1. Introduction
An object is chiral when it is non-superimposable with its mirror-image [
1]. Examples presenting this property vary from macroscopic such as screws or spiral staircases to microscopic i.e., nanoparticles [
2] and molecules [
3] among others. Furthermore, two chiral mirror-image molecules are called enantiomers. While the interaction between two enantiomeric systems and any achiral medium is indistinguishable, the behavior may differ when the two enantiomers interact with a chiral medium, giving rise to chiroptical responses when interacting with chiral light [
4]. Electronic and vibronic circular dichroism (ECD and VCD) or optical rotatory dispersion (ORD) are some of the most common spectroscopies arising from such responses [
5]. In the last few decades, these and other chiroptical spectroscopies have been widely employed for the structure determination of chemical systems [
6]. On the other hand, the current interest in chiroptical light-powered molecular motors [
7] or sensors [
8] has triggered the development of efficient chiroptical systems [
9,
10,
11]. Accordingly, the prediction and understanding of the chiroptical responses is essential [
12].
Ab initio simulations have been shown to have great relevance for the prediction of chiroptical responses [
13]. However, even though simplified time-dependent density functional theory (TD-DFT) techniques have been developed for large systems [
14,
15], the necessary computational and time resources may be a limitation. As a qualitative alternative, the application of the exciton chirality method (ECM) [
16] has been considered to be practical for systems incorporating two identical chromophores—for instance, molecular tweezers used as chiroptical proof of natural amines [
17]. When two identical chromophores are in close vicinity, in phase and out of phase excitations arise. If the mutual orientation between the chromophores is chiral, the rotatory strength (RS) of the two transitions may be of opposite sign. The sign of the RS associated with the less energetic transition determines the sense of chirality [
18]. Nevertheless, the presence of more chromophores may be desired for designing powerful chiroptical systems [
19]. In this respect, we previously revised the chiroptical symmetry analysis (CSA) for systems with three and four chromophores [
20]. The main scope of this approach is to serve as a useful tool for developing particular systems rather than predicting experimental spectra of systems. Like EC, CSA also provides the sense of chirality of the responding system. On the other hand, different parameters (i.e., rotatory strength or energy difference between allowed transitions) can be calculated by the CSA. Considering the conformational stability typically presented by trianglimines as well as taking advantage of the experimental and simulated ECD reported by Szymkowiak et al. [
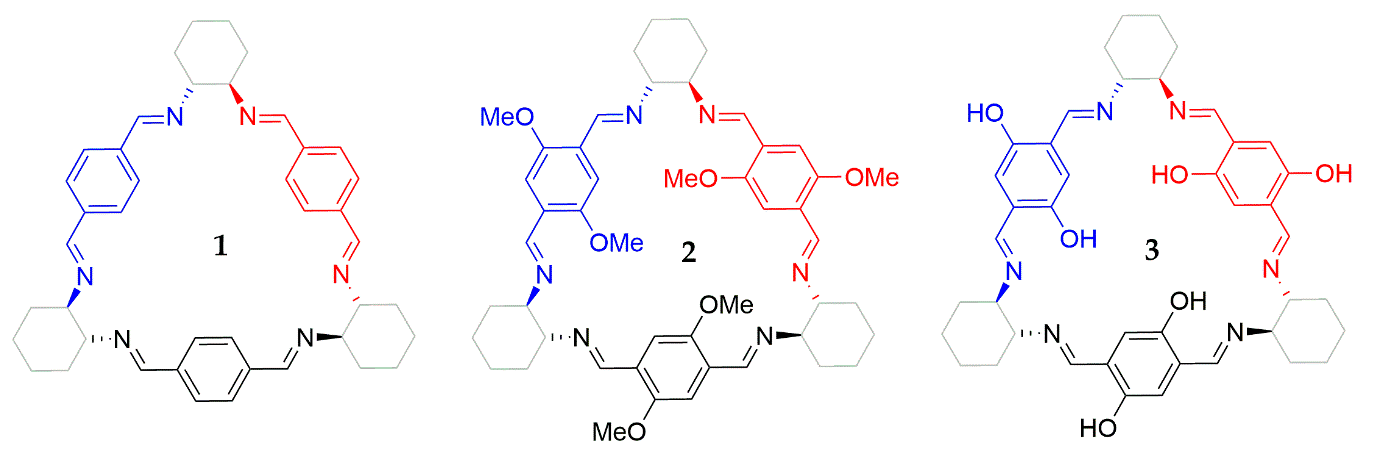
21], in this work, we perform the chiroptical symmetry analysis for 1, 2, and 3 (
Figure 1) as a case study and intend to draw some conclusions about the reliability of this strategy on the design of tailored chiroptical systems.
2. Materials and Methods
All calculations were performed with Gaussian09 program [
22]. Trianglimine structures
1,
2, and
3 (
Figure 1) were optimized at cam-B3LYP [
23,
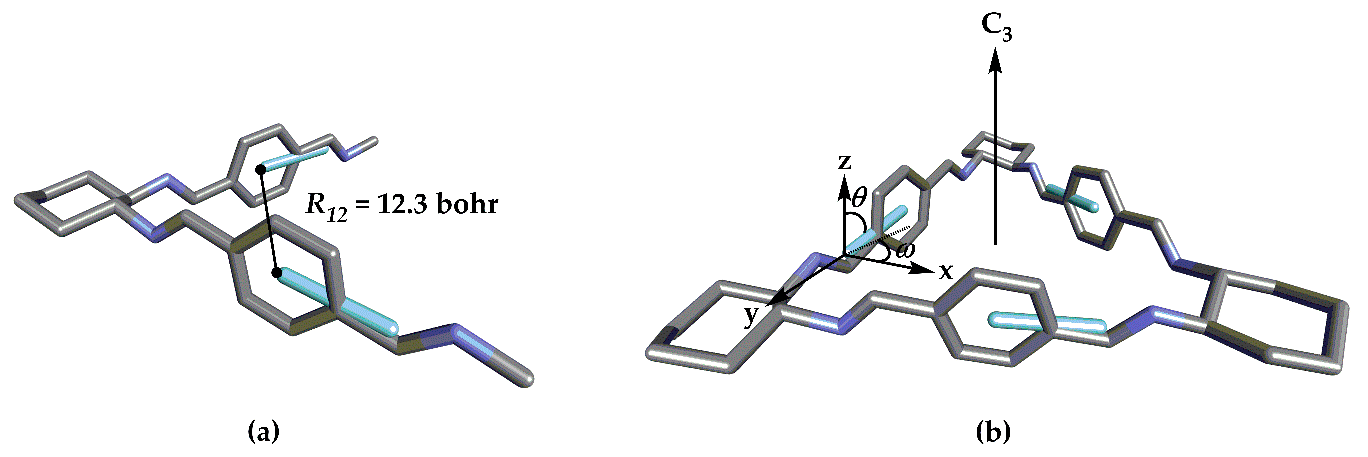
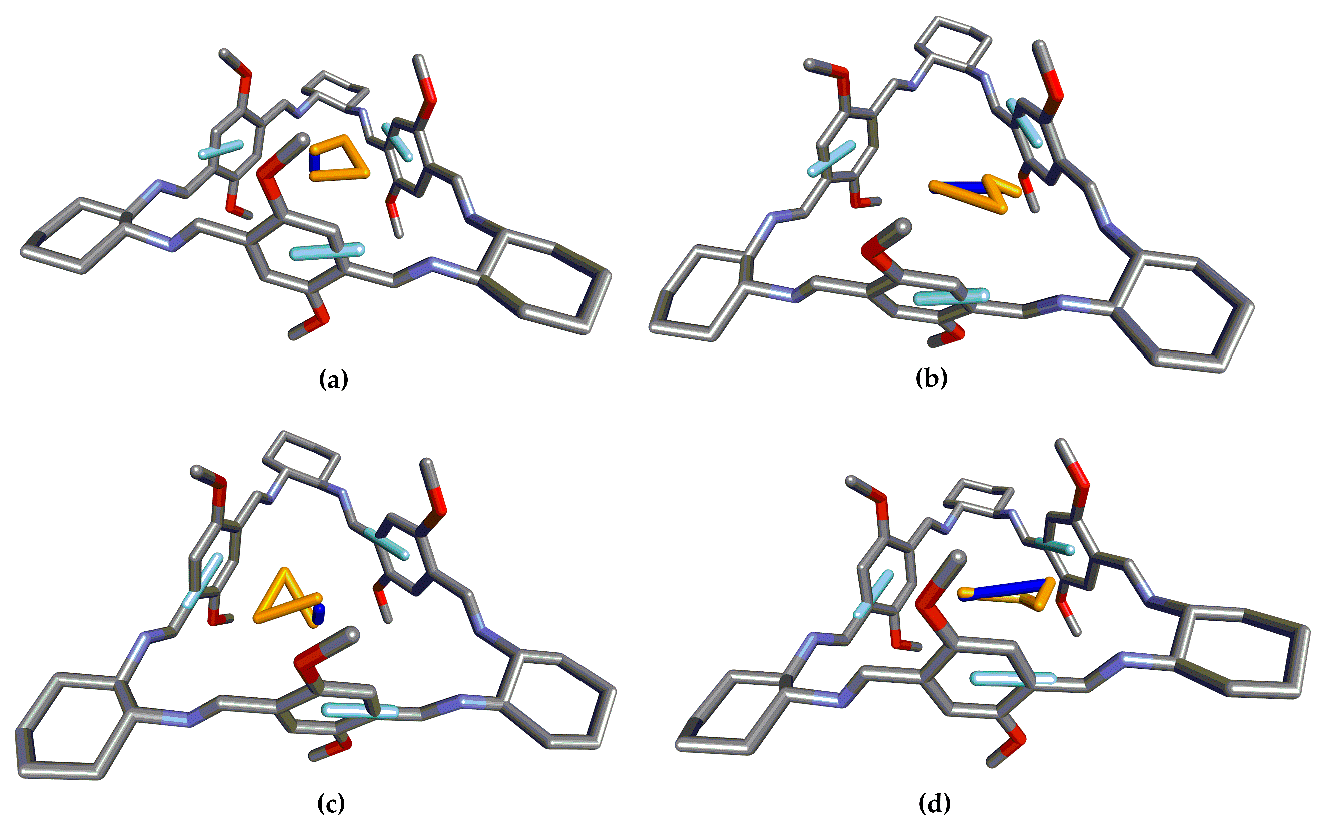
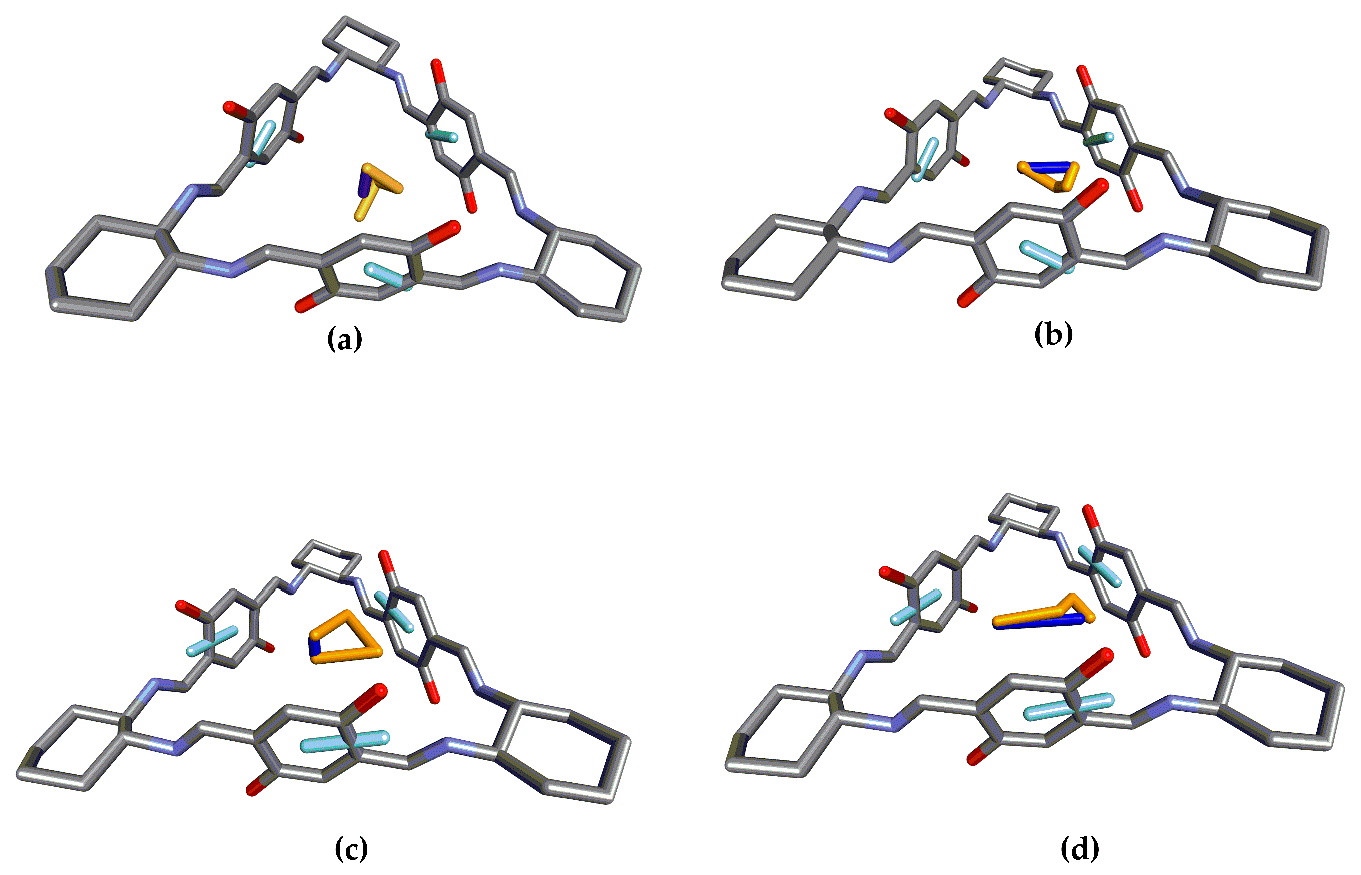
24] level using 6-311G(d,p) basis set. Subsequently, vibrational frequencies were calculated at the same level of theory to confirm that the obtained structures were true minima. The structural measures necessary for the CSA were extracted from the corresponding structures (
Figure 2 and
Figure 3). In addition, TD-DFT calculations were performed to compare the ECD spectra with those predicted by CSA. Particularly, the cam-B3LYP functional was chosen since the simulated spectra showed good agreement with the reported experimental ECD spectra [
21]. On the other hand, UV/Vis and ECD spectra were simulated for analogue structures featuring only two and one chromophores (depicted in
Figure 2 and
Figure 3, respectively) by removing the rest of the structure and adding hydrogen atoms for each analogue without further optimization.
All trianglimines under study have
D3 symmetry; therefore, the expressions obtained previously [
20] for this symmetry were employed to predict the ECD spectra in a qualitative manner. The first step in the application of the CSA involves the identification of the chromophores that are responsible for optical properties in a molecule. Concerning thee trianglimine derivatives under study, three identical chromophores are present in each structure (represented in different colors in
Figure 1). According to the CSA, the chiroptical response of a system mainly originates from the contribution of each chromophore interacting cooperatively with the others due to their relative orientation. This implies that the symmetry of the system determines which chromophores interact simultaneously resulting in the ECD spectra. When a system with
C2 or
D2 symmetry presents two non-conjugated chromophores, the Davydov model allows for obtaining a picture of the approximate ECD spectra: The chromophores are represented by the corresponding Electric Dipole Transition Moment (EDTM),
, and the interaction between them,
V12, is given by Equation (1):
In this equation,
is the vector connecting the origin of
and
(
Figure 2) and
V12 is in au units. The application of the perturbation theory to the originally degenerate first electronically-excited level yields an energy splitting, Δ
E, between these two states and, consequently, the gap between the two main electronic excitations given by Equation (2):
The CSA seeks to obtain the splitting between those first electronically-excited states attainable from the ground state by absorption of electromagnetic radiation allowed by electric dipole transition. Additionally, the corresponding RS for systems with two or more independent chromophores could be revealed by this method. It should be noted that the application of CSA for systems bearing two chromophores is equal to that of ECM.
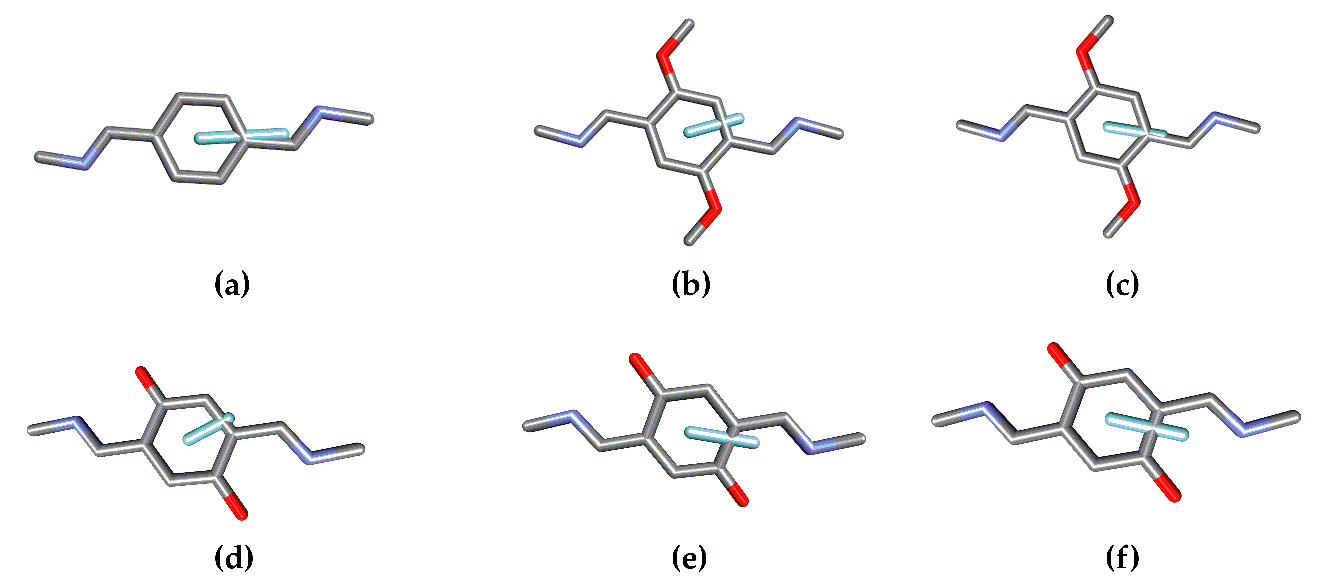
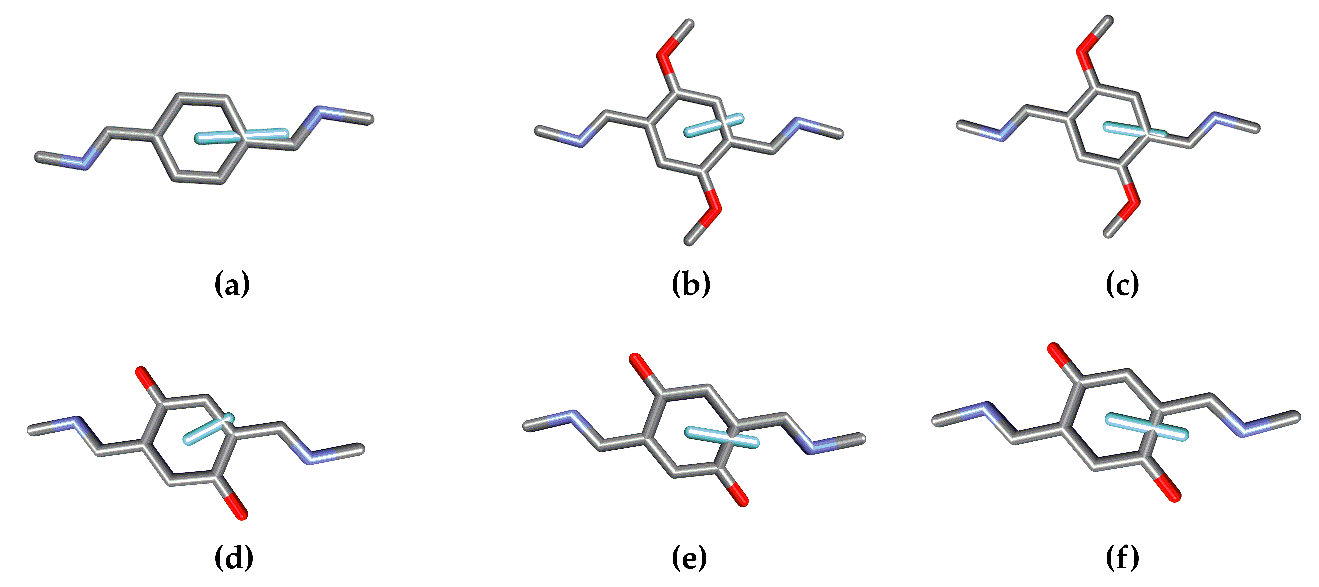
The structural parameters can be obtained after determining the location of the EDTM in each chromophore. This will depend on the nature of the chromophore and may present more than one EDTM at different energies and orientations.
Figure 3 presents the different EDTMs for the chromophores in 1, 2, and 3. While there is only one significant electronic transition for the chromophoric units in 1 (a), there are two in the case of 2 (2.1, b) and (2.2, c) and three for 3 (3.1, d), (3.2, e), and (3.3, f).
For systems presenting
D3 symmetry, the ground electronic state belongs to the totally-symmetric representation (A1) and there are three equivalent monoexcitations. With this basis set, we are able to obtain the
D3 Symmetry-Adapted Linear Combinations for the first electronically-excited states. As we have shown earlier [
20], the reducible representation of this basis can decompose into A1 and E when all the molecular orbitals (MOs) describing the excited state are σ or A2 and E when the excited state of the chromophore contains one π MO. In any case, the transition from the ground state will be only orbitally allowed for states A1 and E or A2 and E with 3
V12 energy difference between A1 or A2 and E.
The EDTM corresponding to the A2 transition is along the main symmetry axis, whereas the EDTMs for E transitions are located on the orthogonal plane. The rotatory strengths are obtained from Equations (3) and (4). It is noteworthy that this approximation is only valid when the electronic transition of the independent chromophore presents negligible magnetic transition dipole moment (MTDM, for a more thorough explanation see reference [
20]). Other studies have developed ab initio based exciton chirality methods [
25]:
After this brief description of the CSA for molecules with D3 symmetry, we need to know the structural parameters of the molecule as well as the EDTM of the monomer in order to use CSA on each particular case. By this way, after calculation of V12 and the rotatory strengths for each allowed transition, we will be able to predict the main features of the ECD spectra originated from the exciton coupling between the interacting independent chromophores. Therefore, we will predict one A2 and two degenerated E transitions per electronic transition of the independent chromophores. Since the chromophoric units in 1, 2, and 3 present respectively one, two, and three significant electronic transitions, by the CSA, we could simulate one, two, and three A2 along with one, two, and three pairs of E transitions, respectively.
4. Discussion
The application of the CSA analysis for the truncated system 1 with two chromophores helps to visualize how the two independent chromophores interact as parallel or antiparallel to split into two electronic transitions with a defined orientation of the EDTM (
Figure S1). The comparison of CSA and TD-DFT shows the reliability of the exciton coupling for the prediction of the main features of the ECD. In addition, CSA of 1 assist in understanding how the EDTM of the allowed transitions are oriented with respect to the system. This is of special interest when looking for chiroptical applications, for instance on the development of ordered monolayers for the construction of chiroptical surfaces [
11]. In this study, for instance, we can observe from the CSA that the A2 transition oriented parallel to the
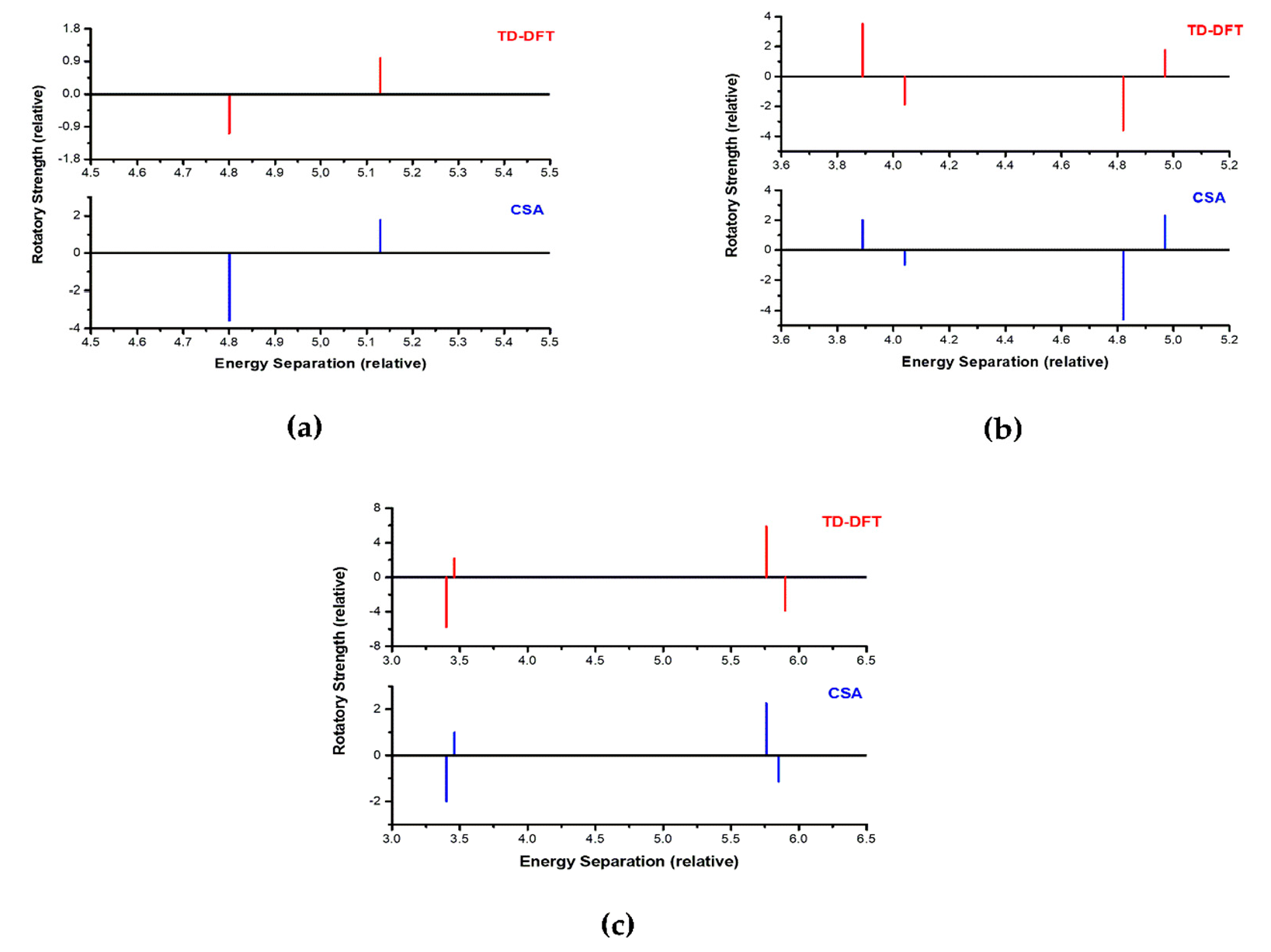
C3-axis has no comparable partner in the truncated system with only two chromophores. Notably, the EDTM and RS predicted with CSA are sufficient to reproduce the TD-DFT calculations well.
The chromophores in structure 2 present two significant electronic transitions with very distinct energies, 3.8879 eV and 4.8181 eV and relative orientations
θ = 99.7 and
θ = 77.4. Thus, CSA of the two transitions independently allows the prediction of the main features in two regions of the spectra. The CSA represented in
Figure 5 shows how the module of the EDTMs associated with each transition can be tuned by the relative orientation of the electronic transition of the independent chromophore with respect to the molecular system
θ.
Table 4 demonstrates how well the CSA compares with the TD-DFT predicted main parameters for the ECD.
Macrocycle 3 presents three significant electronic transitions with very distinct energies; however, the electronic transition 3.2 has a
θ c.a. 90°. As the sign of the RS changes by exceeding this angle and CSA is only an approximation, in this case, prediction by the CSA is not recommended. Taking this into account, the prediction was performed considering the other two electronic transitions 3.1 and 3.3. Finally, the main features of the ECD could be obtained by the CSA as depicted in
Table 6.
Remarkably, the
θ values for the two transitions under consideration for both
2 and
3 are one higher and the other lower than 90°. Since this fact renders a flip in the sign of the RS, the main features of the predicted ECD spectra present a positive and a negative exciton couplet at the same time in the same spectra at different energies (
Figure 7).

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}