
Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward?


{kind=link}
{kind=link}
Abstract
:1. Introduction
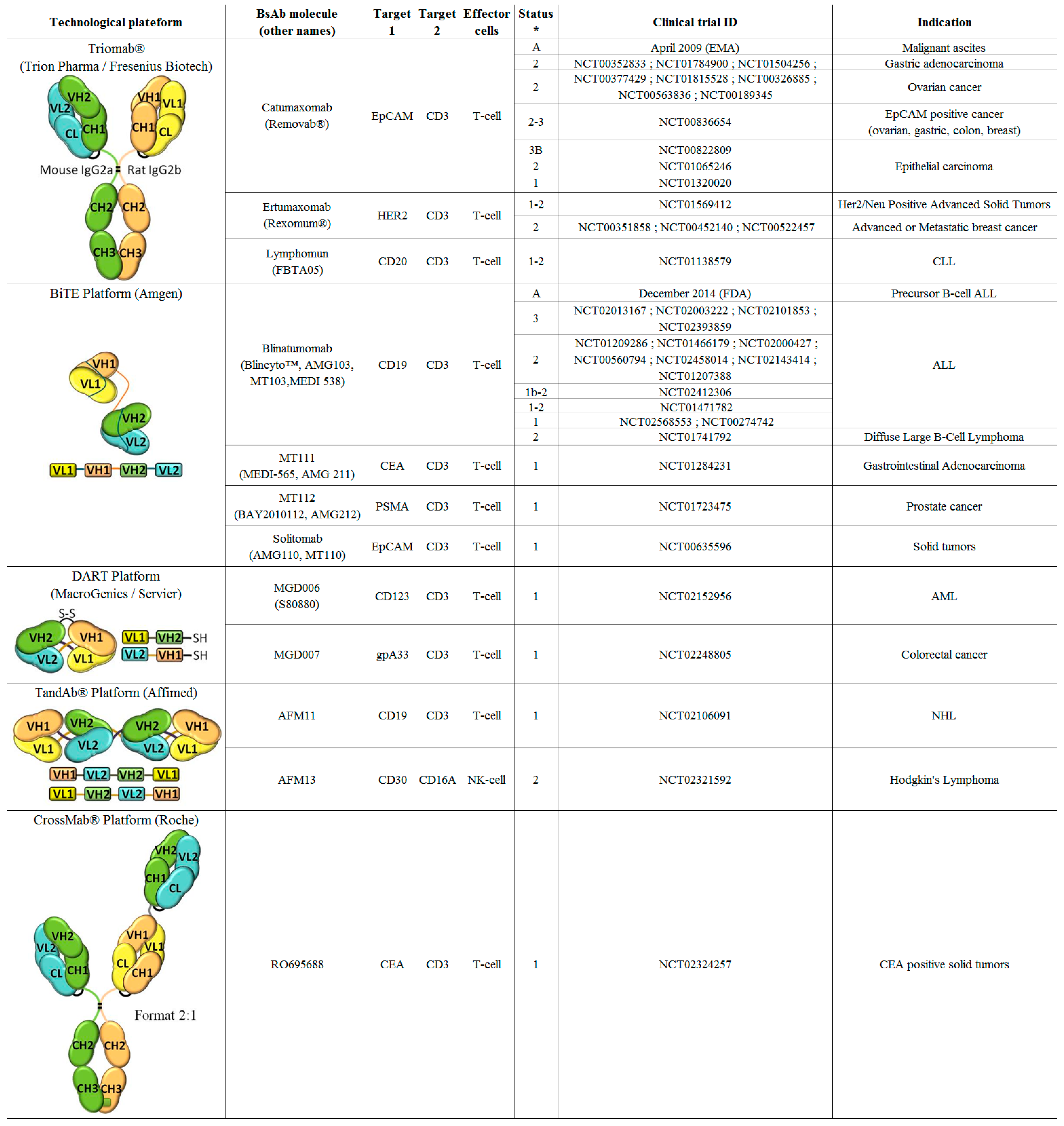
2. Bispecific Antibody Technological Platforms
2.1. Trifunctional Hybrid Antibodies Platform—Triomab® Format (Trion Pharma (Fresenius Biotech, Planegg, Germany))

2.2. Bispecific T-Cell Engager (BiTE) Platform (Amgen, Thousand Oaks, CA, US)
2.3. Dual-Affinity Re-Targeting (DART) Platform MacroGenics (Rockville, MD, US)
2.4. TandAb® Platform (Affimed, Heidelberg, Germany)
2.5. CrossMAb (Roche, Basel, Switzerland)
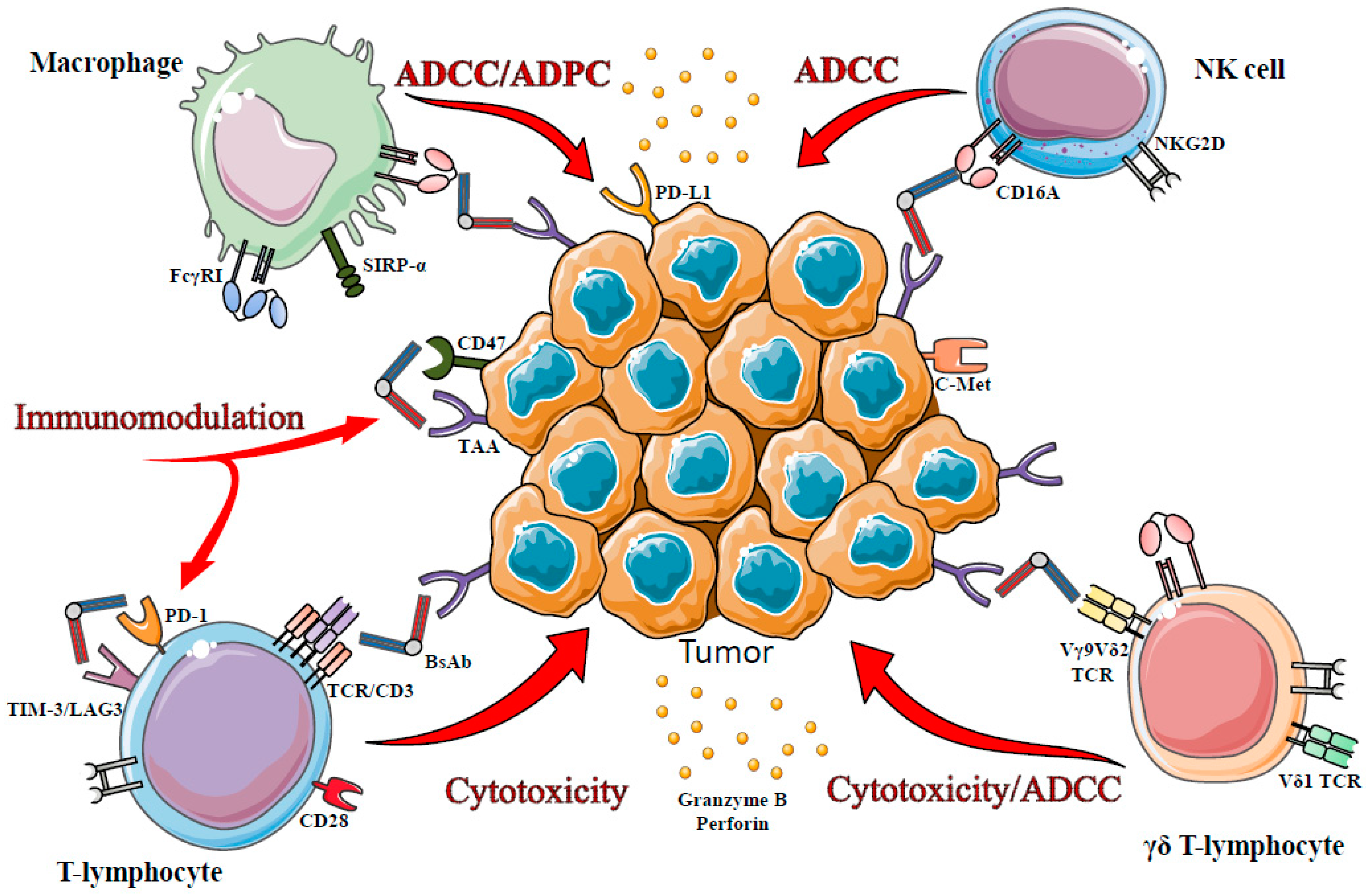
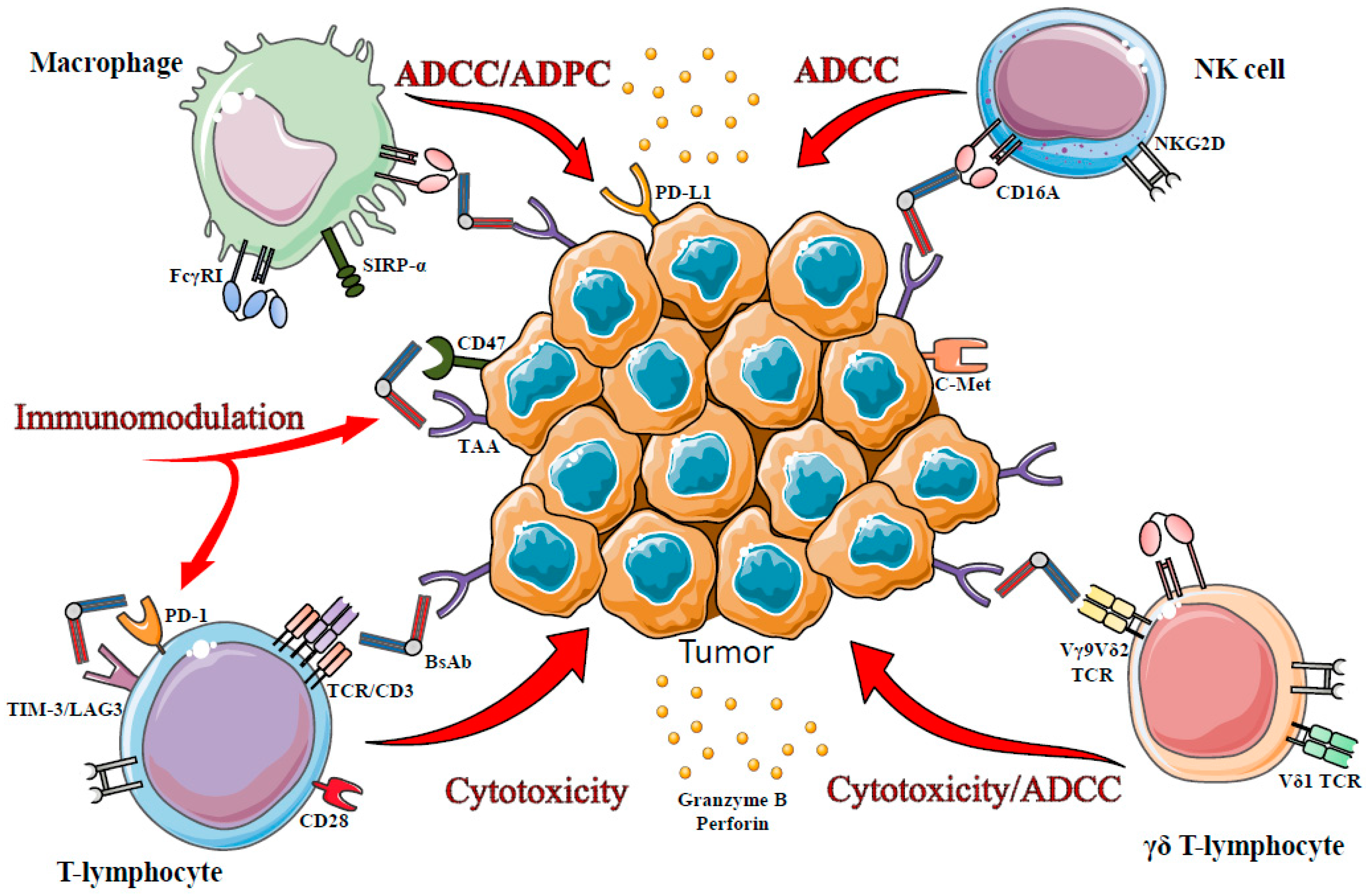
3. T-Cells in bsAb-Mediated Immunotherapy
3.1. CD3-TCR Targeting
3.1.1. From Bench to Bed Side: Two Bispecific Antibodies Approved for Human Use
Catumaxomab/Removab®
Blinatumomab/Blincyto™
3.1.2. In the Pipeline
3.2. Other Ways to Recruit Effector T Cells
3.2.1. γδ T-Lymphocyte, Potential Targets?
3.2.2. Targeting Inhibitory Receptors?
4. Innate Immunity in bsAb-Mediated Immunotherapy
4.1. NK Cells
4.1.1. From Bench to Clinic: Only One Bispecific Antibody
4.1.2. In the Pipeline
4.2. Macrophages
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coley, W.B. Contribution to the knowledge of sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Dr william coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [PubMed]
- Burnet, M. Cancer a biological approach. I. The processes of control. II. The Significance of Somatic Mutation. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Discussion. In Cellular and Humoral Aspects of the Hypersensitive States; Lawrence, H.S., Ed.; Hoeber-Harper: New York, NY, USA, 1959; pp. 529–533. [Google Scholar]
- Stutman, O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science 1974, 183, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Stutman, O. Chemical carcinogenesis in nude mice: Comparison between nude mice from homozygous matings and heterozygous matings and effect of age and carcinogen dose. J. Natl. Cancer Inst. 1979, 62, 353–358. [Google Scholar] [PubMed]
- Smyth, M.J.; Godfrey, D.I.; Trapani, J.A. A fresh look at tumor immunosurveillance and immunotherapy. Nat. Immunol. 2001, 2, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Dighe, A.S.; Richards, E.; Old, L.J.; Schreiber, R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFNγ receptors. Immunity 1994, 1, 447–456. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.H.; Elder, D.E.; Guerry, D.; Braitman, L.E.; Trock, B.J.; Schultz, D.; Synnestvedt, M.; Halpern, A.C. Model predicting survival in stage I melanoma based on tumor progression. J. Natl. Cancer Inst. 1989, 81, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.G.; Mihm, M.C.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Sandel, M.H.; Speetjens, F.M.; Menon, A.G.; Albertsson, P.A.; Basse, P.H.; Hokland, M.; Nagelkerke, J.F.; Tollenaar, R.A.E.M.; van de Velde, C.J.H.; Kuppen, P.J.K. Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol. Immunol. 2005, 42, 541–546. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Goc, J.; Germain, C.; Giraldo, N.A.; Dieu-Nosjean, M.C.; Sautes-Fridman, C.; Fridman, W.H. Shaping of an effective immune microenvironment to and by cancer cells. Cancer Immunol. Immunother. 2014, 63, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pages, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Broussard, E.K.; Disis, M.L. TNM staging in colorectal cancer: T is for T cell and M is for memory. J. Clin. Oncol. 2011, 29, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Angell, H.; Galon, J. From the immune contexture to the immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Kreamer, K.M. Immune checkpoint blockade: A new paradigm in treating advanced cancer. J. Adv. Pract. Oncol. 2014, 5, 418–431. [Google Scholar] [PubMed]
- Antonia, S.; Mule, J.J.; Weber, J.S. Current developments of immunotherapy in the clinic. Curr. Opin. Immunol. 2004, 16, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Yee, C. Adoptive T-cell therapy for cancer: Boutique therapy or treatment modality? Clin. Cancer Res. 2013, 19, 4550–4552. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Savoldo, B.; Brenner, M. Fifteen years of gene therapy based on chimeric antigen receptors: “Are we nearly there yet?”. Hum. Gene Ther. 2009, 20, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Kloss, C.C.; Gunset, G.; Sadelain, M. CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS ONE 2013, 8, e61338. [Google Scholar] [CrossRef] [PubMed]
- Reff, M.E.; Carner, K.; Chambers, K.S.; Chinn, P.C.; Leonard, J.E.; Raab, R.; Newman, R.A.; Hanna, N.; Anderson, D.R. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood 1994, 83, 435–445. [Google Scholar] [PubMed]
- Tokuda, Y. Antibodies as molecular target-based therapy: Trastuzumab. Int. J. Clin. Oncol. 2003, 8, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the brake on the immune system: Ipilimumab in melanoma and other tumors. Cancer Biother. Radiopharm. 2010, 25, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Grillo-Lopez, A.J.; White, C.A.; Dallaire, B.K.; Varns, C.L.; Shen, C.D.; Wei, A.; Leonard, J.E.; McClure, A.; Weaver, R.; Cairelli, S.; et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- The Antibody Society. Available online: https://www.Antibodysociety.Org/news/approved_mabs.Pdf (accessed on 4 February 2015).
- Oflazoglu, E.; Audoly, L.P. Evolution of anti-CD20 monoclonal antibody therapeutics in oncology. mAbs 2010, 2, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.; Stewart, D.A. Obinutuzumab for the treatment of lymphoproliferative disorders. Expert Opin. Biol. Ther. 2012, 12, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Riethmuller, G. Symmetry breaking: Bispecific antibodies, the beginnings, and 50 years on. Cancer Immun. 2012, 12, 12–18. [Google Scholar] [PubMed]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy: The light at the end of the tunnel? mAbs 2009, 1, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Prado, N.; Compte, M.; Harwood, S.; Alvarez-Mendez, A.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. The coming of age of engineered multivalent antibodies. Drug Discov. Today 2015, 20, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Carter, P. Bispecific human IgG by design. J. Immunol. Methods 2001, 248, 7–15. [Google Scholar] [CrossRef]
- Marvin, J.S.; Zhu, Z. Bispecific antibodies for dual-modality cancer therapy: Killing two signaling cascades with one stone. Curr. Opin. Drug Discov. Dev. 2006, 9, 184–193. [Google Scholar]
- Weidle, U.H.; Kontermann, R.E.; Brinkmann, U. Tumor-antigen-binding bispecific antibodies for cancer treatment. Semin. Oncol. 2014, 41, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Karpovsky, B.; Titus, J.A.; Stephany, D.A.; Segal, D.M. Production of target-specific effector cells using hetero-cross-linked aggregates containing anti-target cell and anti-Fc gamma receptor antibodies. J. Exp. Med. 1984, 160, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. mAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Chatal, J.F.; Barbet, J.; Boerman, O.; Sharkey, R.M. Cancer imaging and therapy with bispecific antibody pretargeting. Update Cancer Ther. 2007, 2, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar] [PubMed]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy light-chain pairing in rat mouse quadromas-implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar] [PubMed]
- Zeidler, R.; Reisbach, G.; Wollenberg, B.; Lang, S.; Chaubal, S.; Schmitt, B.; Lindhofer, H. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J. Immunol. 1999, 163, 1246–1252. [Google Scholar] [PubMed]
- Zeidler, R.; Mysliwietz, J.; Csanady, M.; Walz, A.; Ziegler, I.; Schmitt, B.; Wollenberg, B.; Lindhofer, H. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br. J. Cancer 2000, 83, 261–266. [Google Scholar] [PubMed]
- Mack, M.; Riethmuller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.L.; Zhang, W.J.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; von der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bahner, M.; Schanzer, J.; Croasdale, R.; Durr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- 51st Asco Annual Meeting, Chicago. Available online: http://www.roche.com/irp150601.pdf (accessed on 31 May 2015).
- Friedl, P.; den Boer, A.T.; Gunzer, M. Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 2005, 5, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.V. T cell motility as modulator of interactions with dendritic cells. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L.; Meuer, S.; Fitzgerald, K.A.; Hussey, R.E.; Levine, H.; Schlossman, S.F. Antigen recognition by human T lymphocytes is linked to surface expression of the T3 molecular complex. Cell 1982, 30, 735–743. [Google Scholar] [CrossRef]
- Nicolls, M.R.; Aversa, G.G.; Pearce, N.W.; Spinelli, A.; Berger, M.F.; Gurley, K.E.; Hall, B.M. Induction of long-term specific tolerance to allografts in rats by therapy with an anti-CD3-like monoclonal antibody. Transplantation 1993, 55, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Cilag. Orthoclone®OKT3 muromonab-CD3—Worldwide Discontinuation. 2010. Available online: www.cochrane-renal.org/docs/Newsletter_October_2010.pdf (accessed on 4 January 2015).
- Manzke, O.; Titzer, S.; Tesch, H.; Diehl, V.; Bohlen, H. CD3 × CD19 bispecific antibodies and CD28 costimulation for locoregional treatment of low-malignancy non-Hodgkin’s lymphoma. Cancer Immunol. Immunother. 1997, 45, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on mhc class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM × anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Passebosc-Faure, K.; Li, G.; Lambert, C.; Cottier, M.; Gentil-Perret, A.; Fournel, P.; Perol, M.; Genin, C. Evaluation of a panel of molecular markers for the diagnosis of malignant serous effusions. Clin. Cancer Res. 2005, 11, 6862–6867. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Buley, I.D.; Heryet, A.; Gray, W. Immunocytochemical staining of serous effusions with the monoclonal antibody Ber-EP4. Cytopathology 1992, 3, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arias, A.A.; Loy, T.S.; Bickel, J.T.; Chapman, R.K. Utility of Ber-EP4 in the diagnosis of adenocarcinoma in effusions: An immunocytochemical study of 232 cases. Diagn. Cytopathol. 1993, 9, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Van der Gun, B.T.F.; Melchers, L.J.; Ruiters, M.H.J.; de Leij, L.F.M.H.; McLaughlin, P.M.J.; Rots, M.G. EpCAM in carcinogenesis: The good, the bad or the ugly. Carcinogenesis 2010, 31, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Schoberth, A.; Ruf, P.; Hess, J.; Hennig, M.; Schmalfeldt, B.; Wimberger, P.; Strohlein, M.; Theissen, B.; Heiss, M.M.; et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM × anti-CD3). Cancer Res. 2012, 72, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Wimberger, P.; Kumper, C.; Gorbounova, V.; Sommer, H.; Schmalfeldt, B.; Pfisterer, J.; Lichinitser, M.; Makhson, A.; Moiseyenko, V.; et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM × anti-CD3 antibody: A phase I/II study. Clin. Cancer Res. 2007, 13, 3899–3905. [Google Scholar] [CrossRef] [PubMed]
- Marme, A.; Strauss, G.; Bastert, G.; Grischke, E.M.; Moldenhauer, G. Intraperitoneal bispecific antibody (HEA125 × OKT3) therapy inhibits malignant ascites production in advanced ovarian carcinoma. Int. J. Cancer 2002, 101, 183–189. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, G.L.; Mirick, G.R.; Kroger, L.A.; Bradt, B.M.; Lamborn, K.R.; de Nardo, S.J. Characterization of human IgG antimouse antibody in patients with B-cell malignancies. Clin. Cancer Res. 2003, 9, 4013S–4021S. [Google Scholar]
- Miotti, S.; Negri, D.R.; Valota, O.; Calabrese, M.; Bolhuis, R.L.; Gratama, J.W.; Colnaghi, M.I.; Canevari, S. Level of anti-mouse-antibody response induced by bi-specific monoclonal antibody OC/TR in ovarian-carcinoma patients is associated with longer survival. Int. J. Cancer 1999, 84, 62–68. [Google Scholar] [CrossRef]
- Linke, R.; Klein, A.; Seimetz, D. Catumaxomab: Clinical development and future directions. mAbs 2010, 2, 129–136. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.; Thomas, D.; Ravandi, F.; Faderl, S.; Cortes, J.; Borthakur, G.; Pierce, S.; Garcia-Manero, G.; Kantarjian, H.M. Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer 2008, 113, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, D.; Ravandi, F.; Faderl, S.; Jabbour, E.; Garcia-Manero, G.; Pierce, S.; Shan, J.; Cortes, J.; O’Brien, S. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer 2010, 116, 5568–5574. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [PubMed]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gokbuget, N.; Zugmaier, G.; Klappers, P.; Stelljes, M.; Neumann, S.; Viardot, A.; Marks, R.; Diedrich, H.; Faul, C.; et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, Z.; Maniar, T.; Nagorsen, D. Unleashing the clinical power of T cells: CD19/CD3 Bi-specific T cell engager (BiTE®) antibody construct blinatumomab as a potential therapy. Int. Immunol. 2015, 27, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Frankel, S.R.; Baeuerle, P.A. Targeting T cells to tumor cells using bispecific antibodies. Curr. Opin. Chem. Biol. 2013, 17, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; Gall, F.L.; et al. A tetravalent bispecific TandAB (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. mAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, B.; Quadt, C.; Dreier, T.; Kufer, P.; Lorenczewski, G.; Prang, N.; Brandl, C.; Lippold, S.; Cobb, K.; Brasky, K.; et al. T-cell activation and B-cell depletion in chimpanzees treated with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Cancer Immunol. Immunother. 2006, 55, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Macrogenics. Available online: http://www.Macrogenics.Com/products-mgd011.html (accessed on 18 December 2015).
- Moore, P.A.; Zhang, W.J.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.H.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Affimed. Available online: http://www.Affimed.Com/products.php (accessed on 18 December 2015).
- Bhutani, D.; Lum, L.G. Activated T cells armed with bispecific antibodies kill tumor targets. Curr. Opin. Hematol. 2015, 22, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, P.; Zhou, C.; Jing, L.; Dong, B.; Chen, S.; Zhang, N.; Liu, Y.; Miao, J.; Wang, Z.; et al. A novel bispecific antibody, S-FAB, induces potent cancer cell killing. J. Immunother. 2015, 38, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.L.; Rossi, E.A.; Cardillo, T.M.; Goldenberg, D.M.; Chang, C.H. A new class of bispecific antibodies to redirect t cells for cancer immunotherapy. mAbs 2014, 6, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Olson, K.; Haber, L.J.; Varghese, B.; Duramad, P.; Tustian, A.D.; Oyejide, A.; Kirshner, J.R.; Canova, L.; Menon, J.; et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Ellerman, D.; Mathieu, M.; Hristopoulos, M.; Chen, X.; Li, Y.; Yan, X.; Clark, R.; Reyes, A.; Stefanich, E.; et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Hayday, A.C. γβ cells: A right time and a right place for a conserved third way of protection. Ann. Rev. Immunol. 2000, 18, 975–1026. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, S.; el Daker, S.; Dieli, F.; Martini, F.; Martino, A. γδ T cells cross-link innate and adaptive immunity in Mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Nuotio-Antar, A.M.; Smith, C.W. Gammadelta t cells promote inflammation and insulin resistance during high fat diet-induced obesity in mice. J. Leuk. Biol. 2015, 97, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gamma delta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Morita, C.T.; Tanaka, Y.; Nieves, E.; Brenner, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Ding, Y.P.; Tanaka, Y.; Shen, L.W.; Wei, C.H.; Minato, N.; Zhang, W. Gammadelta T cells and their potential for immunotherapy. Int. J. Biol. Sci. 2014, 10, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Braza, M.S.; Klein, B.; Fiol, G.; Rossi, J.F. γδ T-cell killing of primary follicular lymphoma cells is dramatically potentiated by GA101, a type II glycoengineered anti-CD20 monoclonal antibody. Haematol. Hematol. J. 2011, 96, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Gertner-Dardenne, J.; Bonnafous, C.; Bezombes, C.; Capietto, A.H.; Scaglione, V.; Ingoure, S.; Cendron, D.; Gross, E.; Lepage, J.F.; Quillet-Mary, A.; et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood 2009, 113, 4875–4884. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, H.; Hagi, T.; Mattarollo, S.R.; Morley, J.; Wang, Q.; Fai-So, H.; Moriyasu, F.; Nieda, M.; Nicol, A.J. Vγ9Vδ2 T cell cytotoxicity against tumor cells is enhanced by monoclonal antibody drugs—Rituximab and trastuzumab. Int. J. Cancer 2008, 122, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Peipp, M.; Peters, C.; Sebens, S.; Kabelitz, D.; Wesch, D. Gammadelta T cell activation by bispecific antibodies. Cell Immunol. 2015, 296, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.H.; Peipp, M.; Kellner, C.; Sebens, S.; Krause, S.; Petrick, D.; Adam-Klages, S.; Rocken, C.; Becker, T.; Vogel, I.; et al. Novel bispecific antibodies increase gammadelta T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 2014, 74, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Page, D.B.; Postow, M.A.; Callahan, M.K.; Allison, J.P.; Wolchok, J.D. Immune modulation in cancer with antibodies. Ann. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Anaptysbio. Available online: http://www.Anaptysbio.Com/platform/shm-xel-platform/ (accessed on 18 December 2015).
- Sorrento Therapeutics. Available online: http://sorrentotherapeutics.com/wp-content/uploads/2014/12/SABC-2014-Poster-FINAL____Zhang-et-al.pdf (accessed on 18 December 2015).
- Sorrento Therapeutics. Available online: http://www.prnewswire.com/news-releases/sorrento-therapeutics-announces-poster-presentations-on-its-anti-c-metpd-l1-bispecific-antibodies-and-anti-c-met-antibody-drug-conjugate-at-the-37th-annual-san-antonio-breast-cancer-symposium-300005983.html (accessed on 18 December 2015).
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Hersey, P. Natural killer cells—A new cytotoxic mechanism against tumours? Aust. N. Z. J. Med. 1979, 9, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Feng, H.L.; Zhang, S.H.; Liang, J.; Squiban, P.; Farag, S. Enhancing natural killer (NK) cell mediated killing of non-hodgkin’s lymphoma. Blood 2009, 114, 1059–1060. [Google Scholar]
- Romagne, F.; Andre, P.; Spee, P.; Zahn, S.; Anfossi, N.; Gauthier, L.; Capanni, M.; Ruggeri, L.; Benson, D.M.; Blaser, B.W.; et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009, 114, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Gras Navarro, A.; Bjorklund, A.T.; Chekenya, M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front. Immunol. 2015, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.; Canter, R.J.; Grossenbacher, S.K.; Mac, S.; Chen, M.; Smith, R.C.; Hagino, T.; Perez-Cunningham, J.; Sckisel, G.D.; Urayama, S.; et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. J. Immunol. 2015, 195, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Johnston, J.V.; Shufford, W.W.; Mittler, R.S.; Chen, L.P. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell. Immunol. 1998, 190, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S. Therapeutic applications: Natural killer cells in the clinic. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immunother. 2010, 59, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Eguizabal, C.; Zenarruzabeitia, O.; Monge, J.; Santos, S.; Vesga, M.A.; Maruri, N.; Arrieta, A.; Rinon, M.; Tamayo-Orbegozo, E.; Amo, L.; et al. Natural killer cells for cancer immunotherapy: Pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front. Immunol. 2014, 5, 439. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16 × CD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Zhang, B.; Gleason, M.K.; Oh, S.; Weiner, L.M.; Kaufman, D.S.; McCullar, V.; Miller, J.S.; Verneris, M.R. Heterodimeric bispecific single-chain variable-fragment antibodies against epcam and CD16 induce effective antibody-dependent cellular cytotoxicity against human carcinoma cells. Cancer Biother. Radiopharm. 2013, 28, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Schubert, I.; Kellner, C.; Stein, C.; Kugler, M.; Schwenkert, M.; Saul, D.; Mentz, K.; Singer, H.; Stockmeyer, B.; Hillen, W.; et al. A single-chain triplebody with specificity for CD19 and CD33 mediates effective lysis of mixed lineage leukemia cells by dual targeting. mAbs 2011, 3, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Shi, M.; Feng, J.; Yu, M.; Sun, Y.; Shen, B.; Guo, N. A trivalent anti-erbB2/anti-CD16 bispecific antibody retargeting NK cells against human breast cancer cells. Biochem. Biophys. Res. Commun. 2003, 311, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, R.; Willems, A.; Schoonooghe, S.; Fiers, W.; Grooten, J.; Mertens, N. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J. Immunol. 2000, 165, 7050–7057. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Shi, M.; Wang, M.; Xie, Z.; Hu, M.; Yu, M.; Shen, B.; Ma, Y.; Guo, N. In vitro and in vivo antitumor effect of a trivalent bispecific antibody targeting ERBB2 and CD16. Cancer Biol. Ther. 2008, 7, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Turini, M.; Chames, P.; Bruhns, P.; Baty, D.; Kerfelec, B. A FcγRIII-engaging bispecific antibody expands the range of HER2-expressing breast tumors eligible to antibody therapy. Oncotarget 2014, 5, 5304–5319. [Google Scholar] [CrossRef] [PubMed]
- Rozan, C.; Cornillon, A.; Petiard, C.; Chartier, M.; Behar, G.; Boix, C.; Kerfelec, B.; Robert, B.; Pelegrin, A.; Chames, P.; et al. Single-domain antibody-based and linker-free bispecific antibodies targeting FcγRIII induce potent antitumor activity without recruiting regulatory T cells. Mol. Cancer Ther. 2013, 12, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Nguyen, V.K.; Nei, M. Adaptive evolution of variable region genes encoding an unusual type of immunoglobulin in camelids. Mol. Biol. Evol. 2002, 19, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, M.; Conrath, K.; Van Meirhaeghe, A.; Meersman, F.; Heremans, K.; Frenken, L.G.; Muyldermans, S.; Wyns, L.; Matagne, A. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002, 11, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Vincke, C.; Loris, R.; Saerens, D.; Martinez-Rodriguez, S.; Muyldermans, S.; Conrath, K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 2009, 284, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Els Conrath, K.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Gangi, L.; Paul, S.; Schioppa, T.; Saccani, A.; Sironi, M.; Bottazzi, B.; Doni, A.; Vincenzo, B.; Pasqualini, F.; et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 2006, 107, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- Michael, R.; Mallmann, S.V.S.; Joachim, L. Macrophages in human cancer: Current and future aspects. In Atlas of Genetics and Cytogenetics in Oncology and Haematology; INIST-CNRS: Paris, France, 2012. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Utrera-Barillas, D.; Castro-Manrreza, M.; Castellanos, E.; Gutierrez-Rodriguez, M.; Arciniega-Ruiz de Esparza, O.; Garcia-Cebada, J.; Velazquez, J.R.; Flores-Resendiz, D.; Hernandez-Hernandez, D.; Benitez-Bribiesca, L. The role of macrophages and mast cells in lymphangiogenesis and angiogenesis in cervical carcinogenesis. Exp. Mol. Pathol. 2010, 89, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Niino, D.; Komohara, Y.; Murayama, T.; Aoki, R.; Kimura, Y.; Hashikawa, K.; Kiyasu, J.; Takeuchi, M.; Suefuji, N.; Sugita, Y.; et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for angioimmunoblastic T-cell lymphoma (AITL). Pathol. Int. 2010, 60, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Herlyn, D.; Koprowski, H. IgG2a monoclonal antibodies inhibit human tumor growth through interaction with effector cells. Proc. Natl. Acad. Sci. USA 1982, 79, 4761–4765. [Google Scholar] [CrossRef] [PubMed]
- Steplewski, Z.; Lubeck, M.D.; Koprowski, H. Human macrophages armed with murine immunoglobulin G2a antibodies to tumors destroy human cancer cells. Science 1983, 221, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.O.; Hall, T.; Steplewski, Z.; Koprowski, H. Tumors undergoing rejection induced by monoclonal antibodies of the IgG2a isotype contain increased numbers of macrophages activated for a distinctive form of antibody-dependent cytolysis. Proc. Natl. Acad. Sci. USA 1984, 81, 3506–3510. [Google Scholar] [CrossRef] [PubMed]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [PubMed]
- Valone, F.H.; Kaufman, P.A.; Guyre, P.M.; Lewis, L.D.; Memoli, V.; Deo, Y.; Graziano, R.; Fisher, J.L.; Meyer, L.; Mrozek-Orlowski, M.; et al. Phase Ia/Ib trial of bispecific antibody MDX-210 in patients with advanced breast or ovarian cancer that overexpresses the proto-oncogene HER-2/neu. J. Clin. Oncol. 1995, 13, 2281–2292. [Google Scholar] [PubMed]
- Watanabe, M.; Wallace, P.K.; Keler, T.; Deo, Y.M.; Akewanlop, C.; Hayes, D.F. Antibody dependent cellular phagocytosis (ADCP) and antibody dependent cellular cytotoxicity (ADCC) of breast cancer cells mediated by bispecific antibody, MDX-210. Breast Cancer Res. Treat. 1999, 53, 199–207. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Atherton, P.J.; Jones, J.; Howie, A.J.; Tchekmedyian, S.; Curnow, R.T. A phase II study of the bispecific antibody MDX-h210 (anti-HER2 × CD64) with GM-CSF in Her2+ advanced prostate cancer. Br. J. Cancer 2001, 85, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Lipton, A.; Smith, K.M.; Winston, C.B.; Pfister, D.G. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol. Immunother. 2008, 57, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Piccione, E.C.; Juarez, S.; Liu, J.; Tseng, S.; Ryan, C.E.; Narayanan, C.; Wang, L.; Weiskopf, K.; Majeti, R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. mAbs 2015, 7, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Bano, J.; Chames, P.; Baty, D.; Kerfelec, B. Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies 2016, 5, 1. https://doi.org/10.3390/antib5010001
Del Bano J, Chames P, Baty D, Kerfelec B. Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies. 2016; 5(1):1. https://doi.org/10.3390/antib5010001
Chicago/Turabian StyleDel Bano, Joanie, Patrick Chames, Daniel Baty, and Brigitte Kerfelec. 2016. "Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward?" Antibodies 5, no. 1: 1. https://doi.org/10.3390/antib5010001
APA StyleDel Bano, J., Chames, P., Baty, D., & Kerfelec, B. (2016). Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies, 5(1), 1. https://doi.org/10.3390/antib5010001