1. Introduction
The geometrical symmetry of a centrosymmetric linear molecule in its equilibrium geometry is described by the
D point group (see
Table 1). While the molecular vibrational states (assuming a totally symmetric singlet electronic state) span the representations of this point group of infinite order, the symmetry properties of the combined rotation-vibration states must satisfy the nuclear-statistics requirements and transform according to the irreducible representations (irreps) of the finite molecular symmetry group
where, for the centrosymmetric linear molecule A–B–C–…–C–B–A, the permutation operation
is the simultaneous interchange of the two A nuclei, the two B nuclei, the two C nuclei, etc.,
E is the identity operation,
is the spatial inversion operation, which inverts the positions of all particles through the molecular centre of mass, and
is the permutation-inversion operation [
1]. The irreps of
D(M) are given in
Table 2 (see also Table A-18 of Ref. [
1]).
Table 2 presents several alternative notations for the irreducible representations. These alternative notations and multiple names for the same concept are perhaps not quite in agreement with time-honored principles such as Occam’s Razor, but they represent a nice example of the development of spectroscopic notation. We note already here that
D(M) is isomorphic (and, for a triatomic linear molecule A–B–A like CO
, identical) to the group customarily called
C(M) (Table A-5 of Ref. [
1]), the molecular symmetry group of, for example, the H
O molecule, whose equilibrium structure is bent. The molecular symmetry (MS) groups
D(M) and
C(M) are determined by applying the principle of feasibility first introduced by Longuet-Higgins [
2] (see also Ref. [
1]), and one obviously obtains isomorphic MS groups for all chain molecules A–B–C–…–C–B–A, irrespective of these molecules having linear or bent equilibrium structures. Longuet-Higgins [
2] (see also Ref. [
1]) further showed that for a so-called rigid non-linear molecule (in this context, a rigid molecule is defined as one whose vibration can be described as oscillations around a single potential energy minimum), the MS group is isomorphic to the point group describing the geometrical symmetry at the equilibrium geometry. H
O is a rigid non-linear molecule, whose geometrical symmetry at equilibrium is described by the
C point group which is indeed isomorphic to the MS group
C(M). For the rigid linear molecule CO
, however, as already mentioned, the geometrical symmetry at equilibrium is described by the infinite-order point group
D which obviously is not isomorphic to the MS group
D(M) =
C(M) of order four.
One can argue that the MS group as defined by Longuet-Higgins [
2] (see also Ref. [
1]) provides the simplest symmetry description of a molecule required for understanding its energy level pattern and the properties deriving from this pattern. For rigid non-linear molecules, this symmetry description is identical to that arising from the molecular point group at equilibrium, and this explains the many successful, traditional applications of point group symmetry, especially in chemical contexts. For a rigid linear molecule, the infinite-order point group obviously provides a much more detailed symmetry description than the finite MS group. Again, one can argue that the MS group provides the symmetry operations relevant for describing the ‘fully coupled’ rovibronic (rotation-vibration-electronic) wavefunctions of a molecule and that the additional point group symmetry is redundant and unnecessary. In practice, however, the point group symmetry gives rise to useful information, in particular for the electronic, vibrational, and rotational basis functions used to express the fully coupled wavefunctions, and so it is advantageous to also employ the point group symmetry. The particular problems associated with the symmetry description of linear molecules were described early on by Hougen [
3], and by Bunker and Papoušek [
4]. The latter authors introduced the so-called Extended Molecular Symmetry (EMS) Group which, for a centrosymmetric linear molecule, is isomorphic to the
D point group. We discuss the EMS group in more detail below.
The aim of the present work is to illustrate how
D symmetry can be implemented into general nuclear-motion programs. As an example we use TROVE [
5,
6], a numerical variational method to solve for the ro-vibrational (rotational-vibrational) spectra of (small to medium) general polyatomic molecules, which has been used to simulate the hot spectra of various polyatomic molecules [
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19] as part of the ExoMol project [
20,
21], a database of
ab initio spectra of molecules of astrophysical importance. Since calculations of this type are based on electronic properties of the molecule, primarily the potential energy and dipole moment surfaces, obtained by solving the electronic Schrödinger equation, one can claim that such calculations are also rovibronic. However, as the calculations are directly concerned with the rotation and vibration, we characterize them as ro-vibrational. The use of molecular symmetry has applications in diverse fields, including molecular spectroscopy and the construction of molecular wavefunctions, ligand-field theory, material science, and electronic structure calculations [
1,
22,
23,
24,
25]. While the use of a symmetry-adapted basis set has been shown to make calculations of ro-vibrational energies far more efficient by reducing the size of the Hamiltonian matrix blocks to be diagonalized [
22], it is not strictly necessary. This is in contrast to intensity calculations, which would hardly be practicable without knowledge about the symmetry of the ro-vibrational states, mainly due to the selection rules imposed by the nuclear spin statistics associated with different irreducible representations [
17,
22]. A symmetry-free intensity calculation would involve the actual numerical computation of a colossal number of vanishing intensities for transitions that do not satisfy the symmetry selection rules. In this context, it is also important that some energy levels have zero weights and do not exist in nature. Without knowledge of how the eigenvectors transform under the symmetry operations, it is therefore impossible—or at least immensely complicated—to describe the molecular spectrum correctly. We show how a symmetrization procedure similar to that of TROVE can be extended to enable symmetry classification, in particular of vibrational basis functions, in the
D point group and thereby introduce the possibility of labelling these basis functions by the value of the vibrational angular momentum quantum number
ℓ (see, for example, Ref. [
26]). In practice, it turns out that the infinitely many elements in
D represent a problem in numerical calculations; we circumvent this by employing, instead of
D, one of its subgroups
D with a finite value of
n. We discuss below how to choose an adequate
n-value.
In numerical calculations, the vibrational and rotational basis functions are initially symmetry classified before they are combined to form the ro-vibrational basis. For a linear molecule, only combinations with
k =
ℓ are physically meaningful, where
k is the
z-axis-projection of the rotational angular momentum quantum number and
ℓ is the vibrational angular momentum quantum number (see, for example, Refs. [
1,
18,
26,
27,
28]). With the extended symmetrization procedure of the present work, the vibrational basis functions can be labelled by their
ℓ-values and it becomes straightforward to construct the meaningful combinations. In a given ro-vibrational calculation, the required extent of the rotational excitation is defined by the maximum value
of the angular momentum quantum number
J. The maximum values of
and
,
and
, respectively, are then
=
=
. However, in practise the numerical calculations are computationally limited by the total number of quanta representing vibrational bending modes, which controls the maximum value for
, and thus
. We find that the group
D suitable for symmetry classification in numerical calculations has an
n-value determined by
=
.
The TROVE symmetrization approach makes use of a set of simplified, ‘reduced’ vibrational Hamiltonians, each one describing one vibrational mode of the molecule. The symmetrization is achieved by utilizing the fact that each of these Hamiltonian operators commutes with the operations in the symmetry group of the molecule in question [
22], so that eigenfunctions of a reduced vibrational Hamiltonian generate irreducible representations of the symmetry group. Consequently, we obtain a symmetry-adapted ro-vibrational basis set numerically by solving the eigenvalue problems for the reduced Hamiltonians; the vibrational basis functions are products of the eigenfunctions thus obtained. In the course of the present work, we have implemented in TROVE a general subroutine to generate automatically all transformation matrices associated with the irreducible representations of a given symmetry group
D; this can be applied to general nuclear motion routines. The matrices are chosen to describe the transformation of vibrational basis functions that are eigenfunctions of the operator
(with eigenvalues
) representing the vibrational angular momentum.
To the best of our knowledge, no general transformation matrices for
D have been reported in the literature although the corresponding character tables have been published many times (see, for example [
29]). Hegelund et al. [
30] have described the transformation properties of the customary rigid-rotor/harmonic-oscillator basis functions (see, for example, Refs. [
1,
26,
31]) for
D point groups with arbitrary
n ⩾ 3 (see also Section 12.4 of Ref. [
1]). The basis functions span the irreducible representations of
D and the coefficients obtained, defining the transformation properties, can straightforwardly be organized as transformation matrices. The present paper aims at providing the missing information for
D. As an illustration, we present how this symmetry information is implemented in TROVE as part of the automatic symmetry adaptation technique [
22].
The paper is structured as follows.
Section 2 gives an overview of the rotational and vibrational symmetry classifications and groups for a centrosymmetric linear molecule, and
Section 3 presents the corresponding irreducible-representation transformation matrices and character tables. The symmetrization approach implemented in TROVE is outlined in
Section 4, followed by some numerical examples in
Section 5. Our conclusions are given in
Section 6.
4. Symmetrization Using the TROVE Approach
In this section we use the variational nuclear-motion program TROVE as an example of a practical implementation of the symmetrization of ro-vibrational basis states using
D representations. TROVE uses a general numerical symmetrization approach to build a symmetry-adapted ro-vibrational basis set [
22]. The procedure will be outlined here and extended to include classification based on the vibrational angular momentum quantum number,
ℓ, as necessary for dealing with linear molecules of
D point group symmetry, using
C
H
as an example. This classification is general and can be implemented into other similarly constructed variational routines.
The use of a symmetry-adapted basis set can considerably reduce the size of the Hamiltonian matrix to be diagonalized. This is due to the useful property that the matrix elements between basis functions of different symmetry are zero by definition:
where
and
give the irreducible representations (irreps) of
D that the basis functions,
and
, transform according to, and
and
represent their degenerate components (if present). The block diagonal structure of a Hamiltonian matrix in the
D irreducible representation is given in
Figure 1; the symmetry blocks of non-vanishing matrix elements can be diagonalized separately.
TROVE utilises the concept of a sum-of-product basis set, where the primitive basis functions are
With 1D vibrational basis functions (where is a generalised vibrational coordinate) and rigid-rotor (spherical harmonics) rotational basis functions . The 1D vibrational basis functions are either obtained by solving the corresponding reduced 1D Schrödinger equations or are taken as the harmonic or Morse oscillators.
Symmetrization of the Basis Set for CH Using the Coordinate TROVE Implementation
As an illustration of the practical application of the finite
D group being used in place of
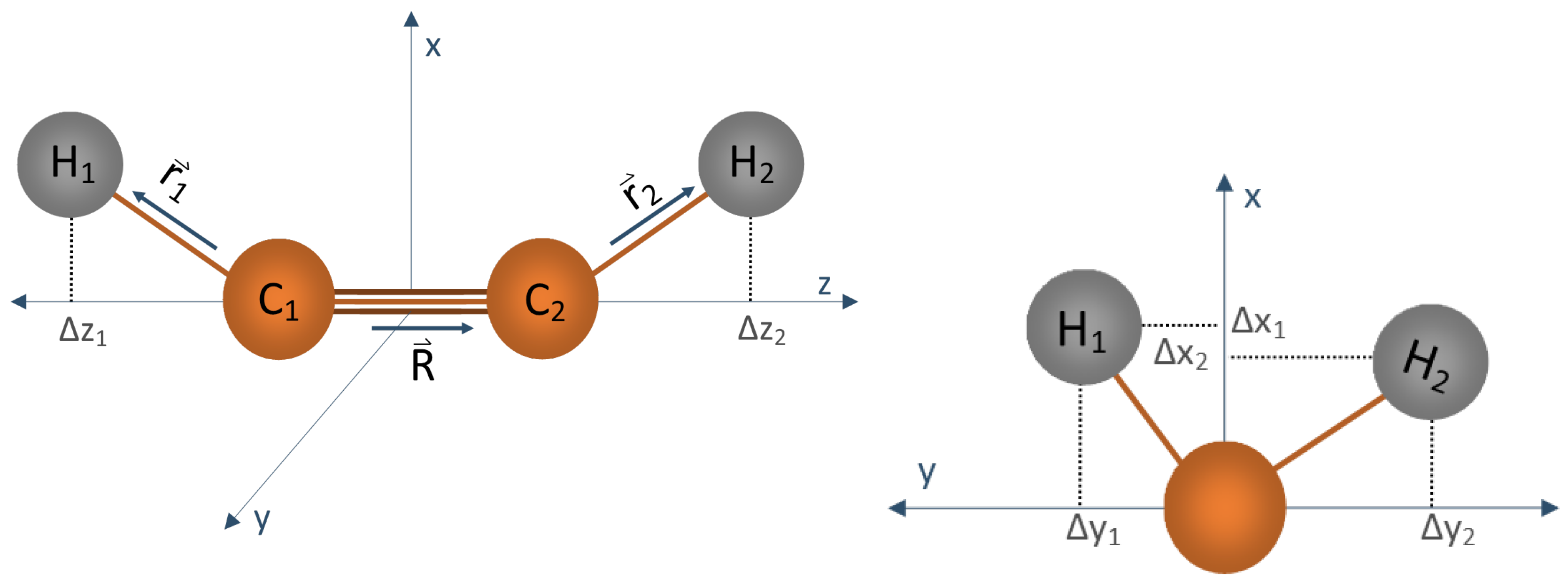
D, we show an example of the construction of the vibrational basis set in case of the linear molecule
C
H
. We use the recent implementation of the
coordinates approach in TROVE (see Ref. [
18]) and select a set of seven vibrational coordinates used for
C
H
:
,
,
,
,
,
,
, as illustrated in
Figure 2. The transformation matrices defining their symmetry properties are listed in
Table 13 (with even
n used in this example). These relate to the symmetry operations of
Table 5, and the general irrep transformation matrices for
D of even
n given in
Table 10.
For the stretching primitive basis functions,
,
and
, we use eigenfunctions of the corresponding 1D reduced stretching Hamiltonian operators
obtained by freezing all other degrees of freedom at their equilibrium values in the
Hamiltonian. We use the Numerov-Cooley approach [
5,
37,
38] to solve these eigenvalue problems. For the bending basis functions
, 1D harmonic oscillators are used. Here
,
,
,
,
,
,
, where
,
, and
are linearized (rectilinear) versions of
,
,
, respectively, as described in Ref. [
18].
According to the TROVE symmetrization technique [
22], the symmetry adapted vibrational basis functions are formed from linear combinations of the products of the 1D vibrational basis functions
as follows, with
C
H
used as an example. For
C
H
, the vibrational part of the basis set of Equation (
23) is divided into three sub-sets:
These sub-sets are then used as basis sets for the corresponding reduced Hamiltonian operators: stretching
and
, and bending
. The reduced Hamiltonians
(
) are constructed by averaging the total vibrational Hamiltonian operator
over the other ground vibrational basis functions. For example,
is given by:
where
and
.
According to the idea of the so-called complete set of commuting operators (CSCO) [
22] which the TROVE symmetrization approach is based on, the eigenfunctions of the reduced operator
must transform according to one of the irreps of the symmetry group of the system, since
commutes with the symmetry operators of this group. Thus the symmetrization of the basis set is generated automatically by solving the appropriate eigenvalue problem, provided that the corresponding irreps have been determined. To this end, the symmetry operators of the appropriate group can be applied to the eigenfunctions and their transformation properties on a set of sampled geometries (usually 40–60 in the case of TROVE) analysed. Some states of the same energy (either with accidental or actual degeneracy) may appear as random mixtures of each other, and have to be processed simultaneously and even further reduced to irreps, if necessary (see
Section 5 for an example).
Applying this procedure to stretching functions gives rise to A-type symmetries: e.g., for D (even n), the eigenfunctions of span the irrep, while the eigenfunctions of span the and irreps.
The 4D bending basis set, based on the 1D harmonic oscillators of Equation (
26), has the disadvantage of being extremely degenerate: combinations of
give rise to large clusters of the same energies. According to the TROVE symmetrization approach these combinations must be processed together, which makes this process extremely slow. In order to facilitate this step we first transform the 4D bending sets (Equation (
26)) to become eigenfunctions of the vibrational angular momentum operator,
where
is a vibrational momentum operator and
are Coriolis coefficients [
39] as described in Ref. [
18].
TROVE is equipped to compute matrix elements of quadratic forms, therefore we use
instead of
. Using the
basis functions we find eigenfunctions of
by diagonalizing the matrix formed by combinations of the 4D bending basis set of Equation (
26):
The eigenfunctions of
are consequently characterized by their vibrational angular momentum quantum number
ℓ =
=
and can thus be divided into independent sub-sets with different symmetry properties: the
sub-set must be a mixture of
A-type functions, while the
sub-sets consist of the
-type irreps (
and
). These mixtures are then further reduced to irreps using the TROVE symmetrization scheme outlined above, in which the reduced 4D-eigenvalue problem, using the eigenfunctions of
as the basis set, is solved for a 4D isotropic harmonic oscillator Hamiltonian:
where
is a related to the harmonic vibrational wavenumber and
are the vibrational momenta, conjugate to
. Thus we obtain eigenfunctions which can be divided into sub-sets of the same energies and values of
ℓ. These sub-sets must transform independently, thereby significantly decreasing the time spent on the symmetry sampling step by breaking the symmetry space into small sets and making numerical calculations more computationally viable. Although the
-diagonalisation step is not strictly necessary for the general symmetrization procedure that follows it, this increase in efficiency is a big advantage.
As mentioned above, in addition to the
ℓ-quantum number being advantageous in building the vibrational basis sets, it is also required for coupling the basis set functions according to the linear molecule angular momentum rule
(see, for example, Refs. [
18,
27,
28]). The maximum value for
is specified as an input into the TROVE numerical routine.
As a result of applying the procedure described above, a symmetry-adapted vibrational basis set is generated. Here is the irrep of the basis function according to D, and indicates a degenerate component in the case of 2D irreps.
The symmetry-adapted rotational basis set in TROVE is represented by:
where
is a special case, given by:
Here
is a rigid rotor wavefunction,
is a parameter used to define the parity of a state, where
for
and
for
(see [
22,
40,
41]). The irreps
of these functions are listed in
Table 14, where
defines their degenerate component. The symmetry properties of
can be derived from those of
using the method described in
Section 3.3.
The symmetrized rotational and vibrational basis functions are then combined to form a full ro-vibrational symmetry-adapted basis set:
where
are symmetrization coefficients with
indicating a degenerate component in the case of 2D irreps,
is a 1D irreps in
D (see
Section 3) and the
condition for linear molecules in the
-approach [
18] was applied. Note that the symmetrized basis functions use
K and
L instead of
k and
ℓ in Equation (
23).






{kind=link}
{kind=link}