaction between Persulfate and Soil Constituents: Activation, Free Radical Formation, Conversion, and Identification. Environ. Sci. Technol. 2018, 52, 14352–14361. [Google Scholar] [CrossRef]
- Johnson, R.L.; Tratnyek, P.G.; Johnson, R.O. Persulfate Persistence under Thermal Activation Conditions. Environ. Sci. Technol. 2008, 42, 9350–9356. [Google Scholar] [CrossRef]
- Oliveira, F.C.; Freitas, J.G.; Furquim, S.A.C.; Rollo, R.M.; Thomson, N.R.; Alleoni, L.R.F.; Nascimento, C.A.O. Persulfate Interaction with Tropical Soils. Water Air Soil Pollut. 2016, 227, 326. [Google Scholar] [CrossRef]
- Sra, K.S.; Thomson, N.R.; Barker, J.F. Persistence of Persulfate in Uncontaminated Aquifer Materials. Environ. Sci. Technol. 2010, 44, 3098–3104. [Google Scholar] [CrossRef]
- Carter, M.R.; Gregorich, E.G. Soil Sampling and Methods of Analysis, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008; ISBN 978-0-8493-3586-0. [Google Scholar]
- Hao, X.; Ball, B.; Culley, J.; Carter, M.; Parkin, G. Soil Density and Porosity. In Soil Sampling and Methods of Analysis; CRC Press: Boca Raton, FL, USA, 2008; pp. 743–759. [Google Scholar]
- ISO 17892-1:2014; Geotechnical Investigation and Testing—Laboratory Testing of Soil—Part 1: Determination of Water Content. International Organization for Standardization: Geneva, Switzerland, 2014.
- Miao, S.; Yu, B.; Ren, Y.; Cai, C. Solubility and Physical Properties of Calcium Sulfate Dihydrate in NaCl and Glycerol Aqueous Solution at 303.15, 323.15, and 343.15 K. J. Chem. Eng. Data 2020, 65, 2703–2711. [Google Scholar] [CrossRef]
- Sokolova, T.A.; Alekseeva, S.A. Adsorption of Sulfate Ions by Soils (A Review). Eurasian Soil Sci. 2008, 41, 140–148. [Google Scholar] [CrossRef]
- Goulden, P.D.; Anthony, D.H.J. Kinetics of uncatalyzed peroxydisulfate oxidation of organic material in fresh water. Anal. Chem. 1978, 50, 953–958. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Miller, I.K. The Chemistry of Persulfate. I. The Kinetics and Mechanism of the Decomposition of the Persulfate Ion in Aqueous Medium1. J. Am. Chem. Soc. 1951, 73, 3055–3059. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Material A | Material B | Material C | Material D | Material E |
|---|---|---|---|---|---|
| Soil texture | |||||
| pH | 7.3 | 7.5 | 6.4 | 6.3 | 7.2 |
| moisture content/% (weight) | 1.01 | 5.02 | 5.55 | 6.69 | 3.56 |
| BET surface area/m2·g−1 | 9.361 | 14.323 | 4.802 | 4.769 | 12.033 |
| Total pore volume/mL·g−1 | 0.024 | 0.024 | 0.027 | 0.017 | 0.016 |
| Bulk density/kg·L−1 | 1.06 | 1.12 | 1.20 | 1.26 | 1.23 |
| Particle density/g·mL−1 | 2.05 | 1.92 | 2.08 | 1.81 | 2.09 |
| Porosity/- | 0.482 | 0.420 | 0.423 | 0.303 | 0.414 |
| Total carbon (TC)/% | 0.071 | 0.080 | 0.289 | 0.820 | 1.630 |
| Total organic carbon (TOC)/% | 0.062 | 0.080 | 0.289 | 0.703 | 1.610 |
| Total inorganic carbon (TIC)/% | 0.007 | 0.008 | 0.014 | 0.117 | 0.026 |
| Pore distribution [nm] | |||||
| <6/% | 13.7 | 45.1 | 7.06 | 10.5 | 50.2 |
| 6–8/% | 6.72 | 11.8 | 1.06 | 4.3 | 7.43 |
| 8–10/% | 6.16 | 7.45 | 2.08 | 3.95 | 5.09 |
| 10–12/% | 6.43 | 6.05 | 2.2 | 4.33 | 4.5 |
| 12–16/% | 8.75 | 5.77 | 2.97 | 6.02 | 5.02 |
| 16–20/% | 9.14 | 4.64 | 3.39 | 6.45 | 4.55 |
| 20–80/% | 39.2 | 15.2 | 47.0 | 44.9 | 18.1 |
| >80/% | 9.91 | 4.02 | 34.3 | 19.5 | 5.09 |
| Metal content [mg·kg−1] | |||||
| Fe | 1347.14 | 4279.55 | 6239.24 | 6932.75 | 1465.61 |
| Mn | 12.82 | 126.95 | 251.50 | 130.29 | 266.31 |
| Cu | 9.59 | 3.50 | 3.53 | 21.48 | 7.80 |
| Al | 3166.12 | 5387.39 | 17,671.09 | 4292.67 | 2992.34 |
| Co | 1.32 | 2.33 | 4.56 | 3.57 | 4.14 |
| As | 6.56 | nd * | 0.85 | nd | 31.23 |
| Mo | 0.28 | nd | 0.26 | nd | 7.20 |
| Zn | 11.15 | 9.39 | 39.47 | 43.30 | 24.62 |
| Pb | 16.30 | 2.69 | 9.22 | 25.25 | 12.76 |
| Cr | 5.07 | 6.19 | 24.21 | 10.72 | 6.55 |
| Na | 50 | 115 | 83 | 164 | 62 |
| Ca | 525 | 510 | 1023 | 4384 | 7571 |
| K | 2556 | 6395 | 20,292 | 725 | 10,910 |
| Mg | 138 | 696 | 1540 | 785 | 1236 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGachy, L.; Škarohlíd, R.; Kostrakiewicz, R.; Kühnl, K.; Těšínská, P.; Müllerová, B.; Šír, M.; Martinec, M. Peroxydisulfate Persistence in ISCO for Groundwater Remediation: Temperature Dependence, Batch/Column Comparison, and Sulfate Fate. Water 2024, 16, 3552. https://doi.org/10.3390/w16243552
McGachy L, Škarohlíd R, Kostrakiewicz R, Kühnl K, Těšínská P, Müllerová B, Šír M, Martinec M. Peroxydisulfate Persistence in ISCO for Groundwater Remediation: Temperature Dependence, Batch/Column Comparison, and Sulfate Fate. Water. 2024; 16(24):3552. https://doi.org/10.3390/w16243552
Chicago/Turabian StyleMcGachy, Lenka, Radek Škarohlíd, Richard Kostrakiewicz, Karel Kühnl, Pavlína Těšínská, Barbora Müllerová, Marek Šír, and Marek Martinec. 2024. "Peroxydisulfate Persistence in ISCO for Groundwater Remediation: Temperature Dependence, Batch/Column Comparison, and Sulfate Fate" Water 16, no. 24: 3552. https://doi.org/10.3390/w16243552
APA StyleMcGachy, L., Škarohlíd, R., Kostrakiewicz, R., Kühnl, K., Těšínská, P., Müllerová, B., Šír, M., & Martinec, M. (2024). Peroxydisulfate Persistence in ISCO for Groundwater Remediation: Temperature Dependence, Batch/Column Comparison, and Sulfate Fate. Water, 16(24), 3552. https://doi.org/10.3390/w16243552