
Development of a Bispecific IgG1 Antibody Targeting BCMA and PDL1

 , , , ,
, , , , 
Highlights
- Using our patented platform, it is feasible to produce a bispecific antibody, bearing a functional IgG1 Fc, and binding bivalently to a tumor antigen (BCMA) and a checkpoint inhibitor (PDL1).
- The BCMAxPDL1 antibody was functionally active in vitro, inhibiting both BCMA and PDL1 at nanomolar concentration, and killing BCMA+ targets through ADCC and CDC.
- Targeting both BCMA and PDL1 may improve antibody therapeutic activity through combined mechanisms and may allow for the localization of the anti-PDL1 effect in the tumor bed.
- The affinities of both moieties have to be carefully studied in vitro, in order to optimize antibody function.
Abstract
:1. Introduction
2. Materials and Methods
2.1. Therapeutic Antibodies
2.2. Cells
2.3. Production of Stable BCMA Transfectants
2.4. Flow Cytometry
2.5. Surface Plasmon Resonance (SPR)
2.6. Inhibition of Binding of APRIL to mBCMA
2.7. Inhibition of PD1-PDL1 Signalling
2.8. Complement-Dependent Cytotoxicity (CDC)
2.9. NK-Mediated ADCC (24 h Killing Assays)
2.10. Long-Term Cell-Mediated Cytotoxicity (7-Day Killing Assays)
2.11. Statistical Analyses
3. Results
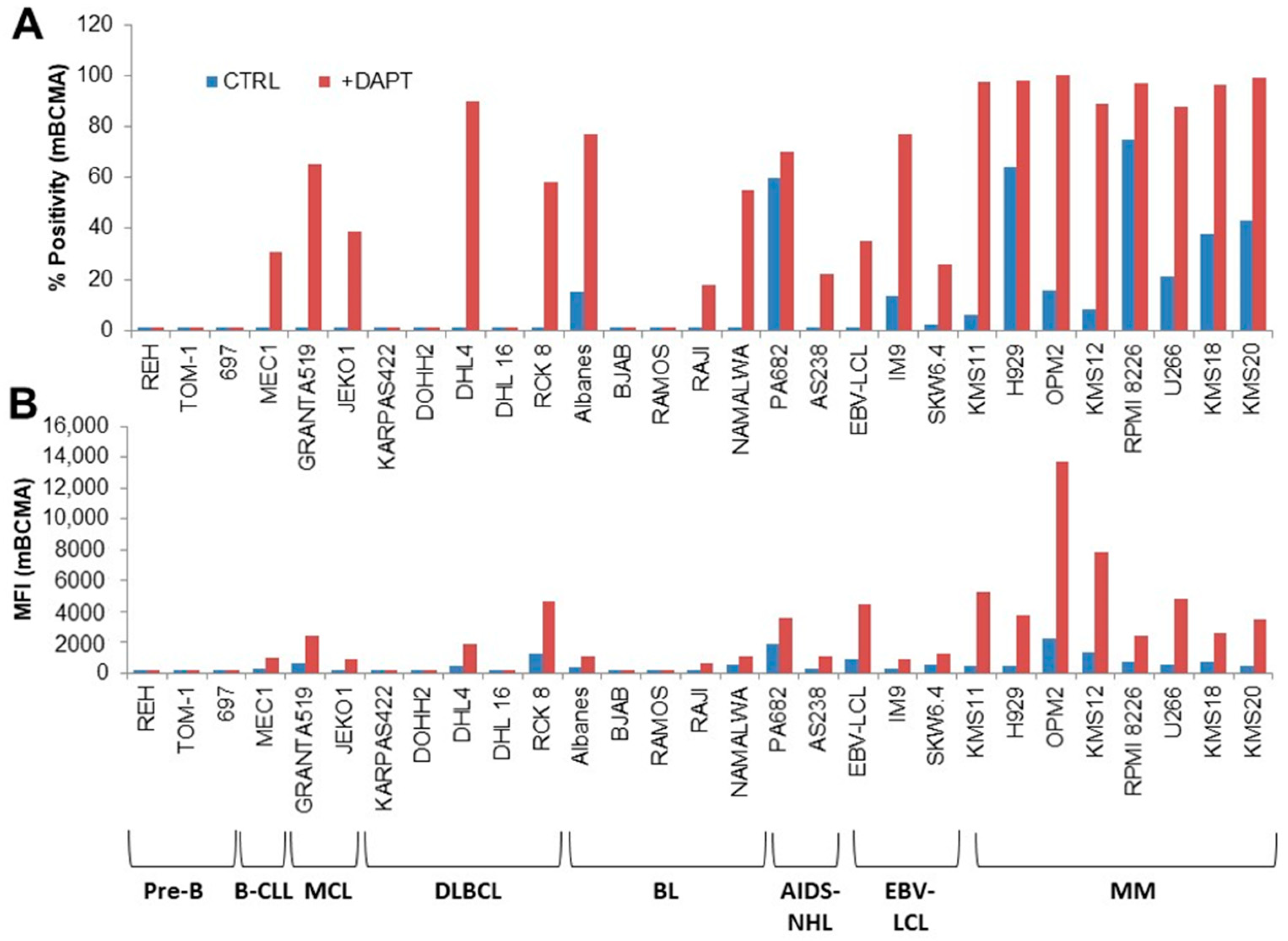
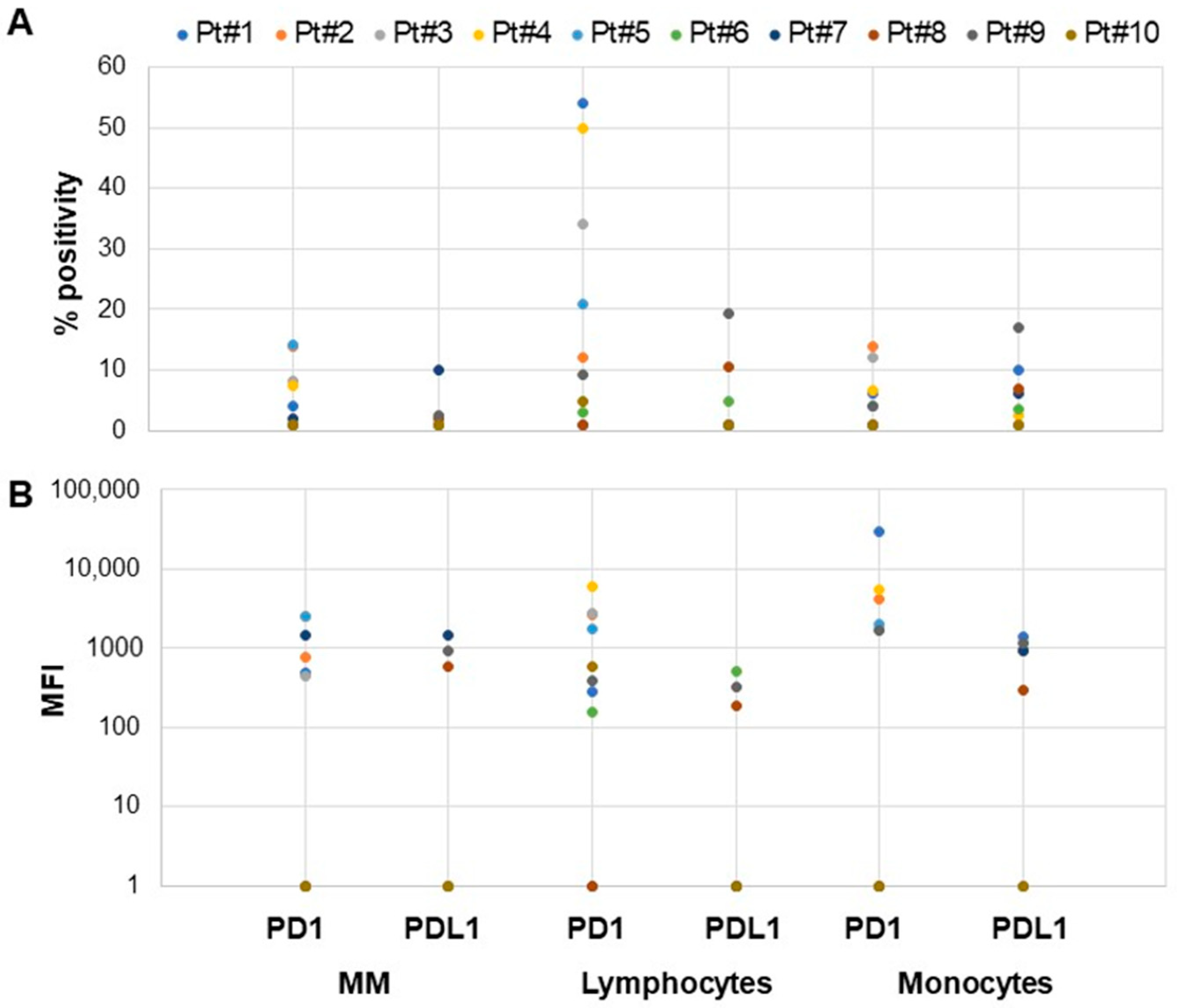
3.1. mBCMA and PD1/PDL1 Expression in Neoplastic B Cells
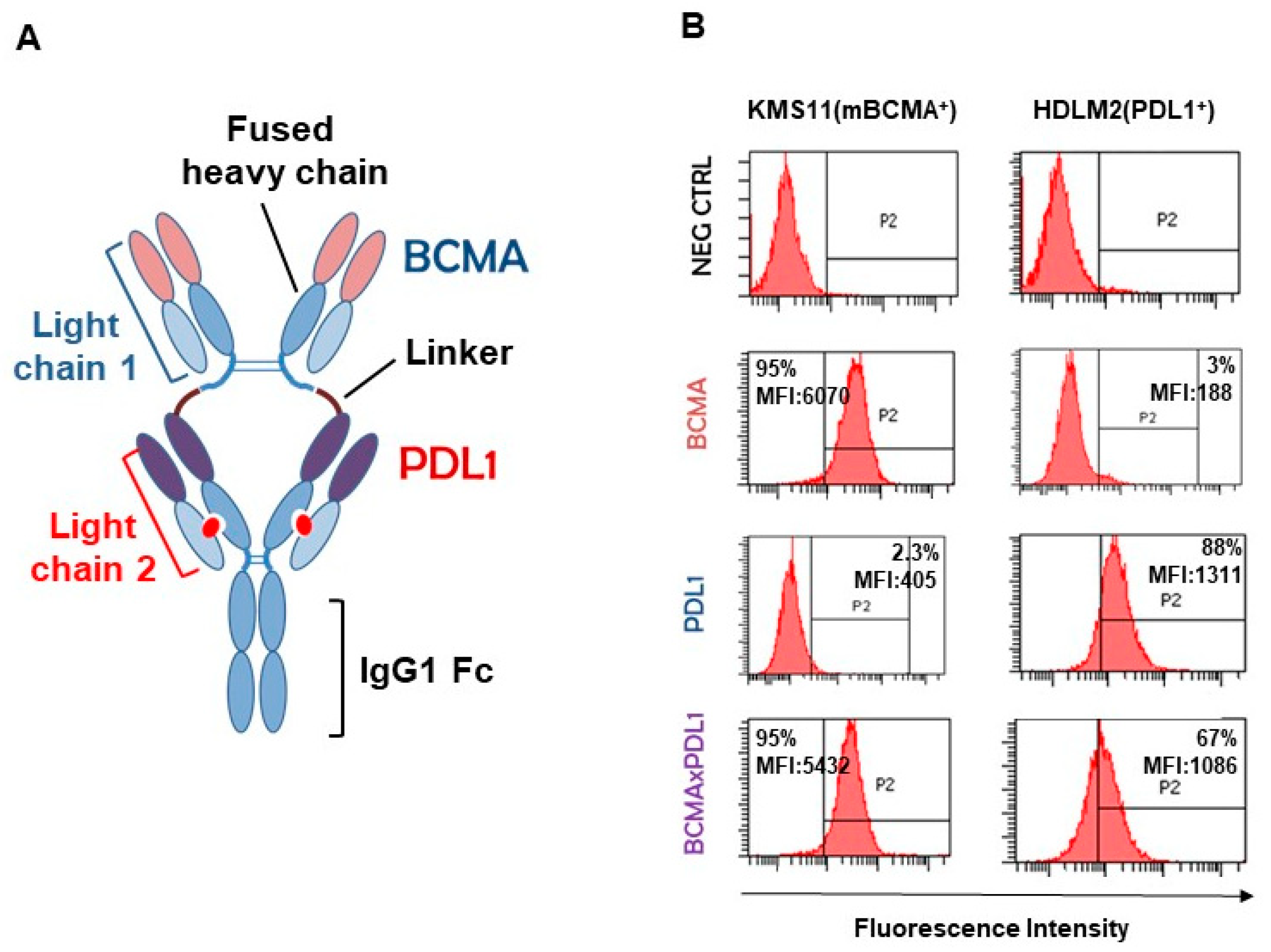
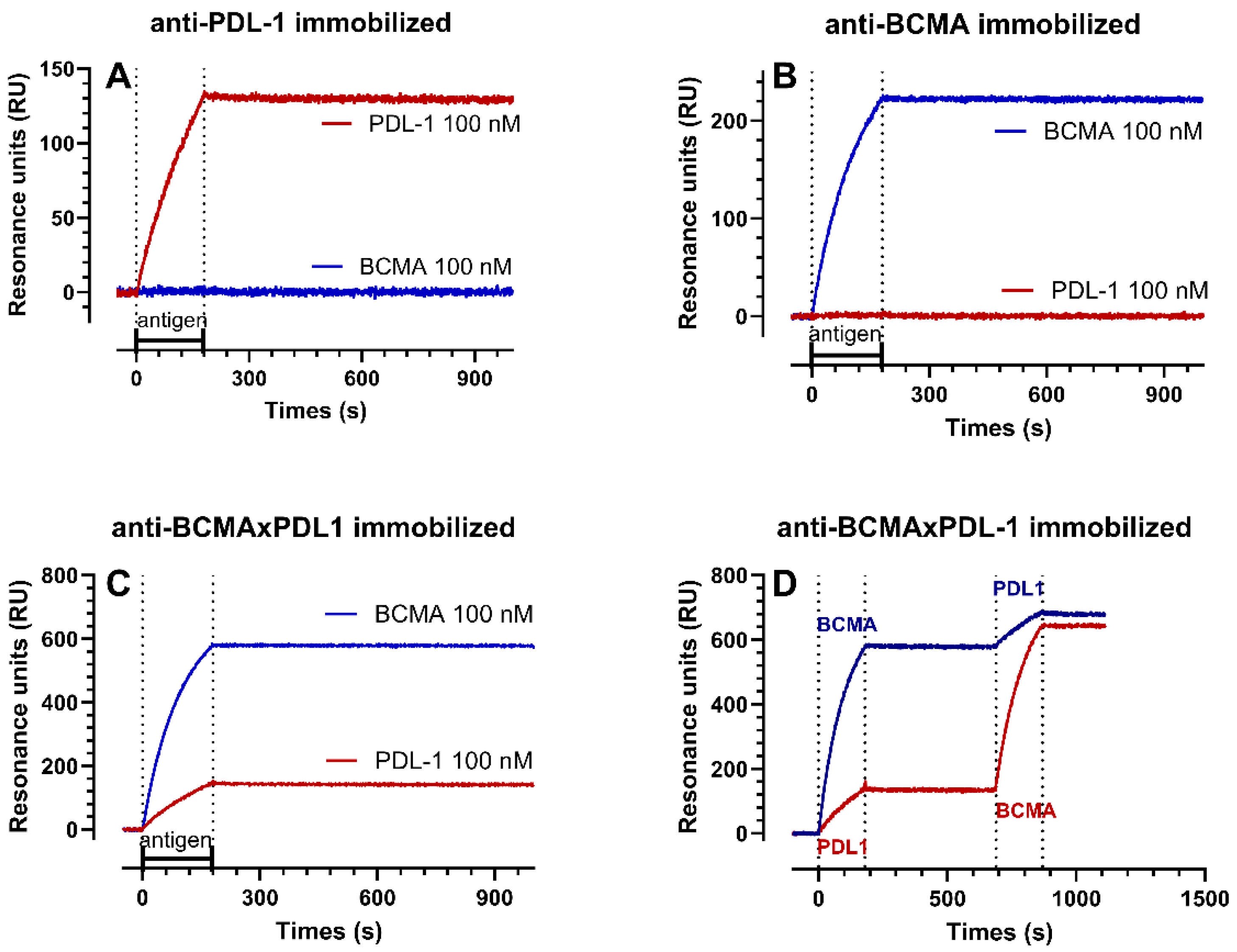
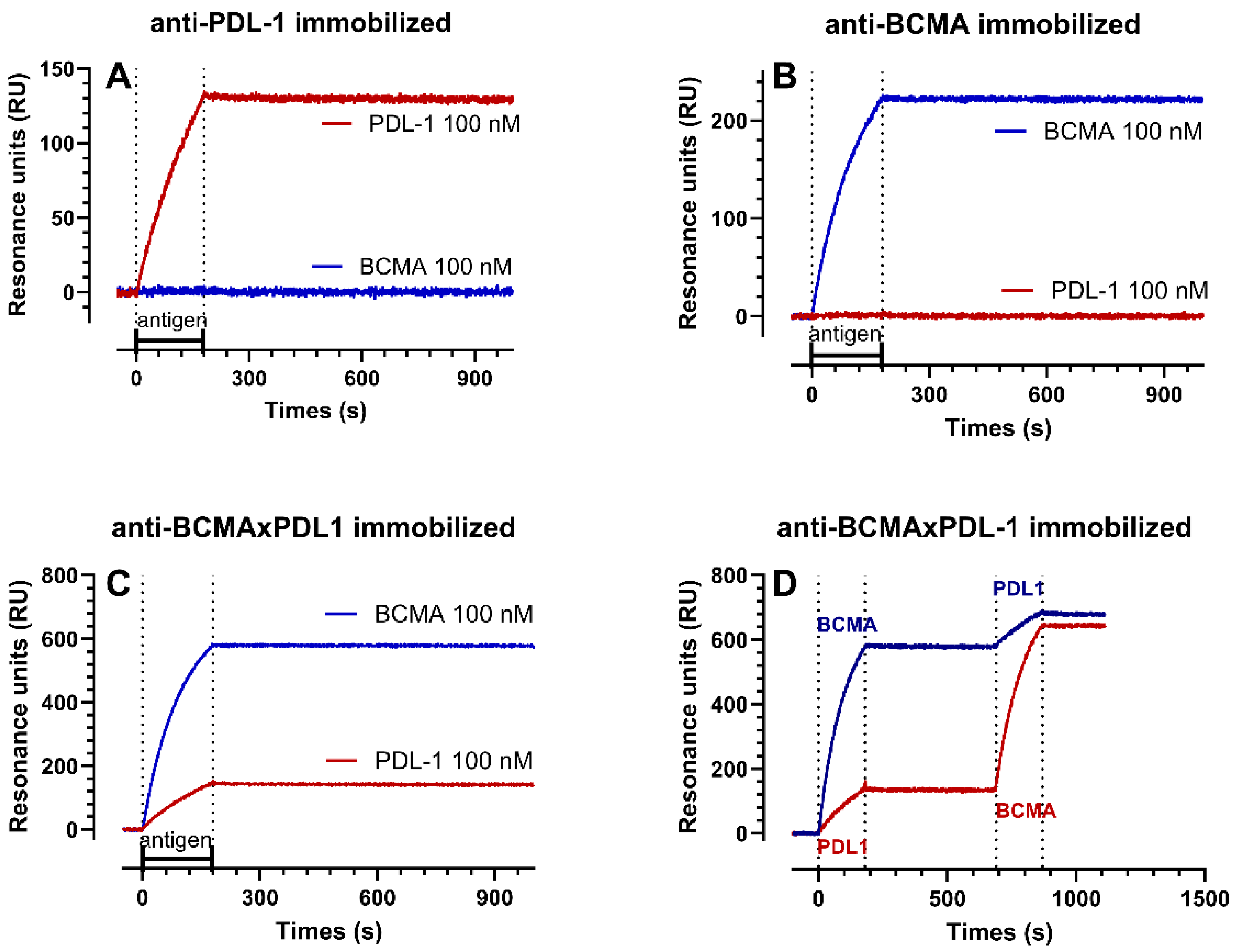
3.2. Structure of the BCMA×PDL1 bsAb and Affinity Measurements
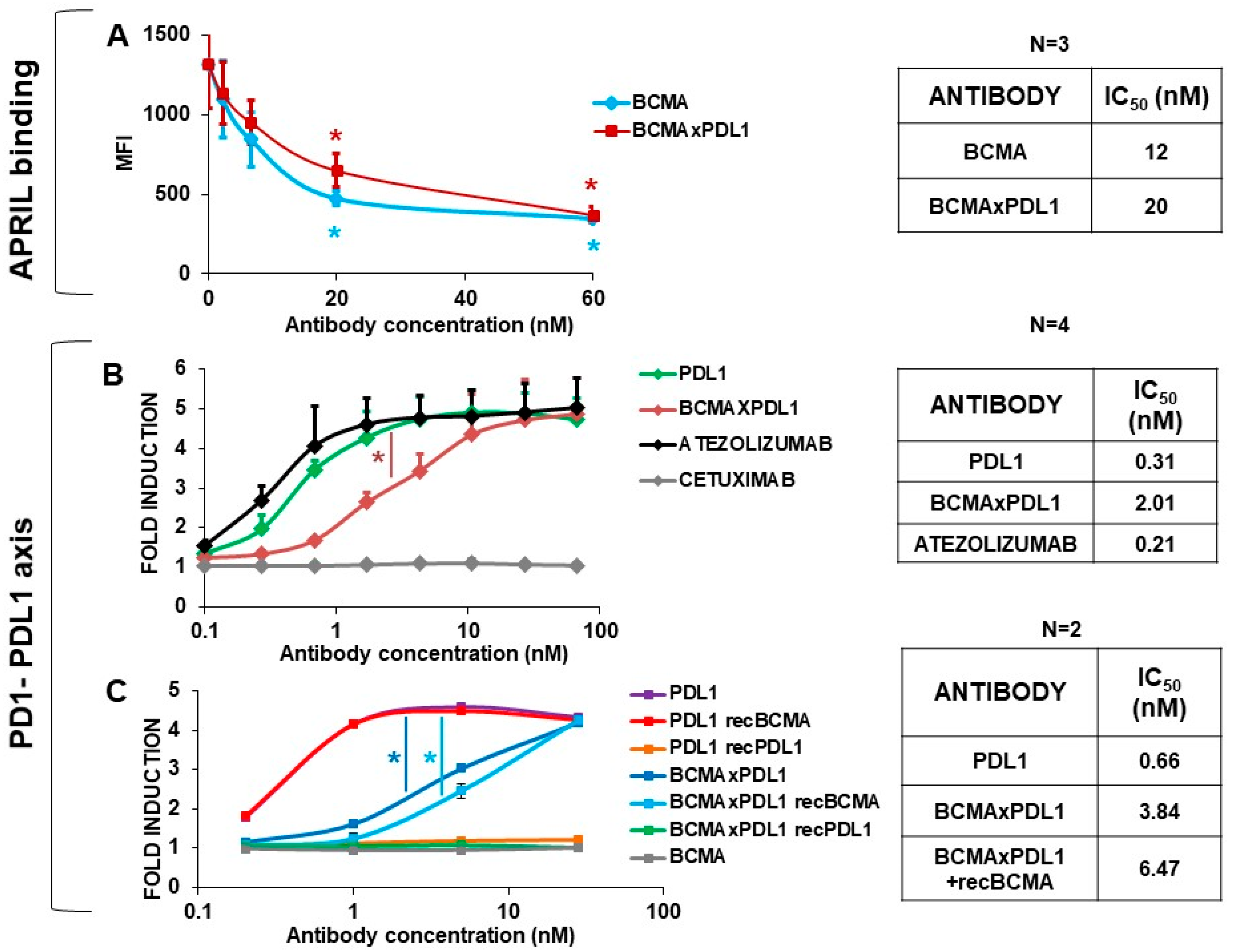
3.3. The BCMA×PDL1 bsAb Blocks PD1-PDL1 Interaction and APRIL Binding to BCMA
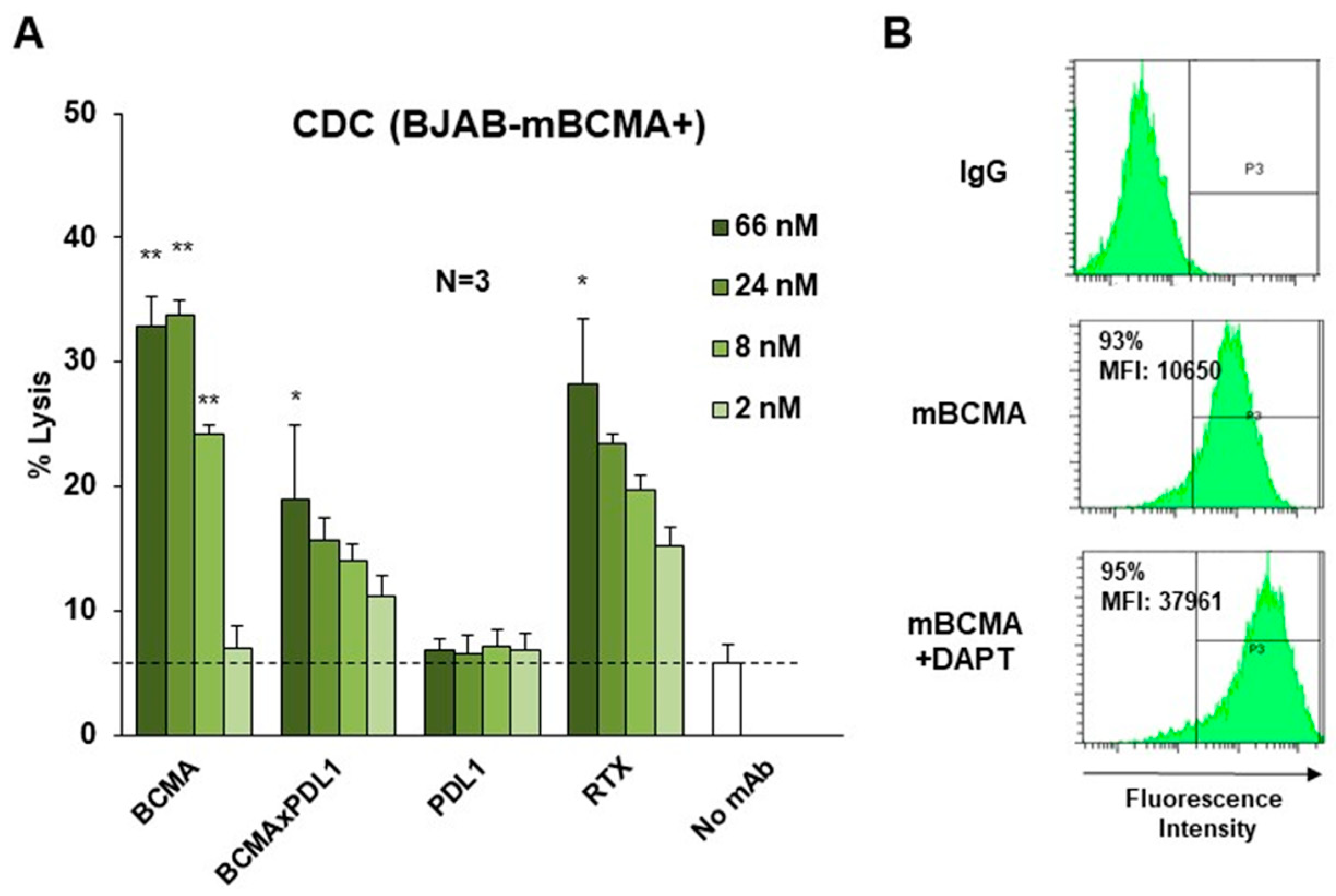
3.4. BCMA×PDL1 Induces CDC
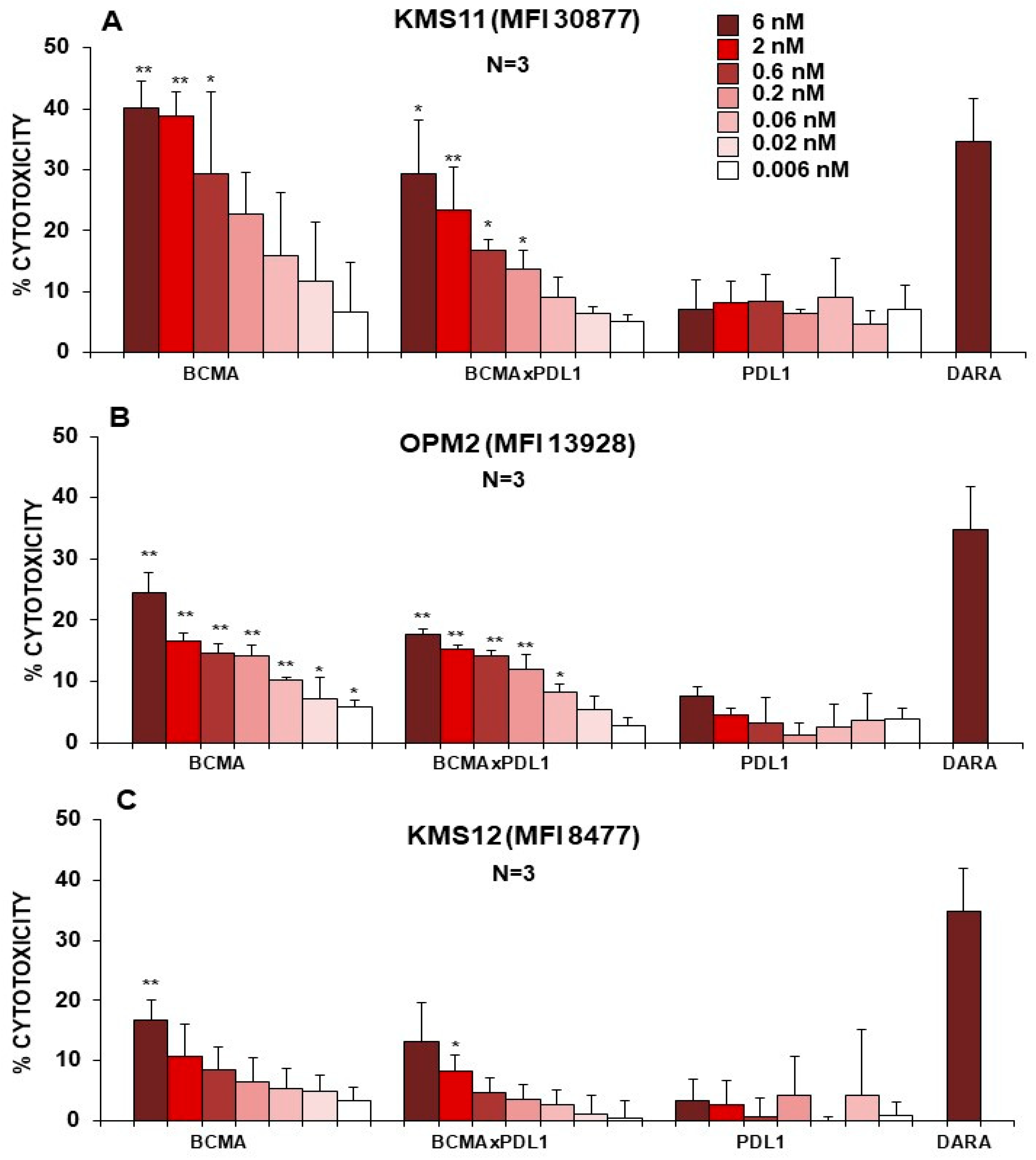
3.5. BCMA×PDL1 BsAb Induces ADCC by NK Cells
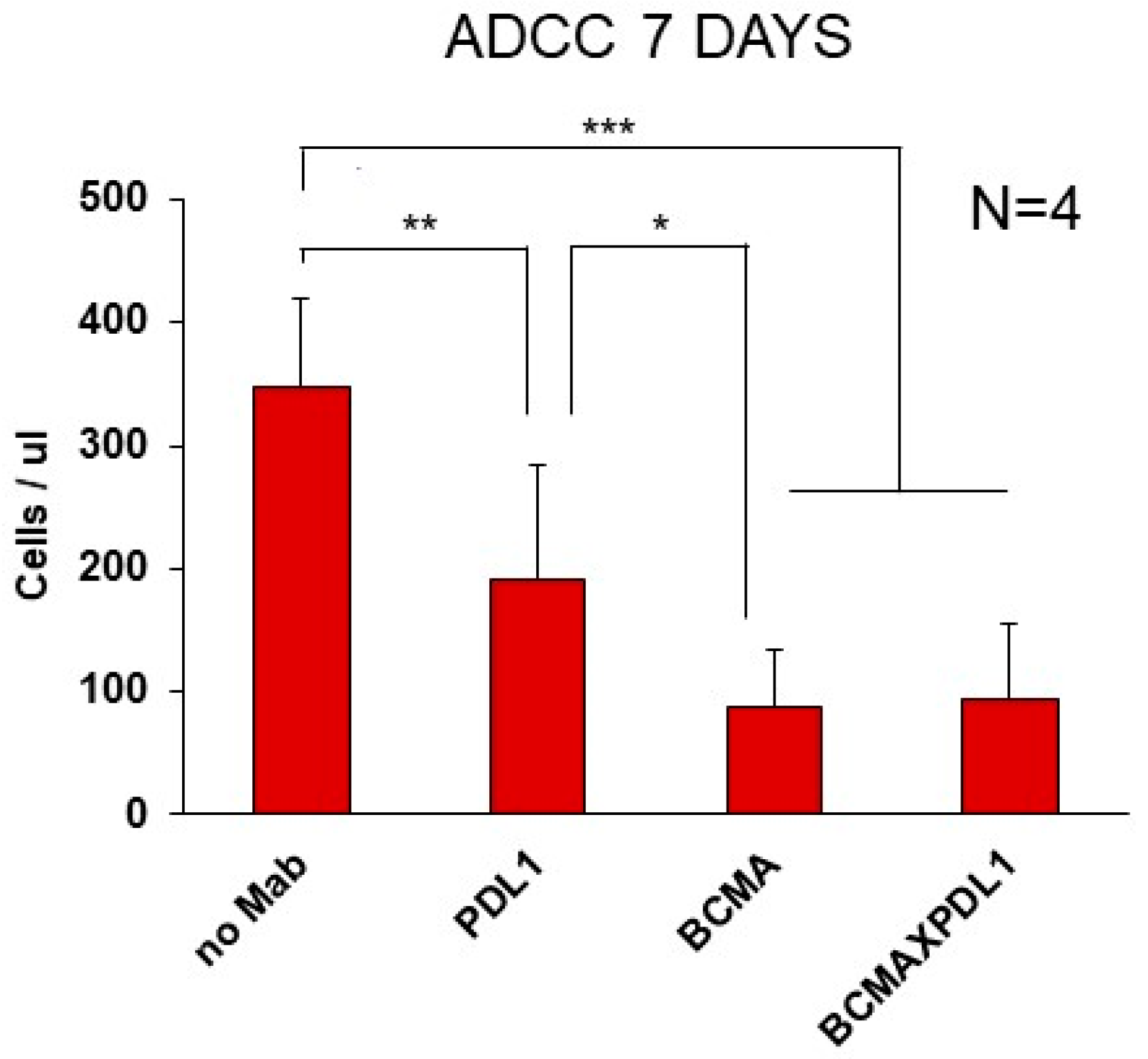
3.6. Cytotoxicity Mediated by BCMA×PDL1 in 7-Day Killing Assays
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Golay, J. Direct Targeting of Cancer Cells with Antibodies: What Can We Learn from the Successes and Failure of Unconjugated Antibodies for Lymphoid Neoplasias? J. Autoimmun. 2017, 85, 6–19. [Google Scholar] [CrossRef]
- Shouse, G. Update on Bi-Specific Monoclonal Antibodies for Blood Cancers. Curr. Opin. Oncol. 2023, 35, 441–445. [Google Scholar] [CrossRef]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic Antibodies: What Have We Learnt from Targeting CD20 and Where Are We Going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.I. Fine-Tuning Bispecific Therapeutics. Pharmacol. Ther. 2020, 212, 107582. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef] [PubMed]
- Salem, D.A.; Maric, I.; Yuan, C.M.; Liewehr, D.J.; Venzon, D.J.; Kochenderfer, J.; Stetler-Stevenson, M. Quantification of B-Cell Maturation Antigen, a Target for Novel Chimeric Antigen Receptor T-Cell Therapy in Myeloma. Leuk. Res. 2018, 71, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gray, F.; Cushman, I.; Wurmser, A.; Chan, H.; Couto, S.; Wang, M.; Nakayama, Y.; Hagner, P.; Al-Masri, H.; et al. A Novel BCMA Immunohistochemistry Assay Reveals a Heterogenous and Dynamic BCMA Expression Profile in Multiple Myeloma. Mod. Pathol. 2023, 36, 100050. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.-H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase Directly Sheds the Survival Receptor BCMA from Plasma Cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Meinl, E.; Thaler, F.S.; Lichtenthaler, S.F. Shedding of BAFF/APRIL Receptors Controls B Cells. Trends Immunol. 2018, 39, 673–676. [Google Scholar] [CrossRef]
- Ray, U.; Orlowski, R.Z. Antibody–Drug Conjugates for Multiple Myeloma: Just the Beginning, or the Beginning of the End? Pharmaceuticals 2023, 16, 590. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Lan, H.; Wu, J.; Xiao, Y. CAR-T Cell Therapy in Multiple Myeloma: Current Limitations and Potential Strategies. Front. Immunol. 2023, 14, 1101495. [Google Scholar] [CrossRef]
- Tanenbaum, B.; Miett, T.; Patel, S.A. The Emerging Therapeutic Landscape of Relapsed/Refractory Multiple Myeloma. Ann. Hematol. 2023, 102, 1–11. [Google Scholar] [CrossRef]
- Interdonato, A.; Choblet, S.; Sana, M.; Valgardsdottir, R.; Cribioli, S.; Alzani, R.; Roth, M.; Duonor-Cerutti, M.; Golay, J. BL-01, an Fc-Bearing, Tetravalent CD20 × CD5 Bispecific Antibody, Redirects Multiple Immune Cells to Kill Tumors in Vitro and in Vivo. Cytotherapy 2022, 24, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Oden, F.; Marino, S.F.; Brand, J.; Scheu, S.; Kriegel, C.; Olal, D.; Takvorian, A.; Westermann, J.; Yilmaz, B.; Hinz, M.; et al. Potent Anti-Tumor Response by Targeting B Cell Maturation Antigen (BCMA) in a Mouse Model of Multiple Myeloma. Mol. Oncol. 2015, 9, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Cérutti, M.; Golay, J. Lepidopteran Cells, an Alternative for the Production of Recombinant Antibodies? MAbs 2012, 4, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “One-Shot” Kinetics and Small Molecule Analysis Using the ProteOn XPR36 Array Biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef]
- Beeg, M.; Burti, C.; Allocati, E.; Ciafardini, C.; Banzi, R.; Nobili, A.; Caprioli, F.; Garattini, S.; Gobbi, M. Surface Plasmon Resonance Unveils Important Pitfalls of Enzyme-Linked Immunoassay for the Detection of Anti-Infliximab Antibodies in Patients’ Sera. Sci. Rep. 2021, 11, 14976. [Google Scholar] [CrossRef]
- Wang, Z.; Chimenti, M.S.; Strouse, C.; Weiner, G.J. T Cells, Particularly Activated CD4+ Cells, Maintain Anti-CD20-Mediated NK Cell Viability and Antibody Dependent Cellular Cytotoxicity. Cancer Immunol. Immunother. 2022, 71, 237–249. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, C.; Lum, L.G.; Simons, A.; Weiner, G.J. Bispecific Antibody-Activated T Cells Enhance NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity. J. Hematol. Oncol. 2021, 14, 204. [Google Scholar] [CrossRef]
- Raghunandan, S.; Pauly, M.; Blum, W.G.; Qayed, M.; Dhodapkar, M.V.; Elkhalifa, M.; Watkins, B.; Schoettler, M.; Horwitz, E.; Parikh, S.; et al. BCMA CAR-T Induces Complete and Durable Remission in Refractory Plasmablastic Lymphoma. J. Immunother. Cancer 2023, 11, e006684. [Google Scholar] [CrossRef]
- Martens, A.W.J.; Rietveld, J.M.; de Boer, R.; Peters, F.S.; Ngo, A.; van Mil, L.W.H.G.; de Heer, K.; Spaargaren, M.; Verkleij, C.P.M.; van de Donk, N.W.C.J.; et al. Redirecting T-Cell Activity with Anti-BCMA/Anti-CD3 Bispecific Antibodies in Chronic Lymphocytic Leukemia and Other B-Cell Lymphomas. Cancer Res. Commun. 2022, 2, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Wallweber, H.J.A.; Compaan, D.M.; Starovasnik, M.A.; Hymowitz, S.G. The Crystal Structure of a Proliferation-Inducing Ligand, APRIL. J. Mol. Biol. 2004, 343, 283–290. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P.; Rennert, P.; Browning, J. BAFF AND APRIL: A Tutorial on B Cell Survival. Annu. Rev. Immunol. 2003, 21, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Dalla Palma, B.; Marchica, V.; Catarozzo, M.T.; Giuliani, N.; Accardi, F. Monoclonal and Bispecific Anti-BCMA Antibodies in Multiple Myeloma. J. Clin. Med. 2020, 9, 3022. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.-F.; Klein, B.; Tarte, K. BAFF and APRIL Protect Myeloma Cells from Apoptosis Induced by Interleukin 6 Deprivation and Dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Reijmers, R.M.; Groen, R.W.J.; Kuil, A.; Weijer, K.; Kimberley, F.C.; Medema, J.P.; van Kuppevelt, T.H.; Li, J.-P.; Spaargaren, M.; Pals, S.T. Disruption of Heparan Sulfate Proteoglycan Conformation Perturbs B-Cell Maturation and APRIL-Mediated Plasma Cell Survival. Blood 2011, 117, 6162–6171. [Google Scholar] [CrossRef] [PubMed]
- Kampa, M.; Notas, G.; Stathopoulos, E.N.; Tsapis, A.; Castanas, E. The TNFSF Members APRIL and BAFF and Their Receptors TACI, BCMA, and BAFFR in Oncology, With a Special Focus in Breast Cancer. Front. Oncol. 2020, 10, 827. [Google Scholar] [CrossRef]
- Golay, J.; Regazzi, M. Key Features Defining the Disposition of Bispecific Antibodies and Their Efficacy In Vivo. Ther. Drug Monit. 2020, 42, 57. [Google Scholar] [CrossRef]
- Golay, J.; Lazzari, M.; Facchinetti, V.; Bernasconi, S.; Borleri, G.; Barbui, T.; Rambaldi, A.; Introna, M. CD20 Levels Determine the in Vitro Susceptibility to Rituximab and Complement of B-Cell Chronic Lymphocytic Leukemia: Further Regulation by CD55 and CD59. Blood 2001, 98, 3383–3389. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.J.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Libby, E.N.; Coffey, D.G.; Tuazon, S.A.; Wood, B.; Gooley, T.; et al. γ-Secretase Inhibitor in Combination with BCMA Chimeric Antigen Receptor T-Cell Immunotherapy for Individuals with Relapsed or Refractory Multiple Myeloma: A Phase 1, First-in-Human Trial. Lancet Oncol. 2023, 24, 811–822. [Google Scholar] [CrossRef]
- Bednarska, K.; Nath, K.; Nicol, W.; Gandhi, M.K. Immunity Reloaded: Deconstruction of the PD-1 Axis in B Cell Lymphomas. Blood Rev. 2021, 50, 100832. [Google Scholar] [CrossRef] [PubMed]
- Desantis, V.; Savino, F.D.; Scaringella, A.; Potenza, M.A.; Nacci, C.; Frassanito, M.A.; Vacca, A.; Montagnani, M. The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value. J. Clin. Med. 2022, 11, 2513. [Google Scholar] [CrossRef]
- Zhang, T.; Lin, Y.; Gao, Q. Bispecific Antibodies Targeting Immunomodulatory Checkpoints for Cancer Therapy. Cancer Biol. Med. 2023, 20, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, Y.; Yang, Y.; Lin, B.; Zhu, M.; Xu, J.; Chen, Y.; Wu, W.; Chen, B.; Chen, X.; et al. Anti-PD-1/Her2 Bispecific Antibody IBI315 Enhances the Treatment Effect of Her2-Positive Gastric Cancer through Gasdermin B-Cleavage Induced Pyroptosis. Adv. Sci. 2023, 10, e2303908. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Pérez, L.; Lázaro-Gorines, R.; Harwood, S.L.; Compte, M.; Navarro, R.; Tapia-Galisteo, A.; Bonet, J.; Blanco, B.; Lykkemark, S.; Ramírez-Fernández, Á.; et al. A PD-L1/EGFR Bispecific Antibody Combines Immune Checkpoint Blockade and Direct Anti-Cancer Action for an Enhanced Anti-Tumor Response. Oncoimmunology 2023, 12, 2205336. [Google Scholar] [CrossRef]
- Li, T.; Wang, X.; Niu, M.; Wang, M.; Zhou, J.; Wu, K.; Yi, M. Bispecific Antibody Targeting TGF-β and PD-L1 for Synergistic Cancer Immunotherapy. Front. Immunol. 2023, 14, 1196970. [Google Scholar] [CrossRef]
- Yin, L.; Wang, X.-J.; Chen, D.-X.; Liu, X.-N.; Wang, X.-J. Humanized Mouse Model: A Review on Preclinical Applications for Cancer Immunotherapy. Am. J. Cancer Res. 2020, 10, 4568–4584. [Google Scholar]
- Vrohlings, M.; Müller, J.; Jungmichel, S.; Senn, D.; Howald, A.B.; Schleier, T.; Scheifele, F.; Wendelspiess, S.; Richle, P.; Merten, H.; et al. Preclinical Assessment of CDR101—A BCMAxCD3xPD-L1 Trispecific Antibody with Superior Anti-Tumor Efficacy. Blood 2021, 138 (Suppl. S1), 1583. [Google Scholar] [CrossRef]
- Boross, P.; Jansen, J.H.M.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.J.; Parren, P.W.H.I.; et al. The in Vivo Mechanism of Action of CD20 Monoclonal Antibodies Depends on Local Tumor Burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Cittera, E.; Di Gaetano, N.; Manganini, M.; Mosca, M.; Nebuloni, M.; van Rooijen, N.; Vago, L.; Introna, M. The Role of Complement in the Therapeutic Activity of Rituximab in a Murine B Lymphoma Model Homing in Lymph Nodes. Haematologica 2006, 91, 176–183. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ka | kd | KD | Rmax | |
|---|---|---|---|---|
| 1/Ms | 1/s | nM | RU | |
| recPDL1 on PDL1 mAb | 4.9–5.5 × 104 | 1.1–5.9 × 10−6 | 0.02–0.12 | 222–227 |
| recPDL1 on BCMA×PDL1 bsAb | 3.8–6.2 × 104 | 0.4–1.8 × 10−5 | 0.11–0.29 | 212–271 |
| recBCMA on BCMA mAB | 5.9–8.1 × 104 | 5.9–8.6 × 10−6 | 0.11–0.10 | 289–312 |
| recBCMA on BCMA×PDL1 bsAb | 1.0–1.0 × 105 | 1.3–2.7 × 10−6 | 0.01–0.03 | 683–700 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattaneo, I.; Choblet, S.; Valgardsdottir, R.; Roth, M.; Massafra, A.; Beeg, M.; Gobbi, M.; Duonor-Cerutti, M.; Golay, J. Development of a Bispecific IgG1 Antibody Targeting BCMA and PDL1. Antibodies 2024, 13, 15. https://doi.org/10.3390/antib13010015
Cattaneo I, Choblet S, Valgardsdottir R, Roth M, Massafra A, Beeg M, Gobbi M, Duonor-Cerutti M, Golay J. Development of a Bispecific IgG1 Antibody Targeting BCMA and PDL1. Antibodies. 2024; 13(1):15. https://doi.org/10.3390/antib13010015
Chicago/Turabian StyleCattaneo, Irene, Sylvie Choblet, Rut Valgardsdottir, Muriel Roth, Annamaria Massafra, Marten Beeg, Marco Gobbi, Martine Duonor-Cerutti, and Josée Golay. 2024. "Development of a Bispecific IgG1 Antibody Targeting BCMA and PDL1" Antibodies 13, no. 1: 15. https://doi.org/10.3390/antib13010015
APA StyleCattaneo, I., Choblet, S., Valgardsdottir, R., Roth, M., Massafra, A., Beeg, M., Gobbi, M., Duonor-Cerutti, M., & Golay, J. (2024). Development of a Bispecific IgG1 Antibody Targeting BCMA and PDL1. Antibodies, 13(1), 15. https://doi.org/10.3390/antib13010015