Complement and Cancer—A Dysfunctional Relationship?


{kind=link}
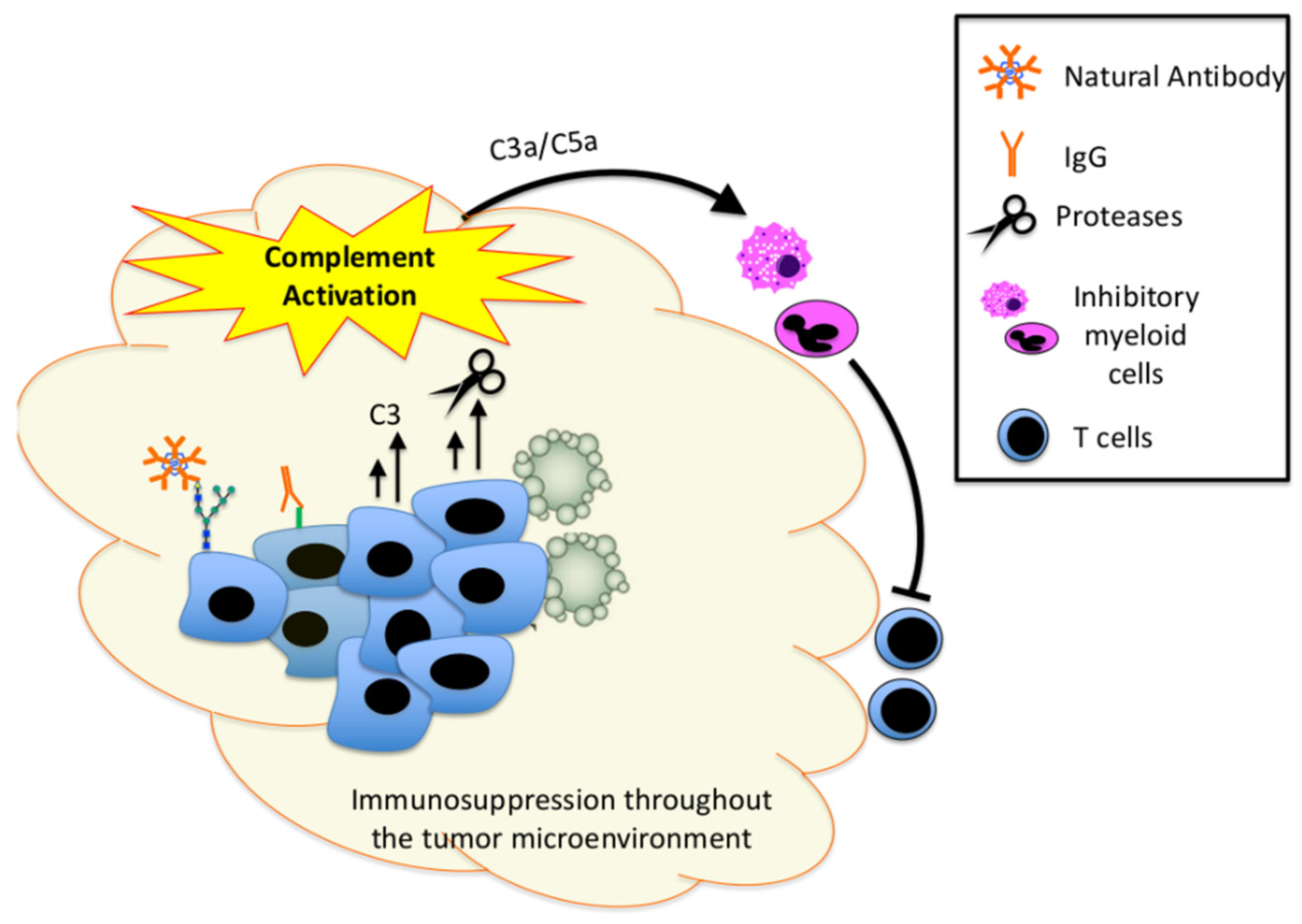{kind=link}
Abstract
:1. Introduction
2. How Does Carcinogenesis Occur?
3. Cancer, Inflammation, and Immunity
4. The Role of the Complement System in Carcinogenesis
4.1. Complement Activation as a Cause of Cancer
4.2. Complement as a Mechanism of Immune Evasion
4.3. Complement Activation and Metastatic Spread of Cancer
5. Cancer as a Cause of Complement Activation
6. Complement Regulatory Proteins
7. Complement Inhibitors as Cancer Treatment
8. Conclusions
Funding
Conflicts of Interest
References
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martel, C.; Franceschi, S. Infections and cancer: Established associations and new hypotheses. Crit. Rev. Oncol./Hematol. 2009, 70, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Doerner, S.K.; Reis, E.S.; Leung, E.S.; Ko, J.S.; Heaney, J.D.; Berger, N.A.; Lambris, J.D.; Nadeau, J.H. High-Fat Diet-Induced Complement Activation Mediates Intestinal Inflammation and Neoplasia, Independent of Obesity. Mol. Cancer Res. 2016, 14, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Waldner, M.J.; Neurath, M.F. Colitis-associated cancer: The role of T cells in tumor development. Semin. Immunopathol. 2009, 31, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Nonresolving macrophage-mediated inflammation in malignancy. FEBS J. 2018, 285, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol./Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Laskowski, J.; Renner, B.; Pickering, M.C.; Serkova, N.J.; Smith-Jones, P.M.; Clambey, E.T.; Nemenoff, R.A.; Thurman, J.M. Complement factor H-deficient mice develop spontaneous hepatic tumors. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, S.; Arber, N. Inflammation and colorectal cancer. Curr. Opin. Pharmacol. 2009, 9, 405–410. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 11037. [Google Scholar] [CrossRef]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; LaFace, I.; et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell 2015, 160, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieweke, M.H.; Thompson, N.L.; Sporn, M.B.; Bissell, M.J. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-beta. Science 1990, 248, 1656–1660. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M.; Lenderink, A.M.; Royer, P.A.; Coleman, K.E.; Zhou, J.; Lambris, J.D.; Nemenoff, R.A.; Quigg, R.J.; Holers, V.M. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ishemia/reperfusion. J. Immunol. 2007, 178, 1819–1828. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.-D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Qin, J.; Wang, D.; Geng, S. Complement C3a promotes proliferation, migration and stemness in cutaneous squamous cell carcinoma. J. Cell. Mol. Med. 2019, 23, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Hu, X.B. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol. Rep. 2014, 32, 2817–2823. [Google Scholar] [CrossRef]
- Lara-Astiaso, D.; Izarra, A.; Estrada, J.C.; Albo, C.; Moscoso, I.; Samper, E.; Moncayo-Arlandi, J.; Solano, A.; Bernad, A.; Diez-Juan, A. Complement anaphylatoxins C3a and C5a induce a failing regenerative program in cardiac resident cells. Evidence of a role for cardiac resident stem cells other than cardiomyocyte renewal. Springerplus. 2012, 1, 63. [Google Scholar] [CrossRef] [Green Version]
- Daveau, M.; Benard, M.; Scotte, M.; Schouft, M.-T.; Hiron, M.; Francois, A.; Salier, J.-P.; Fontaine, M. Expression of a functional C5a receptor in regenerating hepatocytes and its involvement in a proliferative signaling pathway in rat. J. Immunol. 2004, 173, 3418–3424. [Google Scholar] [CrossRef] [Green Version]
- Strey, C.W.; Markiewski, M.; Mastellos, D.; Tudoran, R.; Spruce, L.A.; Greenbaum, L.E.; Lambris, J.D. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J. Exp. Med. 2003, 198, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Vlaicu, S.I.; Tegla, C.A.; Cudrici, C.D.; Danoff, J.; Madani, H.; Sugarman, A.; Niculescu, F.; Mircea, P.A.; Rus, V.; Rus, H. Role of C5b-9 complement complex and response gene to complement-32 (RGC-32) in cancer. Immunol. Res. 2013, 56, 109–121. [Google Scholar] [CrossRef]
- Towner, L.D.; Wheat, R.A.; Hughes, T.R.; Morgan, B.P. Complement Membrane Attack and Tumorigenesis: A systems biology approach. J. Biol. Chem. 2016, 291, 14927–14938. [Google Scholar] [CrossRef] [Green Version]
- Okroj, M.; Corrales, L.; Stokowska, A.; Pio, R.; Blom, A.M. Hypoxia increases susceptibility of non-small cell lung cancer cells to complement attack. Cancer Immunol. Immunother. CII 2009, 58, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, R.; Yamaoka, K.; Sawamukai, N.; Shimajiri, S.; Oshita, K.; Yukawa, S.; Tokunaga, M.; Iwata, S.; Saito, K.; Chiba, K.; et al. C5a promotes migration, proliferation, and vessel formation in endothelial cells. Inflamm. Res. 2010, 59, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Olcina, M.M.; Balanis, N.G.; Kim, R.K.; Aksoy, B.A.; Kodysh, J.; Thompson, M.J.; Hammerbacher, J.; Graeber, T.G.; Giaccia, A.J. Mutations in an Innate Immunity Pathway Are Associated with Poor Overall Survival Outcomes and Hypoxic Signaling in Cancer. Cell Rep. 2018, 25, 3721–3732.e6. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef]
- Kwak, J.W.; Laskowski, J.; Li, H.Y.; McSharry, M.V.; Sippel, T.R.; Bullock, B.L.; Johnson, A.M.; Poczobutt, J.M.; Neuwelt, A.J.; Malkoski, S.P.; et al. Complement Activation via a C3a Receptor Pathway Alters CD4(+) T Lymphocytes and Mediates Lung Cancer Progression. Cancer Res. 2018, 78, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a Fosters Squamous Carcinogenesis and Limits T Cell Response to Chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef] [Green Version]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Akhir, F.N.M.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The Complement C3a Receptor Contributes to Melanoma Tumorigenesis by Inhibiting Neutrophil and CD4+ T Cell Responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs-Canner, S.; Magge, D.; Ravindranathan, R.; O’Malley, M.E.; Francis, L.; Liu, Z.; Guo, Z.S.; Obermajer, N.; Bartlett, D.L. Complement Inhibition: A Novel Form of Immunotherapy for Colon Cancer. Ann. Surg. Oncol. 2016, 23, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Vadrevu, S.K.; Chintala, N.K.; Sharma, S.K.; Sharma, P.; Cleveland, C.; Riediger, L.; Manne, S.; Fairlie, D.P.; Gorczyca, W.; Almanza, O.; et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014, 74, 3454–3465. [Google Scholar] [CrossRef] [Green Version]
- Llaudo, I.; Fribourg, M.; Medof, M.E.; Conde, P.; Ochando, J.; Heeger, P.S. C5aR1 regulates migration of suppressive myeloid cells required for costimulatory blockade-induced murine allograft survival. Am. J. Transplant. 2019, 19, 633–645. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, Y.; Xu, W.; Zheng, X.; Yi, X.; Huang, L.; Wang, Y.; Wu, K. Activated Hepatic Stellate Cells (HSCs) Exert Immunosuppressive Effects in Hepatocellular Carcinoma by Producing Complement C3. OncoTargets Ther. 2020, 13, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-C.; Chou, H.-S.; Yang, H.-R.; Lin, F.; Bhatt, S.; Qin, J.; Wang, L.; Fung, J.J.; Qian, S.; Lu, L. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood 2013, 121, 1760–1768. [Google Scholar] [CrossRef]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, Q.; Liao, J.-Y.; Song, E.; Xia, Q.; Pan, J.; Li, Y.; Li, J.; Zhou, B.; Ye, Y.; et al. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell 2020, 180, 1081–1097.e24. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Kunz, N.; Kemper, C. Complement and human T cell metabolism: Location, location, location. Immunol. Rev. 2020, 295, 68–81. [Google Scholar] [CrossRef]
- Piao, C.; Zhang, W.-M.; Li, T.-T.; Zhang, C.-C.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.-X.; Song, W.-C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massague, J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 2017, 168, 1101–1113.e13. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.H.; Hu, Z.; Shen, X.; Dong, L.Y.; Zhou, W.Z.; Yu, X.X. C5a receptor enhances hepatocellular carcinoma cell invasiveness via activating ERK1/2-mediated epithelial-mesenchymal transition. Exp. Mol. Pathol. 2016, 100, 101–108. [Google Scholar] [CrossRef]
- Kaida, T.; Nitta, H.; Kitano, Y.; Yamamura, K.; Arima, K.; Izumi, D.; Higashi, T.; Kurashige, J.; Imai, K.; Hayashi, H.; et al. C5a receptor (CD88) promotes motility and invasiveness of gastric cancer by activating RhoA. Oncotarget 2016, 7, 84798–84809. [Google Scholar] [CrossRef] [Green Version]
- Nitta, H.; Wada, Y.; Kawano, Y.; Murakami, Y.; Irie, A.; Taniguchi, K.; Kikuchi, K.; Yamada, G.; Suzuki, K.; Honda, J.; et al. Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin. Cancer Res. 2013, 19, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Ding, J.-Y.; Lu, C.; Lin, Z.-W.; Chu, Y.; Zhao, G.-Y.; Guo, J.; Ge, D. Overexpression of CD88 predicts poor prognosis in non-small-cell lung cancer. Lung Cancer 2013, 81, 259–265. [Google Scholar] [CrossRef]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Garaud, S.; Zayakin, P.; Buisseret, L.; Rulle, U.; Siliņa, K.; De Wind, A.; Eyden, G.V.D.; Larsimont, D.; Willard-Gallo, K.; Linē, A. Antigen Specificity and Clinical Significance of IgG and IgA Autoantibodies Produced in situ by Tumor-Infiltrating B Cells in Breast Cancer. Front. Immunol. 2018, 9, 2660. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Peng, B.; Lu, Y.; Xu, W.; Qian, W.; Zhang, J.Y. Autoantibodies to tumor-associated antigens as biomarkers in cancer immunodiagnosis. Autoimmun. Rev. 2011, 10, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Noe, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Le Clec’H, B.; A Giraldo, N.; et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 10346. [Google Scholar] [CrossRef] [Green Version]
- Ling, G.S.; Crawford, G.; Buang, N.; Bartok, I.; Tian, K.; Thielens, N.M.; Bally, I.; Harker, J.A.; Ashton-Rickardt, P.G.; Rutschmann, S.; et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science 2018, 360, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Ehrenstein, M.R.; Notley, C.A. The importance of natural IgM: Scavenger, protector and regulator. Nat. Rev. Immunol. 2010, 10, 778–786. [Google Scholar] [CrossRef]
- Vollmers, H.P.; Brandlein, S. Natural antibodies and cancer. New Biotechnol. 2009, 25, 294–298. [Google Scholar] [CrossRef]
- Hensel, F.; Hermann, R.; Schubert, C.; Abe, N.; Schmidt, K.; Franke, A.; Shevchenko, A.; Mann, M.; Müller-Hermelink, H.K.; Vollmers, H.P. Characterization of glycosylphosphatidylinositol-linked molecule CD55/decay-accelerating factor as the receptor for antibody SC-1-induced apoptosis. Cancer Res. 1999, 59, 5299–5306. [Google Scholar]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautes-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.S.; Rupaimoole, R.; Choi, H.J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement Component 3 Is Regulated by TWIST1 and Mediates Epithelial-Mesenchymal Transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef] [Green Version]
- Pratt, J.R.; Basheer, S.A.; Sacks, S.H. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat. Med. 2002, 8, 582–587. [Google Scholar] [CrossRef]
- Gocheva, V.; Joyce, J.A. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 2007, 6, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Frade, R.; Rodrigues-Lima, F.; Huang, S.; Xie, K.; Guillaume, N.; Bar-Eli, M. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998, 58, 2733–2736. [Google Scholar] [CrossRef]
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.; Conway, E.M. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddel, C.J.; Tan, C.W.; Chen, V.M. Thrombin Generation and Cancer: Contributors and Consequences. Cancers 2019, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Manning, M.L.; Williams, S.A.; Jelinek, C.A.; Kostova, M.B.; Denmeade, S.R. Proteolysis of complement factors iC3b and C5 by the serine protease prostate-specific antigen in prostatic fluid and seminal plasma. J. Immunol. 2013, 190, 2567–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.Q.; Lambris, J.D.; Ricklin, D. Protection of host cells by complement regulators. Immunol. Rev. 2016, 274, 152–171. [Google Scholar] [CrossRef] [Green Version]
- Ravindranath, N.M.; Shuler, C. Expression of complement restriction factors (CD46, CD55 & CD59) in head and neck squamous cell carcinomas. J. Oral Pathol. Med. 2006, 35, 560–567. [Google Scholar]
- Goslings, W.R.; Blom, D.J.; De Waard-Siebinga, I.; Van Beelen, E.; Claas, F.H.; Jager, M.J.; Gorter, A. Membrane-bound regulators of complement activation in uveal melanomas. CD46, CD55, and CD59 in uveal melanomas. Investig. Ophthalmol. Visual Sci. 1996, 37, 1884–1891. [Google Scholar]
- Bjorge, L.; Vedeler, C.A.; Ulvestad, E.; Matre, R. Expression and function of CD59 on colonic adenocarcinoma cells. Eur. J. Immunol. 1994, 24, 1597–1603. [Google Scholar] [CrossRef]
- Simpson, K.L.; Jones, A.; Norman, S.; Holmes, C.H. Expression of the complement regulatory proteins decay accelerating factor (DAF, CD55), membrane cofactor protein (MCP, CD46) and CD59 in the normal human uterine cervix and in premalignant and malignant cervical disease. Am. J. Pathol. 1997, 151, 1455–1467. [Google Scholar]
- Murray, K.P.; Mathure, S.; Kaul, R.; Khan, S.; Carson, L.F.; Twiggs, L.B.; Martens, M.G.; Kaul, A. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59-in benign and malignant endometrial tissue. Gynecol. Oncol. 2000, 76, 176–182. [Google Scholar] [CrossRef]
- Geis, N.; Zell, S.; Rutz, R.; Li, W.; Giese, T.; Mamidi, S.; Schultz, S.; Kirschfink, M. Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Curr. Cancer Drug Targets 2010, 10, 922–931. [Google Scholar] [CrossRef]
- Blok, V.T.; Daha, M.R.; Tijsma, O.M.; Weissglas, M.G.; van den Broek, L.J.; Gorter, A. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab. Investig. 2000, 80, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Surowiak, P.; Materna, V.; Maciejczyk, A.; Kaplenko, I.; Spaczyński, M.; Dietel, M.; Lage, H.; Zabel, M. CD46 expression is indicative of shorter revival-free survival for ovarian cancer patients. Anticancer Res. 2006, 26, 4943–4948. [Google Scholar]
- Ajona, D.; Castaño, Z.; Garayoa, M.; Zudaire, E.; Pajares, M.J.; Martínez, A.; Cuttitta, F.; Montuenga, L.M.; Pio, R. Expression of complement factor H by lung cancer cells: Effects on the activation of the alternative pathway of complement. Cancer Res. 2004, 64, 6310–6318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilczek, E.; Rzepko, R.; Nowis, D.; Legat, M.; Golab, J.; Glab, M.; Gorlewicz, A.; Konopacki, F.; Mazurkiewicz, M.; Śladowski, D.; et al. The possible role of factor H in colon cancer resistance to complement attack. Int. J. Cancer 2008, 122, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Junnikkala, S.; Hakulinen, J.; Jarva, H.; Manuelian, T.; Bjørge, L.; Bützow, R.; Zipfel, P.F.; Meri, S. Secretion of soluble complement inhibitors factor H and factor H-like protein (FHL-1) by ovarian tumour cells. Br. J. Cancer 2002, 87, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajona, D.; Hsu, Y.F.; Corrales, L.; Montuenga, L.M.; Pio, R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J. Immunol. 2007, 178, 5991–5998. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Farshchian, M.; Kivisaari, A.; Ala-Aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2015, 135, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Fedarko, N.S.; Fohr, B.; Robey, P.G.; Young, M.F.; Fisher, L.W. Factor H binding to bone sialoprotein and osteopontin enables tumor cell evasion of complement-mediated attack. J. Biol. Chem. 2000, 275, 16666–16672. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.W.; Liu, M.C.; Hong, H.J.; Du, Q.; Chen, Y.L. Expression and prognostic value of soluble CD97 and its ligand CD55 in intrahepatic cholangiocarcinoma. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Q.; Zhang, L.; Jiang, Y.; Ni, X.; Chen, S.; Ye, F.; Du, Y.; Huang, L.; Ding, P.; Wang, N.; et al. The membrane complement regulatory protein CD59 promotes tumor growth and predicts poor prognosis in breast cancer. Int. J. Oncol. 2016, 48, 2015–2024. [Google Scholar] [CrossRef]
- Niehans, G.A.; Cherwitz, D.L.; Staley, N.A.; Knapp, D.J.; Dalmasso, A.P. Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin). Am. J Pathol. 1996, 149, 129–142. [Google Scholar]
- Bjorge, L.; Jensen, T.S.; Ulvestad, E.; Vedeler, C.A.; Matre, R. The influence of tumour necrosis factor-alpha, interleukin-1 beta and interferon-gamma on the expression and function of the complement regulatory protein CD59 on the human colonic adenocarcinoma cell line HT29. Scand. J. Immunol. 1995, 41, 350–356. [Google Scholar] [CrossRef]
- Spiller, O.B.; Criado-Garcia, O.; Rodriguez De Cordoba, S.; Morgan, B.P. Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin. Exp. Immunol. 2000, 121, 234–241. [Google Scholar] [CrossRef]
- Janelle, V.; Langlois, M.P.; Tarrab, E.; Lapierre, P.; Poliquin, L.; Lamarre, A. Transient Complement Inhibition Promotes a Tumor-Specific Immune Response through the Implication of Natural Killer Cells. Cancer Immunol. Res. 2014, 2, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zhang, F.; Tang, C.; Tang, M.; Liu, C.-G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajona, D.; Ortiz-Espinosa, S.; Moreno, H.; Lozano, T.; Pajares, M.J.; Agorreta, J.; Bértolo, C.; Lasarte, J.J.; Vicent, S.; Hoehlig, K.; et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discov. 2017, 7, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naive to complement inhibitor treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef] [PubMed]
- Fishelson, Z.; Donin, N.; Zell, S.; Schultz, S.; Kirschfink, M. Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 2003, 40, 109–123. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, S.-N.; Liu, Q.; Yu, Y.-Y.; Guo, J.; Wang, K.; Xing, B.-C.; Zheng, Q.-F.; Campa, M.J.; Patz, E.F.; et al. Autocrine Complement Inhibits IL10-Dependent T-cell-Mediated Antitumor Immunity to Promote Tumor Progression. Cancer Discov. 2016, 6, 1022–1035. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, S.; Thurman, J.M. Tissue-targeted complement therapeutics. Mol. Immunol. 2018, 102, 120–128. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thurman, J.M.; Laskowski, J.; Nemenoff, R.A. Complement and Cancer—A Dysfunctional Relationship? Antibodies 2020, 9, 61. https://doi.org/10.3390/antib9040061
Thurman JM, Laskowski J, Nemenoff RA. Complement and Cancer—A Dysfunctional Relationship? Antibodies. 2020; 9(4):61. https://doi.org/10.3390/antib9040061
Chicago/Turabian StyleThurman, Joshua M., Jennifer Laskowski, and Raphael A. Nemenoff. 2020. "Complement and Cancer—A Dysfunctional Relationship?" Antibodies 9, no. 4: 61. https://doi.org/10.3390/antib9040061
APA StyleThurman, J. M., Laskowski, J., & Nemenoff, R. A. (2020). Complement and Cancer—A Dysfunctional Relationship? Antibodies, 9(4), 61. https://doi.org/10.3390/antib9040061