A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys

Abstract
:1. Introduction
2. Methods
2.1. Prediction of Human Concentration–Time Profiles from Rats or Monkeys
2.1.1. Methods I–III
animals) × (weight of the animal/weight of human)b
2.1.2. Methods IV–VI
animals)0.85 × (weight of the animal/weight of human)b
2.1.3. Methods VII–VIII
2.1.4. Prediction of Human Clearance and Half-Life from Monkeys or Rats (Scaling Based on a Single Exponent)
species)0.15
2.1.5. Statistical Analysis
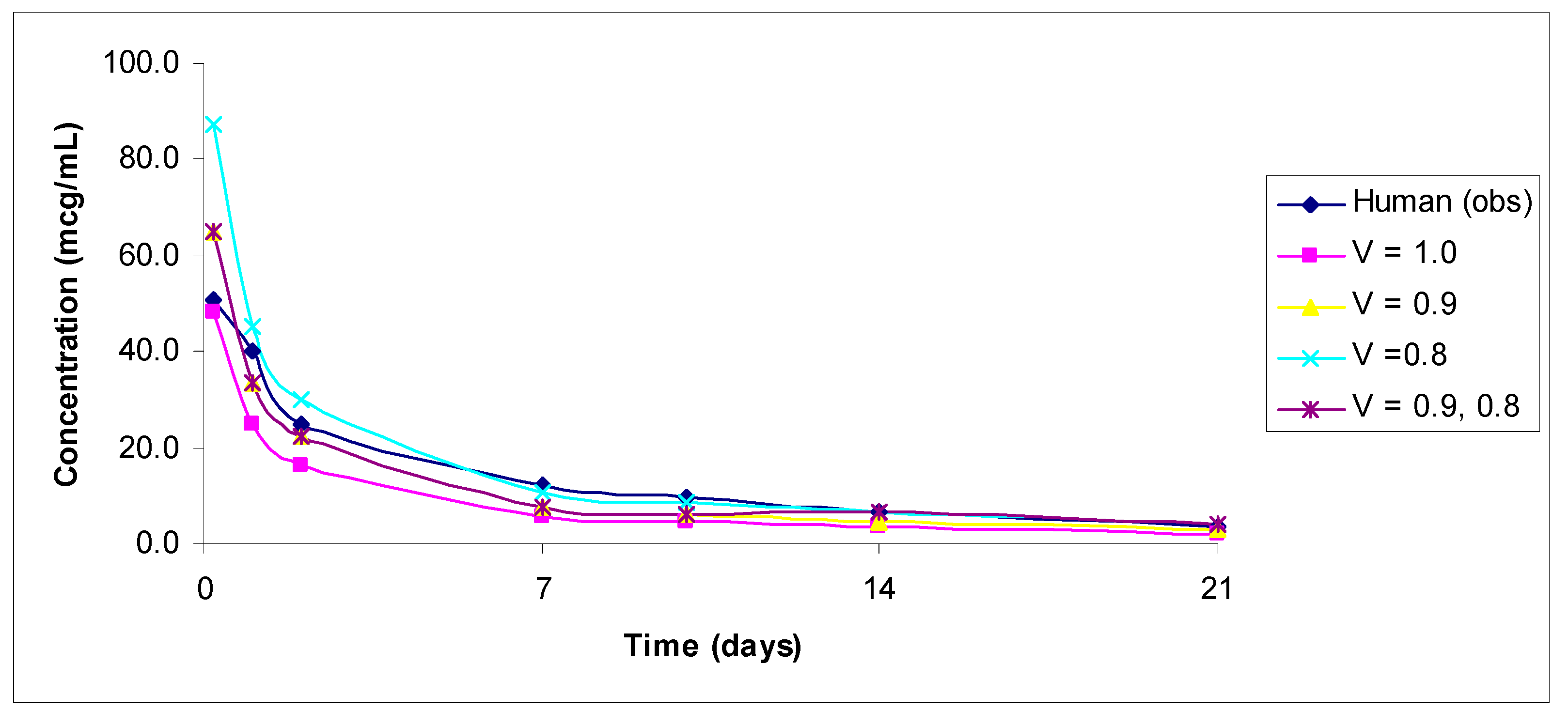
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lambert, J.M.; Charles, Q.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wang, R.E.; Wang, F. Antibody-drug conjugates for nononcological indications. Expert Opin. Biol. Ther. 2016, 16, 591–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deslandes, A. Comparative clinical pharmacokinetics of antibody drug conjugates in first-in-human Phase 1 studies. MAbs 2014, 6, 859–870. [Google Scholar] [CrossRef]
- Mahmood, I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv. Drug Deliv. Rev. 2007, 59, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Interspecies Scaling of Antibody-Drug Conjugates (ADC) for the Prediction of Human Clearance. Antibodies 2021, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. A Single Animal Species-Based Prediction of Human Clearance and First-in-Human Dose of Monoclonal Antibodies: Beyond Monkey. Antibodies 2021, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Mordenti, J. Pharmacokinetic scale-up: Accurate prediction of human pharmacokinetic profiles from animal data. J. Pharm. Sci. 1985, 74, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I.; Yuan, R. A comparative study of allometric scaling with plasma concentrations predicted by species invariant time methods. Biopharm. Drug Disp. 1999, 20, 137–144. [Google Scholar] [CrossRef]
- Mahmood, I. Prediction of clearance, volume of distribution and half-life by allometric scaling and by plasma concentrations predicted by pharmacokinetic constants: A comparative study. J. Pharm. Pharmacol. 1999, 51, 905–910. [Google Scholar] [CrossRef]
- Mahmood, I. Prediction of drug concentration-time profiles of therapeutic proteins in humans from animals. Xenobiotica 2013, 43, 153–160. [Google Scholar] [CrossRef]
- Mahmood, I. Prediction of Plasma Concentration-time Profiles of Drugs in Humans from Animals Following Oral Administration: An Allometric Approach. Curr. Drug Metab. 2016, 17, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Wajima, T.; Yano, Y.; Fukumura, K.; Oguma, T. Prediction of human pharmacokinetic profile in animal scale up based on normalizing time course profiles. J. Pharm. Sci. 2004, 93, 1890–1900. [Google Scholar] [CrossRef]
- Li, C.; Zhang, C.; Deng, R. Prediction of Human Pharmacokinetics of Antibody-Drug Conjugates from Nonclinical Data. Clin. Transl. Sci. 2019, 12, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Iyer, S.; Theil, F.P.; Mortensen, D.L.; Fielder, P.J.; Prabhu, S. Projecting human pharmacokinetics of therapeutic antibodies from non-clinical data: What have we learned? MAbs 2011, 3, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oitate, M.; Masubuchi, N.; Ito, T.; Yabe, Y.; Karibe, T.; Aoki, T.; Murayama, N.; Kurihara, A.; Okudaira, N.; Izumi, T. Prediction of human pharmacokinetics of therapeutic monoclonal antibodies from simple allometry of monkey data. Drug Metab. Pharm. 2011, 26, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Zhou, H.; Jiao, Q.; Davis, H.M. Interspecies scaling of therapeutic monoclonal antibodies: Initial look. J. Clin. Pharmacol. 2009, 49, 1382–1402. [Google Scholar] [CrossRef]
- Gerber, D.E.; Infante, J.R.; Gordon, S.M.; Goldberg, S.B.; Martín, M.; Felip, E.; Garcia, M.M.; Schiller, J.H.; Spigel, D.R.; Cordova, J.; et al. Phase Ia Study of Anti-NaPi2b Antibody-Drug Conjugate Lifastuzumab Vedotin DNIB0600A in Patients with Non-Small Cell Lung Cancer and Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Kedan, L.; Rubinfeld, B.; Zhang, C.; Firestein, R.; Harstad, E.; Roth, L.; Tsai, S.P.; Schutten, M.; Xu, K.; Hristopoulos, M.; et al. Preclinical Development of an Anti-NaPi2b (SLC34A2) Antibody-Drug Conjugate as a Therapeutic for Non-Small Cell Lung and Ovarian Cancers. Clin. Cancer Res. 2015, 21, 5139–5150. [Google Scholar]
- Shapiro, G.I.; Vaishampayan, U.; Lorusso, P.; Barton, J.; Hua, S.; Reich, S.D.; Shazer, R.; Taylor, C.T.; Xuan, D.; Borghaei, H. First-in-human trial of an anti-5T4 antibody-monomethylauristatin conjugate, PF-06263507, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.; Wentland, J.; Han, X.; Zhang, Y.; Rago, B.; Duriga, N.; Spriggs, F.; Kadar, E.; Song, W.; McNally, J.; et al. Preclinical Development of an anti-5T4 Antibody−Drug Conjugate: Pharmacokinetics in Mice, Rats, and NHP and Tumor/Tissue Distribution in Mice. Bioconjug. Chem. 2015, 26, 2223–2232. [Google Scholar] [CrossRef]
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522. [Google Scholar] [CrossRef]
- Nagai, Y.; Oitate, M.; Shiozawa, H.; Ando, O. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS-8201a), a HER2-targeting antibody-drug conjugate, in cynomolgus monkeys. Xenobiotica 2019, 49, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Zhou, C.; Li, D.; Cai, H.; Sukumaran, S.; Carrasco-Triguero, M.; Saad, O.; Nazzal, D.; Lowe, C.; Ramanujan, S.; et al. Preclinical and translational pharmacokinetics of a novel THIOMAB™ antibodyantibiotic conjugate against Staphylococcus aureus. MABS 2019, 11, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.; Rothenberg, M.E.; Deng, R.; Lewin-Koh, N.; She, G.; Kamath, A.V.; Carrasco-Triguero, M.; Saad, O.; Castro, A.; Teufel, L.; et al. A Phase 1, Randomized, Single-Ascending-Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus aureus Thiomab Antibody-Antibiotic Conjugate, in Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63, e02588-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girish, S. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Pos. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar]
- Bender, B.; Leipold, D.D.; Xu, K. A Mechanistic Pharmacokinetic Model Elucidating the Disposition of Trastuzumab Emtansine (T-DM1), an Antibody–Drug Conjugate (ADC) for Treatment of Metastatic Breast Cancer. AAPS J. 2014, 16, 994–1008. [Google Scholar] [CrossRef] [Green Version]
- Durisova, M.; Dedik, L. New Mathematical Methods in Pharmacokinetic Modeling. Basic Clin. Pharmacol. Toxicol. 2005, 96, 335–342. [Google Scholar] [CrossRef]
- Boxenbaum, H. Interspecies pharmacokinetic scaling and the evolutionary-comparative paradigm. Drug Metab. Rev. 1984, 15, 1071–1121. [Google Scholar] [CrossRef]
- Boxenbaum, H. Interspecies scaling, allometry, physiological time and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 1982, 10, 201–227. [Google Scholar] [CrossRef]
- Dedrick, R.L. Animal scale-up. J. Pharmacokinet. Biopharm. 1973, 1, 435–461. [Google Scholar] [CrossRef]
- Dedrick, R.L.; Bischoff, K.B.; Zaharko, D.Z. Interspecies correlation of plasma concentration history of methotrexate (NSC-740). Cancer Chemother. Rep. 1970, 54, 95–101. [Google Scholar] [PubMed]
- Hutchaleelaha, A.; Chow, H.; Mayersohn, M. Comparative pharmacokinetics and interspecies scaling of amphotericin B in several mammalian species. J. Pharm. Pharmacol. 1997, 49, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Lave, T.; Saner, A.; Coassolo, P.; Brandt, R.; Schmitt-Hoffmann, A.H.; Chou, R.C. Animal pharmacokinetics and interspecies scaling from animals to man of lamifiban, a new platelet aggregation inhibitor. J. Pharm. Pharmacol. 1996, 48, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.C.; Lu, D.R. Interspecies pharmacokinetic scaling of BSH in mice, rats, rabbits, and humans. Biopharm. Drug Dispos. 1995, 16, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I. Misconceptions and issues regarding allometric scaling during the drug development process. Expert Opin Drug Metab. Toxicol. 2018, 14, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Mordenti, J.; Chen, S.A.; Moore, J.A.; Ferraiolo, B.L.; Green, G.D. Interspecies scaling of clearance and volume of distribution data for five therapeutic proteins. Pharm. Res. 1991, 8, 1351–1359. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V = 1.0 | V = 0.9 | V = 0.8 | V = 1.0 | V = 0.9 | ||||
|---|---|---|---|---|---|---|---|---|
| Range | V = 1.0 | V = 0.9 | V = 0.8 | CL = 0.85 | CL = 0.85 | CL = 0.85 | V = 0.8 | V = 0.8 |
| Monkey (total) n = 38 | ||||||||
| <0.5 | 5 | 2 | 2 | 25 | 16 | 4 | 5 | 2 |
| >2.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5–1.5 | 33 | 34 | 32 | 13 | 22 | 34 | 33 | 35 |
| 0.7–1.3 | 14 | 30 | 22 | 3 | 12 | 14 | 20 | 32 |
| Monkey (conjugate) n = 25 | ||||||||
| <0.5 | 6 | 2 | 0 | 16 | 13 | 6 | 3 | 0 |
| >2.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5–1.5 | 19 | 23 | 20 | 9 | 12 | 16 | 20 | 20 |
| 0.7–1.3 | 9 | 18 | 14 | 6 | 9 | 7 | 10 | 18 |
| Rat (total) n = 25 | ||||||||
| <0.5 | 20 | 3 | 0 | 25 | 24 | 14 | 16 | 2 |
| >2.0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| 0.5–1.5 | 5 | 22 | 20 | 0 | 1 | 8 | 6 | 23 |
| 0.7–1.3 | 1 | 10 | 15 | 0 | 0 | 3 | 4 | 11 |
| Rat (conjugate) n = 17 | ||||||||
| <0.5 | 8 | 2 | 1 | 17 | 15 | 8 | 8 | 1 |
| >2.0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 1 |
| 0.5–1.5 | 9 | 15 | 7 | 0 | 2 | 9 | 7 | 14 |
| 0.7–1.3 | 1 | 10 | 3 | 0 | 0 | 4 | 1 | 9 |
| V = 1.0 | V = 0.9 | V = 0.8 | V = 1.0 | V = 0.9 | |||
|---|---|---|---|---|---|---|---|
| V = 1.0 | V = 0.9 | V = 0.8 | CL = 0.85 | CL = 0.85 | CL = 0.85 | V = 0.8 | V = 0.8 |
| Lifastuzumab vedotin from monkey 1 mg/kg to 2.4 mg/kg human; Total antibody | |||||||
| 0.54 | 0.73 | 0.99 | 0.30 | 0.41 | 0.55 | 0.65 | 0.80 |
| Lifastuzumab vedotin from rat 5 mg/kg to 2.4 mg/kg human | |||||||
| 0.37 | 0.66 | 1.15 | 0.18 | 0.31 | 0.55 | 0.52 | 0.77 |
| PF-06263507 from monkey 3 mg/kg to 3.34 mg/kg human | |||||||
| Total | |||||||
| 0.74 | 1.00 | 1.35 | 0.45 | 0.60 | 0.81 | 0.88 | 1.09 |
| Conjugates | |||||||
| 0.55 | 0.74 | 0.99 | 0.33 | 0.44 | 0.60 | 0.65 | 0.80 |
| Payload | |||||||
| 0.27 | 0.36 | 0.49 | 0.16 | 0.22 | 0.29 | 0.27 | 0.36 |
| Total trastuzumab deruxtecan from monkey 3 mg/kg to 6.4 mg/kg human | |||||||
| 0.62 | 0.84 | 1.13 | 0.35 | 0.48 | 0.64 | 0.62 | 0.93 |
| DXD | |||||||
| 0.04 | 0.06 | 0.07 | 0.03 | 0.04 | 0.05 | 0.04 | 0.06 |
| DSTA4637S from monkey 1 mg/kg to 5 mg/kg human | |||||||
| Total | |||||||
| 0.67 | 0.90 | 1.21 | 0.33 | 0.45 | 0.61 | 0.67 | 0.99 |
| Conjugates | |||||||
| 0.54 | 0.72 | 0.98 | 0.27 | 0.36 | 0.49 | 0.54 | 0.80 |
| Un-conjugated from monkey 150 mg/kg to 150 mg/kg human | |||||||
| 1.16 | 1.28 | 1.93 | 1.47 | 1.23 | 1.16 | 1.16 | 1.28 |
| V = 1.0 | V = 0.9 | V =0.8 | CL = 0.85 | CL = 0.85 | CL = 0.85 | V = 0.8 | V = 0.8 |
| DSTA4637S from rat 1 mg/kg to 5 mg/kg human | |||||||
| Total | |||||||
| 0.45 | 0.80 | 1.40 | 0.15 | 0.27 | 0.47 | 0.45 | 0.96 |
| Conjugates | |||||||
| 0.32 | 0.56 | 0.99 | 0.11 | 0.19 | 0.33 | 0.32 | 0.65 |
| Un-conjugated from rat 50 mg/kg to 150 mg/kg human | |||||||
| 0.58 | 1.03 | 1.80 | 0.21 | 0.37 | 0.66 | 0.58 | 1.03 |
| Trastuzumab-DM1 from monkey 30 mg/kg to 3.6 mg/kg human | |||||||
| Total | |||||||
| 0.71 | 0.95 | 1.28 | 0.62 | 0.83 | 1.13 | 0.81 | 1.02 |
| T-DM1 | |||||||
| 0.83 | 1.13 | 1.52 | 0.73 | 0.99 | 1.33 | 0.95 | 1.20 |
| Trastuzumab-DM1 from rat 3 mg/kg to 3.6 mg/kg human | |||||||
| Total | |||||||
| 0.32 | 0.57 | 1.00 | 0.14 | 0.24 | 0.42 | 0.42 | 0.65 |
| TDM-1 | |||||||
| 0.56 | 0.99 | 1.74 | 0.24 | 0.41 | 0.73 | 0.72 | 1.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, I. A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys. Antibodies 2022, 11, 42. https://doi.org/10.3390/antib11020042
Mahmood I. A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys. Antibodies. 2022; 11(2):42. https://doi.org/10.3390/antib11020042
Chicago/Turabian StyleMahmood, Iftekhar. 2022. "A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys" Antibodies 11, no. 2: 42. https://doi.org/10.3390/antib11020042
APA StyleMahmood, I. (2022). A Simple Method for the Prediction of Human Concentration–Time Profiles and Pharmacokinetics of Antibody–Drug Conjugates (ADC) from Rats or Monkeys. Antibodies, 11(2), 42. https://doi.org/10.3390/antib11020042