
Enhanced Characterization of Lysine-Linked Antibody Drug Conjugates Enabled by Middle-Down Mass Spectrometry and Higher-Energy Collisional Dissociation-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation and Ultraviolet Photodissociation

 , ,
, ,
Abstract
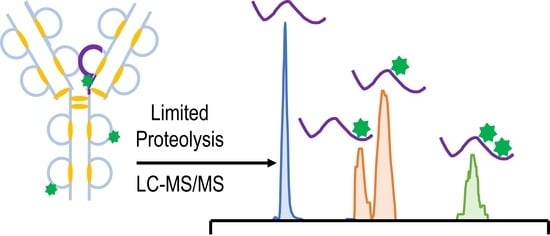
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Liquid Chromatography-Mass Spectrometry
2.3. Data Analysis
3. Results
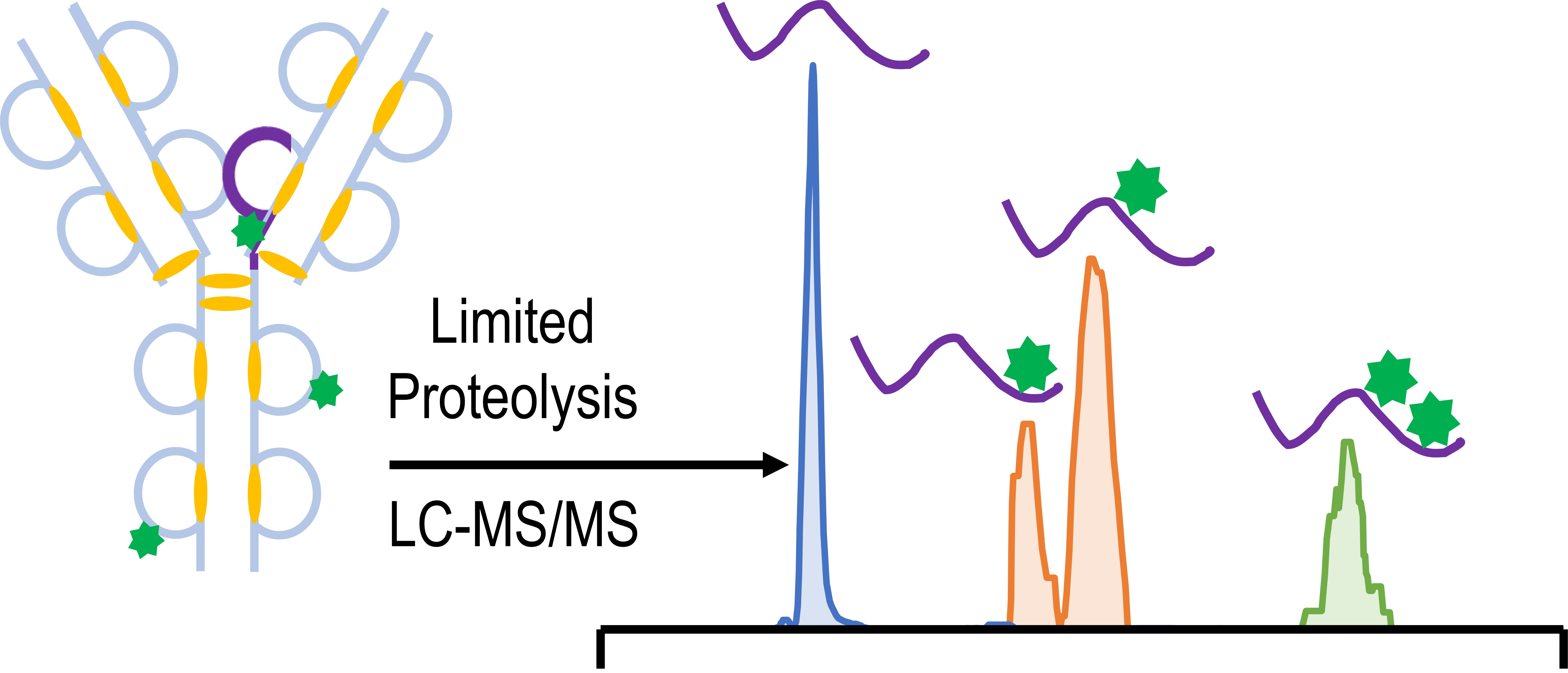
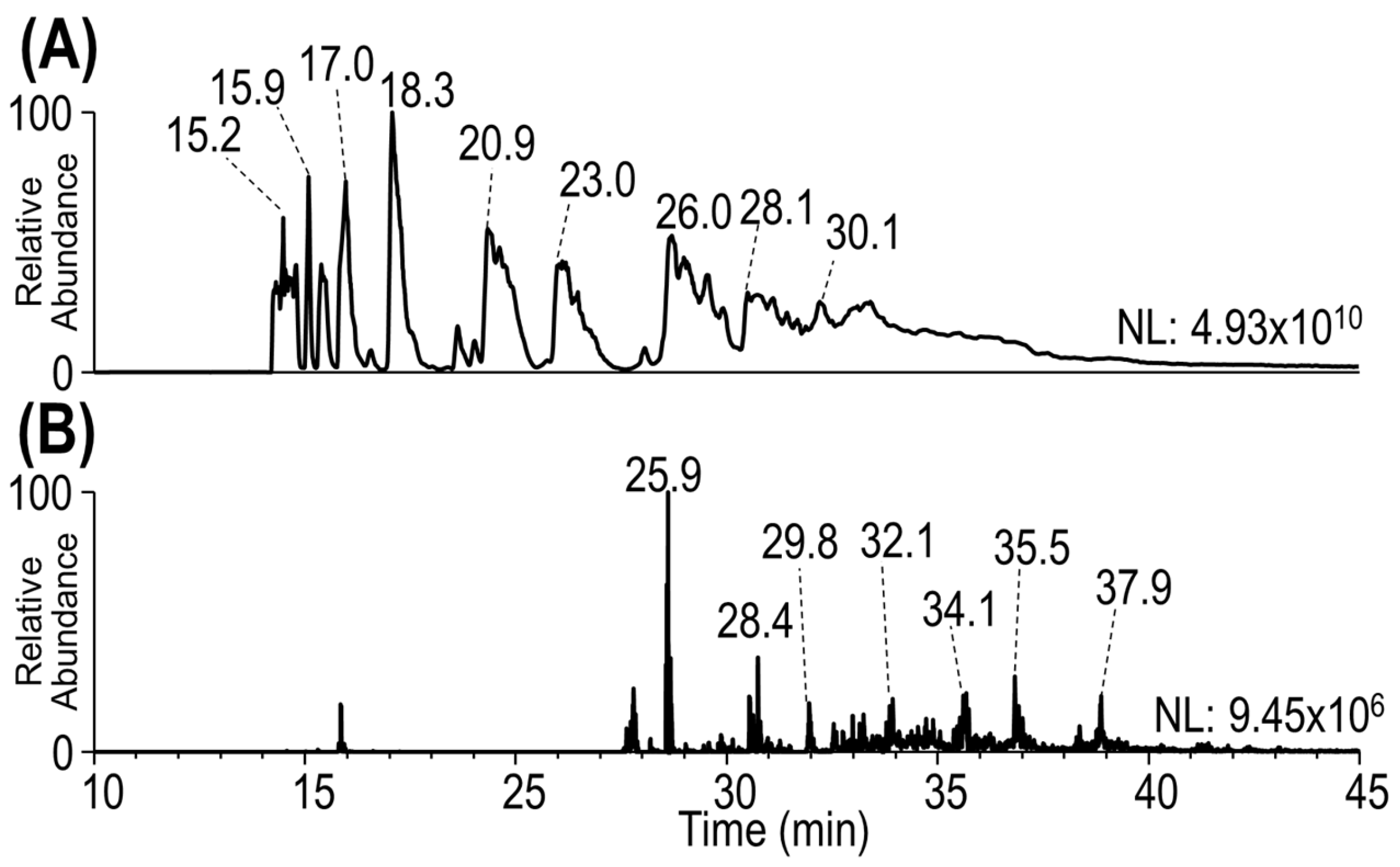
3.1. Development of a Middle-Down Method
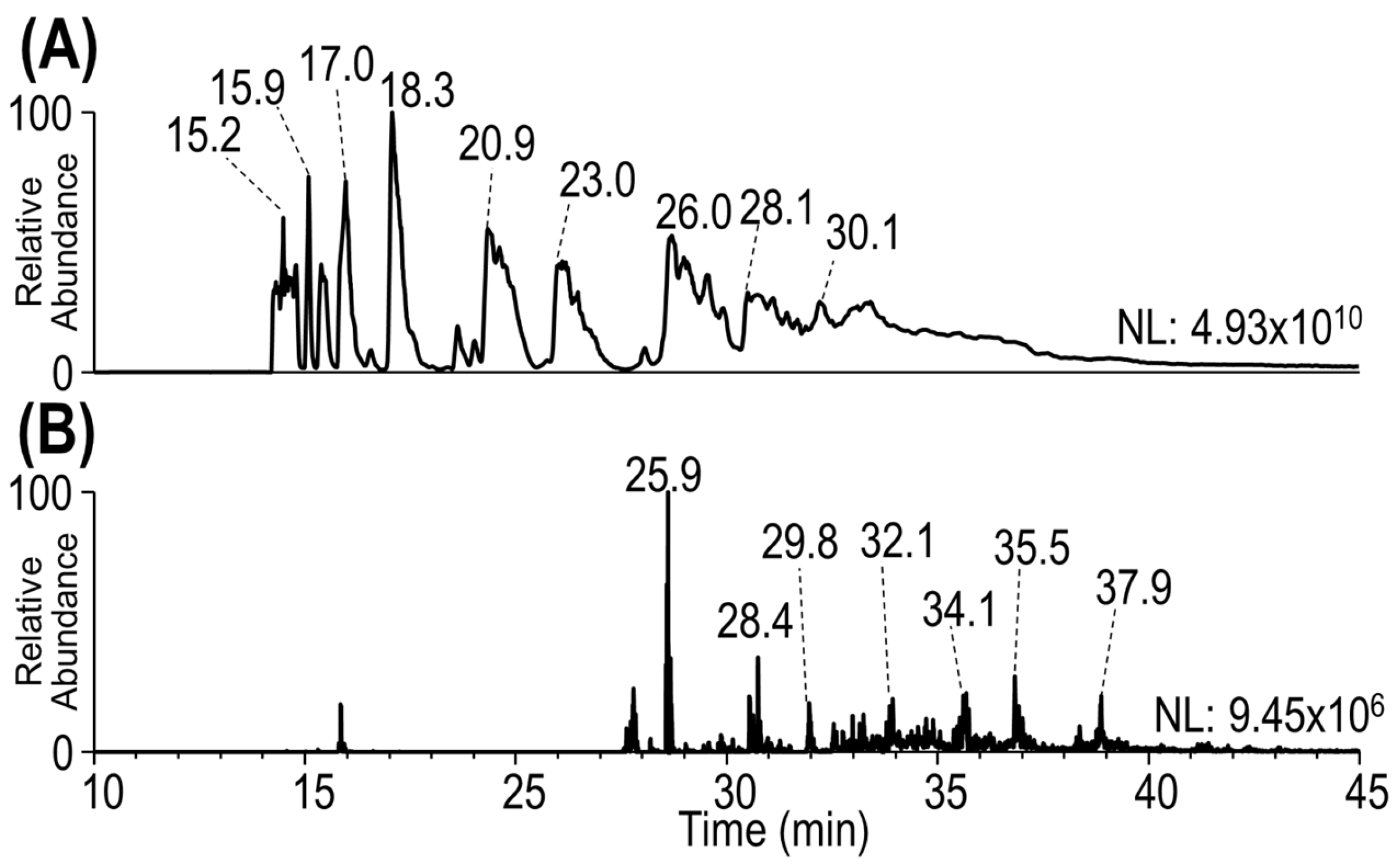
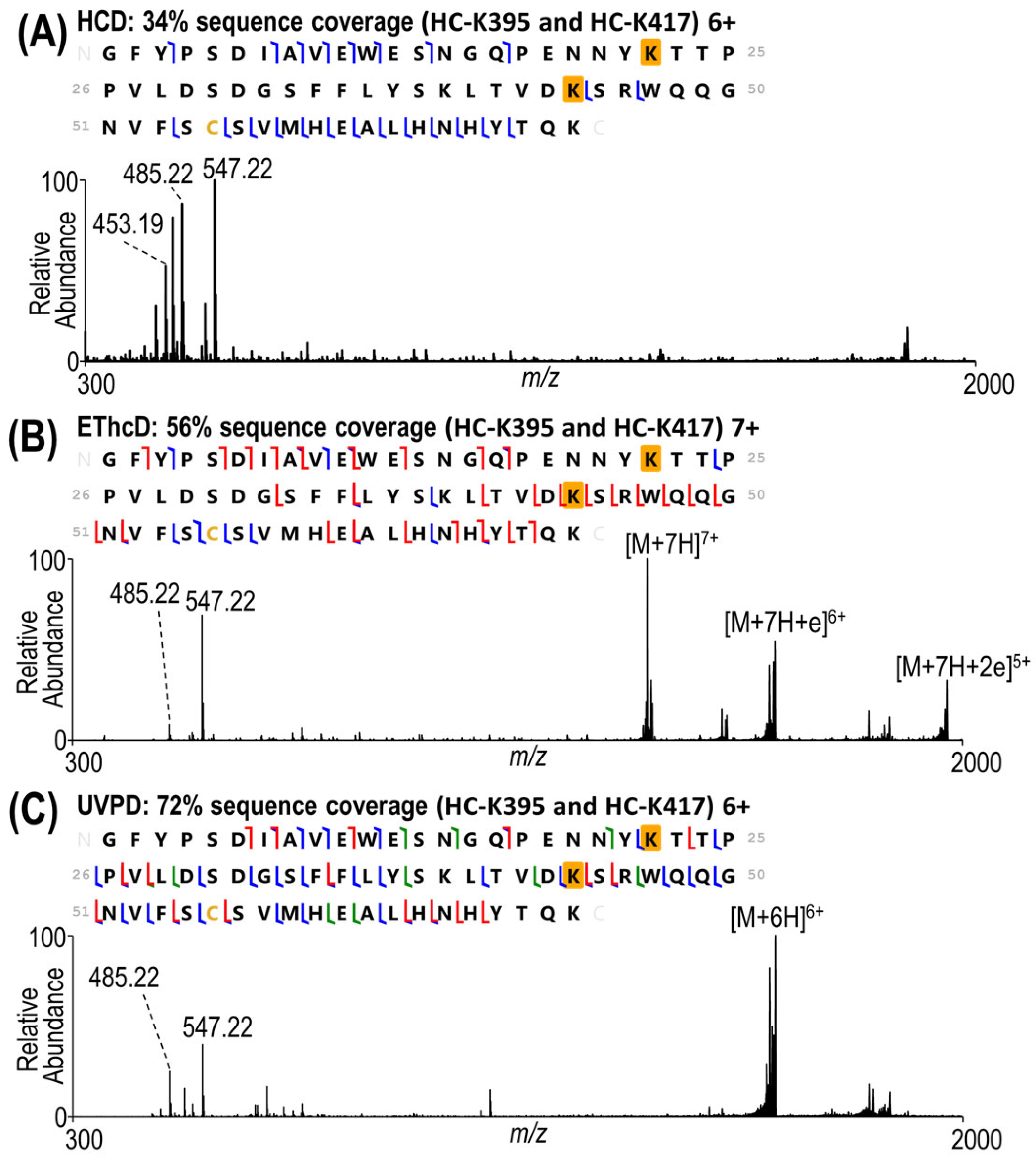
3.2. Characterization of Heterogeneous Species with Multiple Conjugation Sites
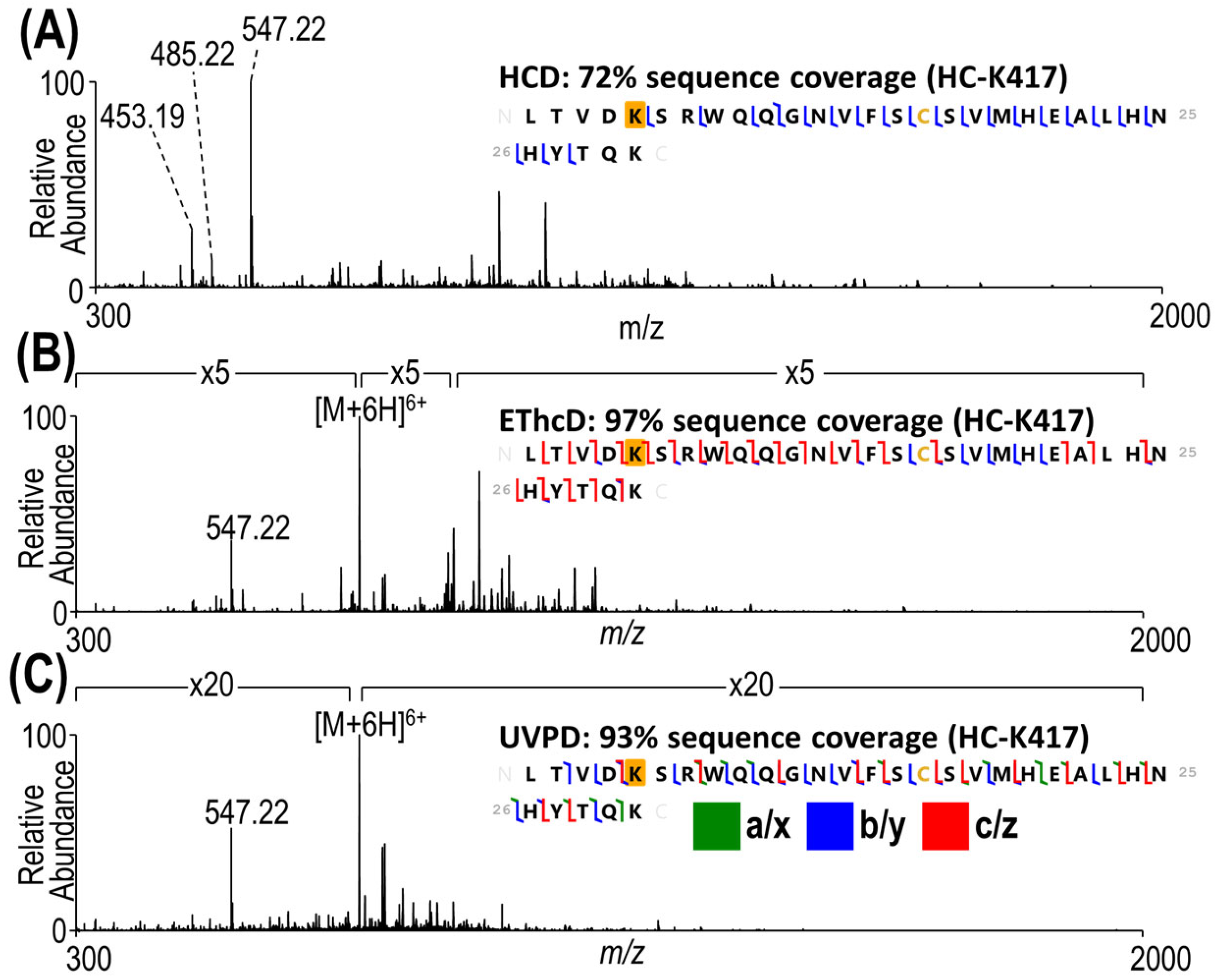
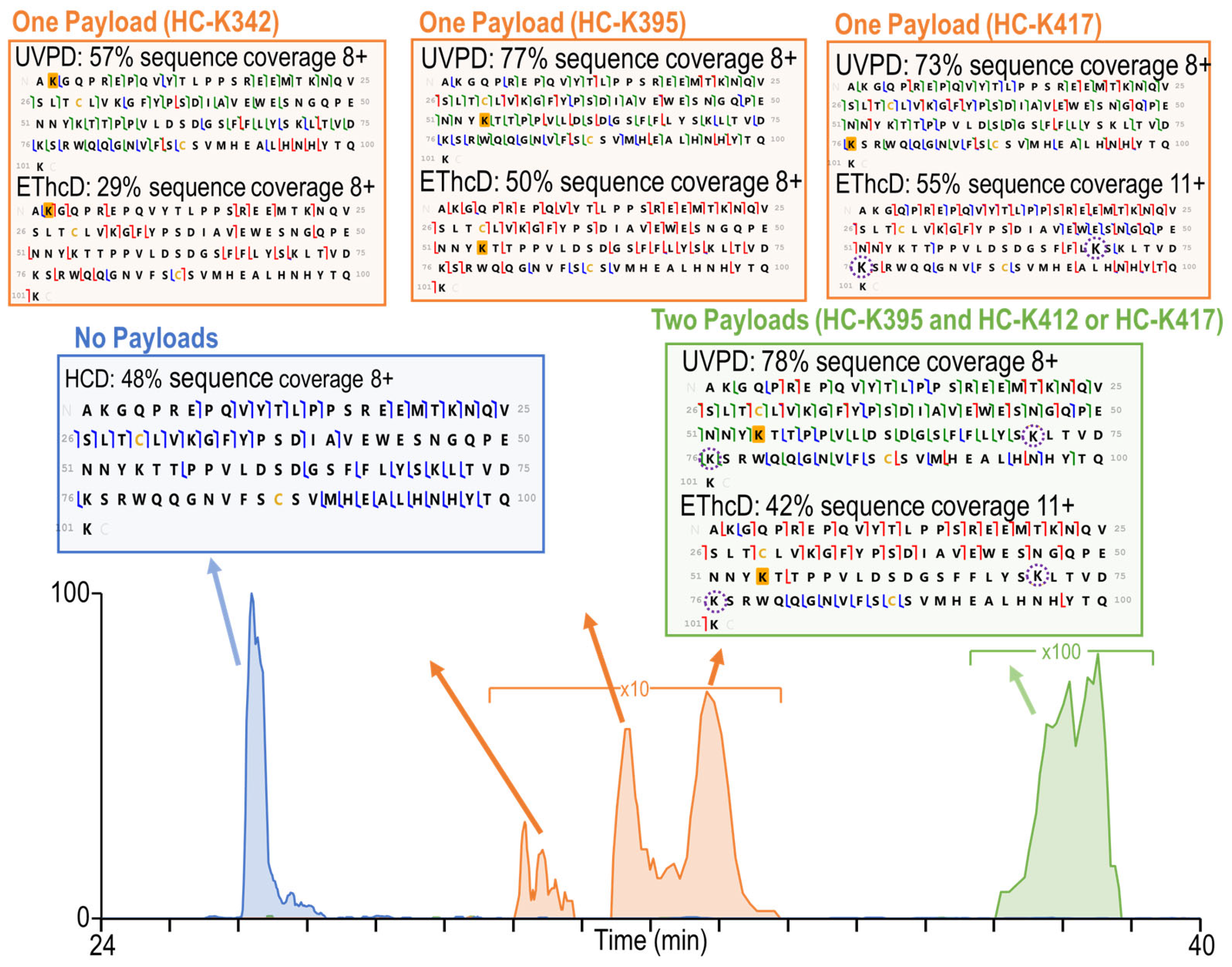
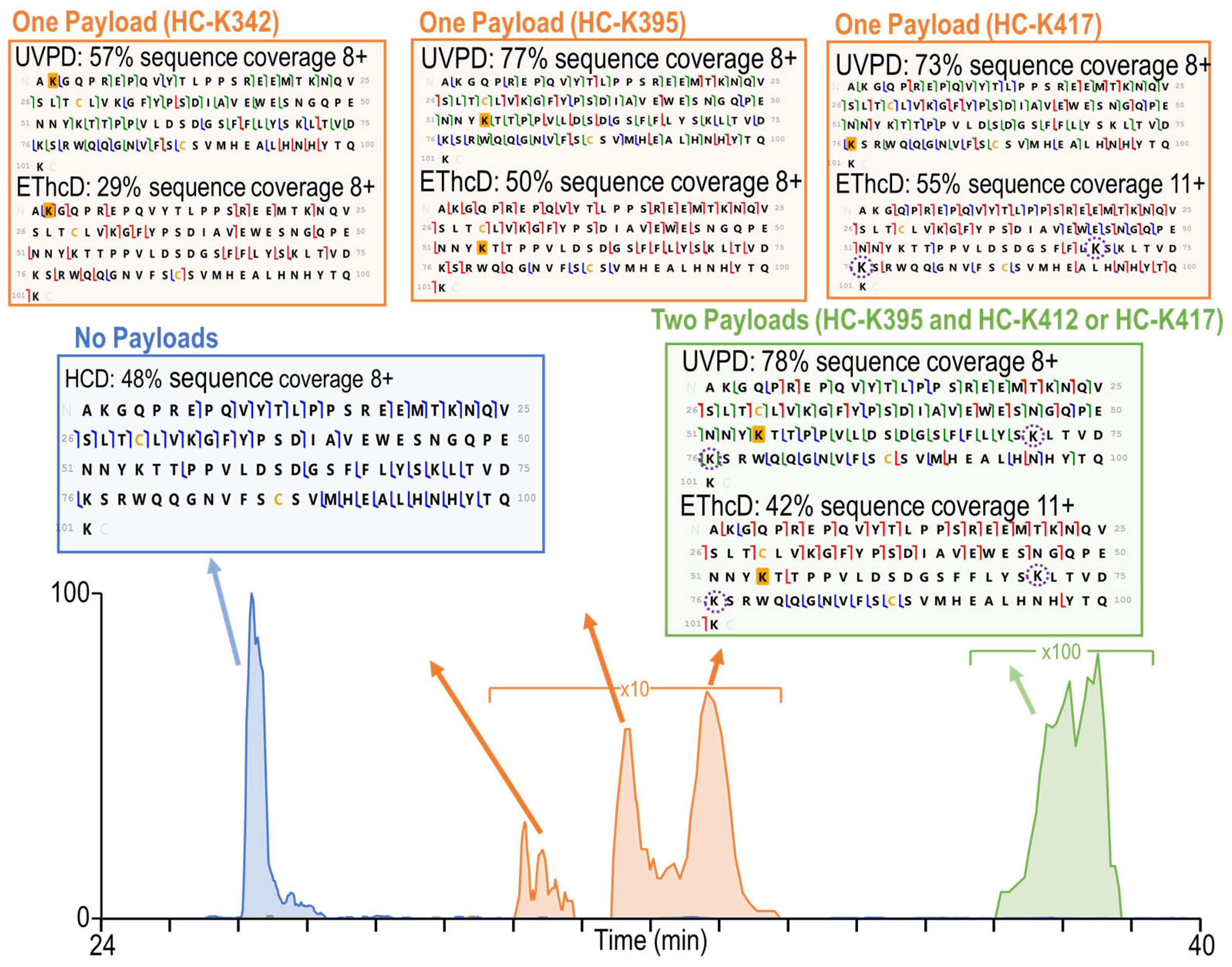
3.3. Complete Characterization of Payload Binding Sites
3.4. Comparison of a Second Batch of T-DM1
3.5. Comparison to Bottom-Up Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Sigal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Watts, E.; Williams, J.D.; Miesbauer, L.J.; Bruncko, M.; Brodbelt, J.S. Comprehensive Middle-Down Mass Spectrometry Characterization of an Antibody–Drug Conjugate by Combined Ion Activation Methods. Anal. Chem. 2020, 92, 9790–9798. [Google Scholar] [CrossRef]
- Chen, B.; Lin, Z.; Zhu, Y.; Jin, Y.; Larson, E.; Xu, Q.; Fu, C.; Zhang, Z.; Zhang, Q.; Pritts, W.A.; et al. Middle-Down Multi-Attribute Analysis of Antibody-Drug Conjugates with Electron Transfer Dissociation. Anal. Chem. 2019, 91, 11661–11669. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Friese, O.V.; Smith, J.N.; Brown, P.W.; Rouse, J.C. Practical Approaches for Overcoming Challenges in Heightened Characterization of Antibody-Drug Conjugates with New Methodologies and Ultrahigh-Resolution Mass Spectrometry. mAbs 2018, 10, 335–345. [Google Scholar] [CrossRef]
- Huang, R.Y.-C.; Chen, G. Characterization of Antibody–Drug Conjugates by Mass Spectrometry: Advances and Future Trends. Drug Discov. Today 2016, 21, 850–855. [Google Scholar] [CrossRef]
- Nagornov, K.O.; Gasilova, N.; Kozhinov, A.N.; Virta, P.; Holm, P.; Menin, L.; Nesatyy, V.J.; Tsybin, Y.O. Drug-to-Antibody Ratio Estimation via Proteoform Peak Integration in the Analysis of Antibody–Oligonucleotide Conjugates with Orbitrap Fourier Transform Mass Spectrometry. Anal. Chem. 2021, 93, 12930–12937. [Google Scholar] [CrossRef]
- Huang, Y.; Mou, S.; Wang, Y.; Mu, R.; Liang, M.; Rosenbaum, A.I. Characterization of Antibody–Drug Conjugate Pharmacokinetics and in Vivo Biotransformation Using Quantitative Intact LC-HRMS and Surrogate Analyte LC-MRM. Anal. Chem. 2021, 93, 6135–6144. [Google Scholar] [CrossRef]
- Jones, J.; Pack, L.; Hunter, J.H.; Valliere-Douglass, J.F. Native Size-Exclusion Chromatography-Mass Spectrometry: Suitability for Antibody–Drug Conjugate Drug-to-Antibody Ratio Quantitation across a Range of Chemotypes and Drug-Loading Levels. mAbs 2020, 12, 1682895. [Google Scholar] [CrossRef]
- He, J.; Su, D.; Ng, C.; Liu, L.; Yu, S.-F.; Pillow, T.H.; Del Rosario, G.; Darwish, M.; Lee, B.-C.; Ohri, R.; et al. High-Resolution Accurate-Mass Mass Spectrometry Enabling In-Depth Characterization of in Vivo Biotransformations for Intact Antibody-Drug Conjugates. Anal. Chem. 2017, 89, 5476–5483. [Google Scholar] [CrossRef]
- Sarrut, M.; Corgier, A.; Fekete, S.; Guillarme, D.; Lascoux, D.; Janin-Bussat, M.-C.; Beck, A.; Heinisch, S. Analysis of Antibody-Drug Conjugates by Comprehensive on-Line Two-Dimensional Hydrophobic Interaction Chromatography x Reversed Phase Liquid Chromatography Hyphenated to High Resolution Mass Spectrometry. I—Optimization of Separation Conditions. J. Chromatogr. B 2016, 1032, 103–111. [Google Scholar] [CrossRef]
- Pacholarz, K.J.; Barran, P.E. Use of a Charge Reducing Agent to Enable Intact Mass Analysis of Cysteine-Linked Antibody-Drug-Conjugates by Native Mass Spectrometry. EuPA Open Proteom. 2016, 11, 23–27. [Google Scholar] [CrossRef]
- Redman, E.A.; Mellors, J.S.; Starkey, J.A.; Ramsey, J.M. Characterization of Intact Antibody Drug Conjugate Variants Using Microfluidic Capillary Electrophoresis–Mass Spectrometry. Anal. Chem. 2016, 88, 2220–2226. [Google Scholar] [CrossRef]
- Birdsall, R.E.; Shion, H.; Kotch, F.W.; Xu, A.; Porter, T.J.; Chen, W. A Rapid On-Line Method for Mass Spectrometric Confirmation of a Cysteine-Conjugated Antibody-Drug-Conjugate Structure Using Multidimensional Chromatography. mAbs 2015, 7, 1036–1044. [Google Scholar] [CrossRef]
- Dyachenko, A.; Wang, G.; Belov, M.; Makarov, A.; de Jong, R.N.; van den Bremer, E.T.J.; Parren, P.W.H.I.; Heck, A.J.R. Tandem Native Mass-Spectrometry on Antibody–Drug Conjugates and Submillion Da Antibody–Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal. Chem. 2015, 87, 6095–6102. [Google Scholar] [CrossRef]
- Chen, T.-H.; Yang, Y.; Zhang, Z.; Fu, C.; Zhang, Q.; Williams, J.D.; Wirth, M.J. Native Reversed-Phase Liquid Chromatography: A Technique for LCMS of Intact Antibody–Drug Conjugates. Anal. Chem. 2019, 91, 2805–2812. [Google Scholar] [CrossRef]
- Barbero, L.M.; Ianni, A.D.; Molinaro, F.; Cowan, K.J.; Sirtori, F.R. Hybrid liquid chromatography high resolution and accuracy mass spectrometry approach for quantification of antibody-drug conjugates at the intact protein level in biological samples. Eur. J. Pharm. Sci. 2023, 188, 106502. [Google Scholar] [CrossRef]
- Larson, E.J.; Roberts, D.S.; Melby, J.A.; Buck, K.M.; Zhu, Y.; Zhou, S.; Han, L.; Zhang, Q.; Ge, Y. High-Throughput Multi-Attribute Analysis of Antibody-Drug Conjugates Enabled by Trapped Ion Mobility Spectrometry and Top-Down Mass Spectrometry. Anal. Chem. 2021, 93, 10013–10021. [Google Scholar] [CrossRef] [PubMed]
- Deslignière, E.; Ehkirch, A.; Duivelshof, B.L.; Toftevall, H.; Sjögren, J.; Guillarme, D.; D’Atri, V.; Beck, A.; Hernandez-Alba, O.; Cianférani, S. State-of-the-Art Native Mass Spectrometry and Ion Mobility Methods to Monitor Homogeneous Site-Specific Antibody-Drug Conjugates Synthesis. Pharmaceuticals 2021, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Botzanowski, T.; Erb, S.; Hernandez-Alba, O.; Ehkirch, A.; Colas, O.; Wagner-Rousset, E.; Rabuka, D.; Beck, A.; Drake, P.M.; Cianférani, S. Insights from Native Mass Spectrometry Approaches for Top- and Middle- Level Characterization of Site-Specific Antibody-Drug Conjugates. mAbs 2017, 9, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-C.; Deyanova, E.G.; Passmore, D.; Rangan, V.; Deshpande, S.; Tymiak, A.A.; Chen, G. Utility of Ion Mobility Mass Spectrometry for Drug-to-Antibody Ratio Measurements in Antibody-Drug Conjugates. J. Am. Soc. Mass Spectrom. 2015, 26, 1791–1794. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Mayhugh, B.M.; Srinivasan, J.M.; Sacha, G.A.; Nail, S.L.; Topp, E.M. Stability of Antibody Drug Conjugate Formulations Evaluated Using Solid-State Hydrogen-Deuterium Exchange Mass Spectrometry. J. Pharm. Sci. 2021, 110, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.Y.; Salas-Solano, O.; Valliere-Douglass, J.F. Antibody Structural Integrity of Site-Specific Antibody-Drug Conjugates Investigated by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2015, 87, 5669–5676. [Google Scholar] [CrossRef]
- Pan, L.Y.; Salas-Solano, O.; Valliere-Douglass, J.F. Conformation and Dynamics of Interchain Cysteine-Linked Antibody-Drug Conjugates as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2014, 86, 2657–2664. [Google Scholar] [CrossRef]
- Debaene, F.; Bœuf, A.; Wagner-Rousset, E.; Colas, O.; Ayoub, D.; Corvaïa, N.; Van Dorsselaer, A.; Beck, A.; Cianférani, S. Innovative Native MS Methodologies for Antibody Drug Conjugate Characterization: High Resolution Native MS and IM-MS for Average DAR and DAR Distribution Assessment. Anal. Chem. 2014, 86, 10674–10683. [Google Scholar] [CrossRef]
- Larson, E.J.; Zhu, Y.; Wu, Z.; Chen, B.; Zhang, Z.; Zhou, S.; Han, L.; Zhang, Q.; Ge, Y. Rapid Analysis of Reduced Antibody Drug Conjugate by Online LC-MS/MS with Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2020, 92, 15096–15103. [Google Scholar] [CrossRef]
- Hernandez-Alba, O.; Houel, S.; Hessmann, S.; Erb, S.; Rabuka, D.; Huguet, R.; Josephs, J.; Beck, A.; Drake, P.M.; Cianférani, S. A Case Study to Identify the Drug Conjugation Site of a Site-Specific Antibody-Drug-Conjugate Using Middle-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huo, S.; Xue, C.; An, B.; Qu, J. Current LC-MS-Based Strategies for Characterization and Quantification of Antibody-Drug Conjugates. J Pharm Anal. 2020, 10, 209–220. [Google Scholar] [CrossRef]
- Arlotta, K.J.; Gandhi, A.V.; Chen, H.-N.; Nervig, C.S.; Carpenter, J.F.; Owen, S.C. In-Depth Comparison of Lysine-Based Antibody-Drug Conjugates Prepared on Solid Support Versus in Solution. Antibodies 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, L.; Shion, H.; Yu, C.; Yu, Y.Q.; Zhu, L.; Li, M.; Chen, W.; Gao, K. In-Depth Structural Characterization of Kadcyla® (Ado-Trastuzumab Emtansine) and Its Biosimilar Candidate. mAbs 2016, 8, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.; Lu, G.; Liu, Y.; Hu, Q.; Xing, W.; Cui, D.; Zhou, F.; Zhang, J.; Hao, H.; Wang, G.; et al. Conjugation Site Analysis of Antibody-Drug-Conjugates (ADCs) by Signature Ion Fingerprinting and Normalized Area Quantitation Approach Using Nano-Liquid Chromatography Coupled to High Resolution Mass Spectrometry. Anal. Chim. Acta 2017, 955, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Gao, Y.; Liu, D.; Tan, X.; Hu, L.; Qiu, Z.; Liu, J.; He, H.; Liu, Y. Study on the Heterogeneity of T-DM1 and the Analysis of the Unconjugated Linker Structure under a Stable Conjugation Process. ACS Omega 2019, 4, 8834–8845. [Google Scholar] [CrossRef] [PubMed]
- Cotham, V.C.; Horton, A.P.; Lee, J.; Georgiou, G.; Brodbelt, J.S. Middle-Down 193-Nm Ultraviolet Photodissociation for Unambiguous Antibody Identification and Its Implications for Immunoproteomic Analysis. Anal. Chem. 2017, 89, 6498–6504. [Google Scholar] [CrossRef] [PubMed]
- Cotham, V.C.; Brodbelt, J.S. Characterization of Therapeutic Monoclonal Antibodies at the Subunit-Level Using Middle-Down 193 Nm Ultraviolet Photodissociation. Anal. Chem. 2016, 88, 4004–4013. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, L.; Ayoub, D.; Srzentic, K.; Nagornov, K.; Kozhinov, A.; Gasilova, N.; Menin, L.; Beck, A.; Tsybin, Y. Structural Analysis of Monoclonal Antibodies with Top-down and Middle-down Electron Transfer Dissociation Mass Spectrometry: The First Decade. CHIMIA 2022, 76, 114. [Google Scholar] [CrossRef]
- Fornelli, L.; Srzentić, K.; Huguet, R.; Mullen, C.; Sharma, S.; Zabrouskov, V.; Fellers, R.T.; Durbin, K.R.; Compton, P.D.; Kelleher, N.L. Accurate Sequence Analysis of a Monoclonal Antibody by Top-Down and Middle-Down Orbitrap Mass Spectrometry Applying Multiple Ion Activation Techniques. Anal. Chem. 2018, 90, 8421–8429. [Google Scholar] [CrossRef]
- Srzentić, K.; Nagornov, K.O.; Fornelli, L.; Lobas, A.A.; Ayoub, D.; Kozhinov, A.N.; Gasilova, N.; Menin, L.; Beck, A.; Gorshkov, M.V.; et al. Multiplexed Middle-Down Mass Spectrometry as a Method for Revealing Light and Heavy Chain Connectivity in a Monoclonal Antibody. Anal. Chem. 2018, 90, 12527–12535. [Google Scholar] [CrossRef]
- Greer, S.M.; Sidoli, S.; Coradin, M.; Schack Jespersen, M.; Schwämmle, V.; Jensen, O.N.; Garcia, B.A.; Brodbelt, J.S. Extensive Characterization of Heavily Modified Histone Tails by 193 Nm Ultraviolet Photodissociation Mass Spectrometry via a Middle–Down Strategy. Anal. Chem. 2018, 90, 10425–10433. [Google Scholar] [CrossRef]
- Sanders, J.D.; Greer, S.M.; Brodbelt, J.S. Integrating Carbamylation and Ultraviolet Photodissociation Mass Spectrometry for Middle-Down Proteomics. Anal. Chem. 2017, 89, 11772–11778. [Google Scholar] [CrossRef] [PubMed]
- Pandeswari, P.B.; Sabareesh, V. Middle-down Approach: A Choice to Sequence and Characterize Proteins/Proteomes by Mass Spectrometry. RSC Adv. 2018, 9, 313–344. [Google Scholar] [CrossRef]
- Riley, N.M.; Coon, J.J. The Role of Electron Transfer Dissociation in Modern Proteomics. Anal. Chem. 2018, 90, 40–64. [Google Scholar] [CrossRef] [PubMed]
- Brodbelt, J.S.; Morrison, L.J.; Santos, I. Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev. 2020, 120, 3328–3380. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.R.; Holden, D.D.; Brodbelt, J.S. Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2016, 88, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Penkert, M.; Yates, L.M.; Schümann, M.; Perlman, D.; Fiedler, D.; Krause, E. Unambiguous Identification of Serine and Threonine Pyrophosphorylation Using Neutral-Loss-Triggered Electron-Transfer/Higher-Energy Collision Dissociation. Anal. Chem. 2017, 89, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Escobar, E.E.; King, D.T.; Serrano-Negrón, J.E.; Alteen, M.G.; Vocadlo, D.J.; Brodbelt, J.S. Precision Mapping of O-Linked N-Acetylglucosamine Sites in Proteins Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc. 2020, 142, 11569–11577. [Google Scholar] [CrossRef] [PubMed]
- Sandra, K.; Vanhoenacker, G.; Vandenheede, I.; Steenbeke, M.; Joseph, M.; Sandra, P. Multiple Heart-Cutting and Comprehensive Two-Dimensional Liquid Chromatography Hyphenated to Mass Spectrometry for the Characterization of the Antibody-Drug Conjugate Ado-Trastuzumab Emtansine. J. Chromatogr. B 2016, 1032, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Martelet, A.; Garrigue, V.; Zhang, Z.; Genet, B.; Guttman, A. Multi-Attribute Method Based Characterization of Antibody Drug Conjugates (ADC) at the Intact and Subunit Levels. J. Pharm. Biomed. Anal. 2021, 201, 114094. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watts, E.; Bashyal, A.; Dunham, S.D.; Crittenden, C.M.; Brodbelt, J.S. Enhanced Characterization of Lysine-Linked Antibody Drug Conjugates Enabled by Middle-Down Mass Spectrometry and Higher-Energy Collisional Dissociation-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation and Ultraviolet Photodissociation. Antibodies 2024, 13, 30. https://doi.org/10.3390/antib13020030
Watts E, Bashyal A, Dunham SD, Crittenden CM, Brodbelt JS. Enhanced Characterization of Lysine-Linked Antibody Drug Conjugates Enabled by Middle-Down Mass Spectrometry and Higher-Energy Collisional Dissociation-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation and Ultraviolet Photodissociation. Antibodies. 2024; 13(2):30. https://doi.org/10.3390/antib13020030
Chicago/Turabian StyleWatts, Eleanor, Aarti Bashyal, Sean D. Dunham, Christopher M. Crittenden, and Jennifer S. Brodbelt. 2024. "Enhanced Characterization of Lysine-Linked Antibody Drug Conjugates Enabled by Middle-Down Mass Spectrometry and Higher-Energy Collisional Dissociation-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation and Ultraviolet Photodissociation" Antibodies 13, no. 2: 30. https://doi.org/10.3390/antib13020030
APA StyleWatts, E., Bashyal, A., Dunham, S. D., Crittenden, C. M., & Brodbelt, J. S. (2024). Enhanced Characterization of Lysine-Linked Antibody Drug Conjugates Enabled by Middle-Down Mass Spectrometry and Higher-Energy Collisional Dissociation-Triggered Electron-Transfer/Higher-Energy Collisional Dissociation and Ultraviolet Photodissociation. Antibodies, 13(2), 30. https://doi.org/10.3390/antib13020030