Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
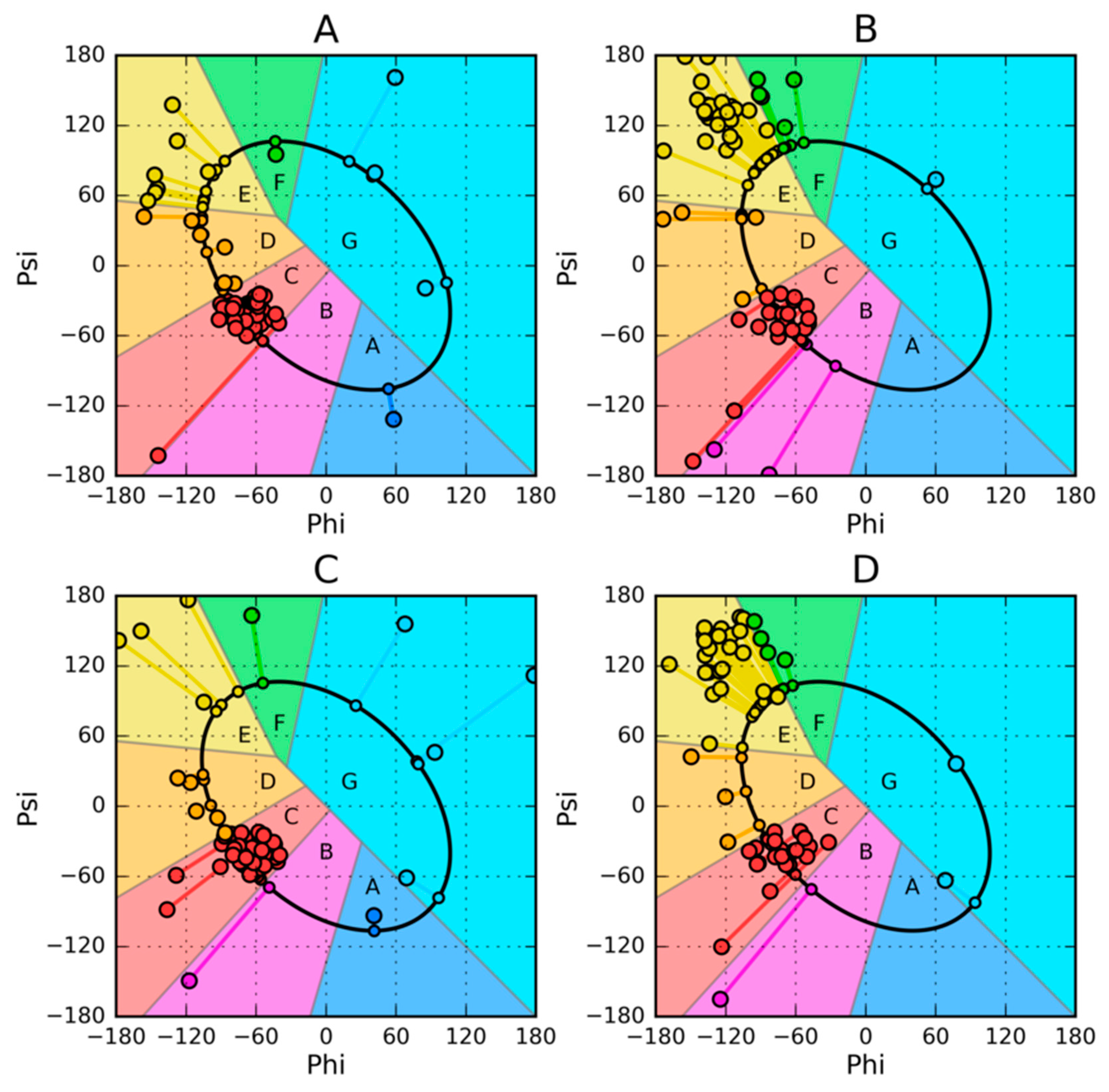
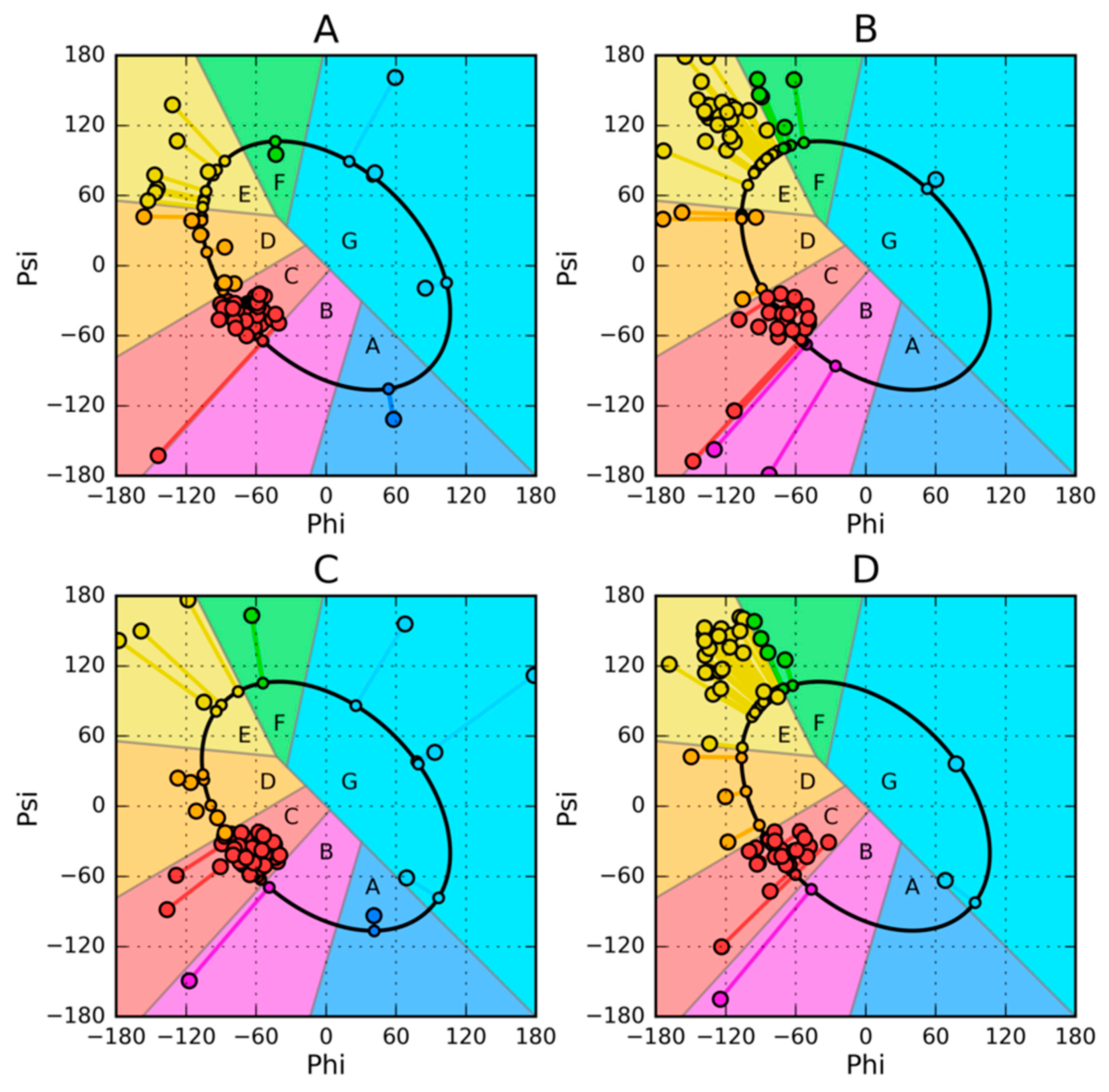
2.2. Model of Amino Acid Conformation Analysis in the Chain
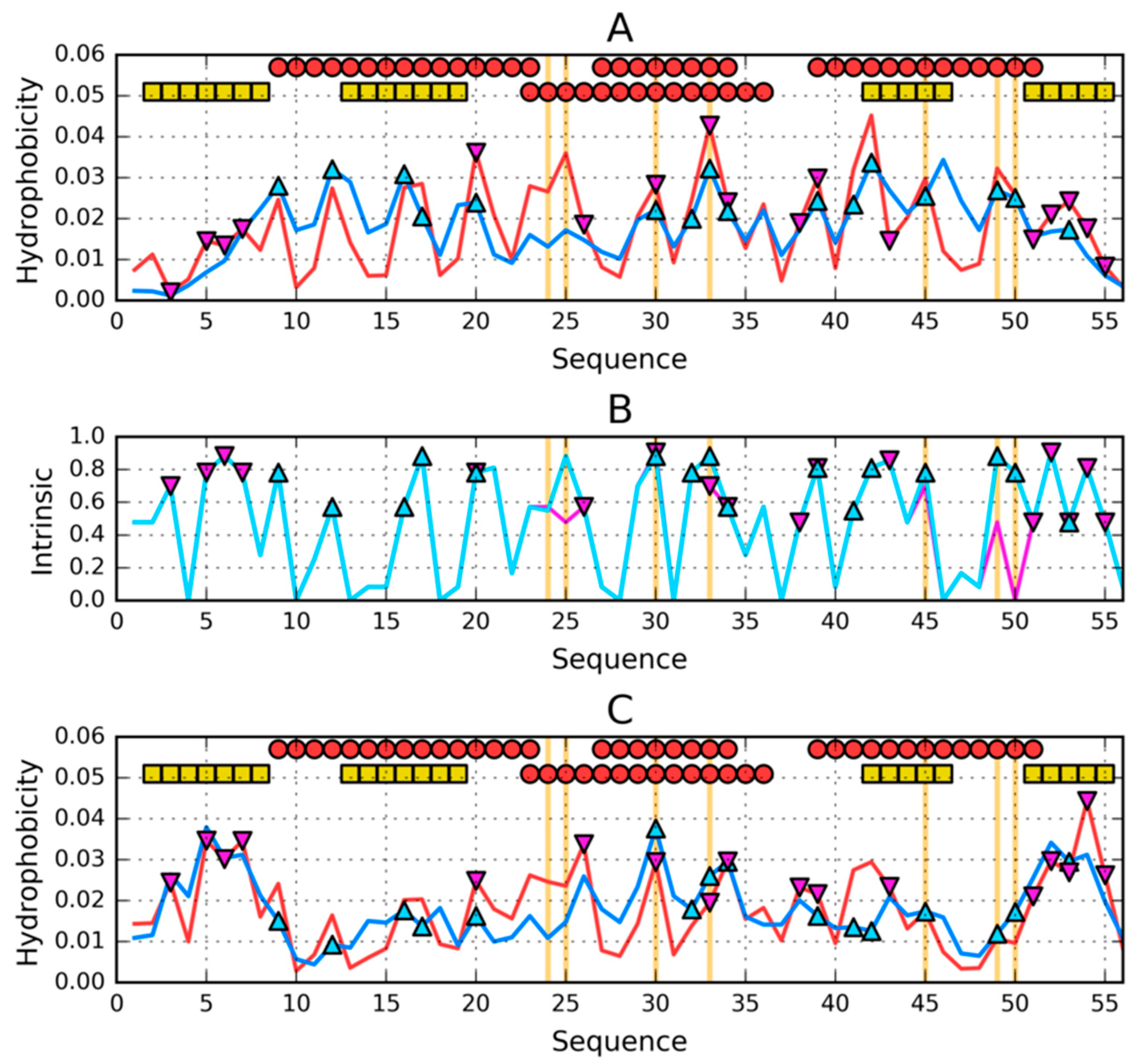
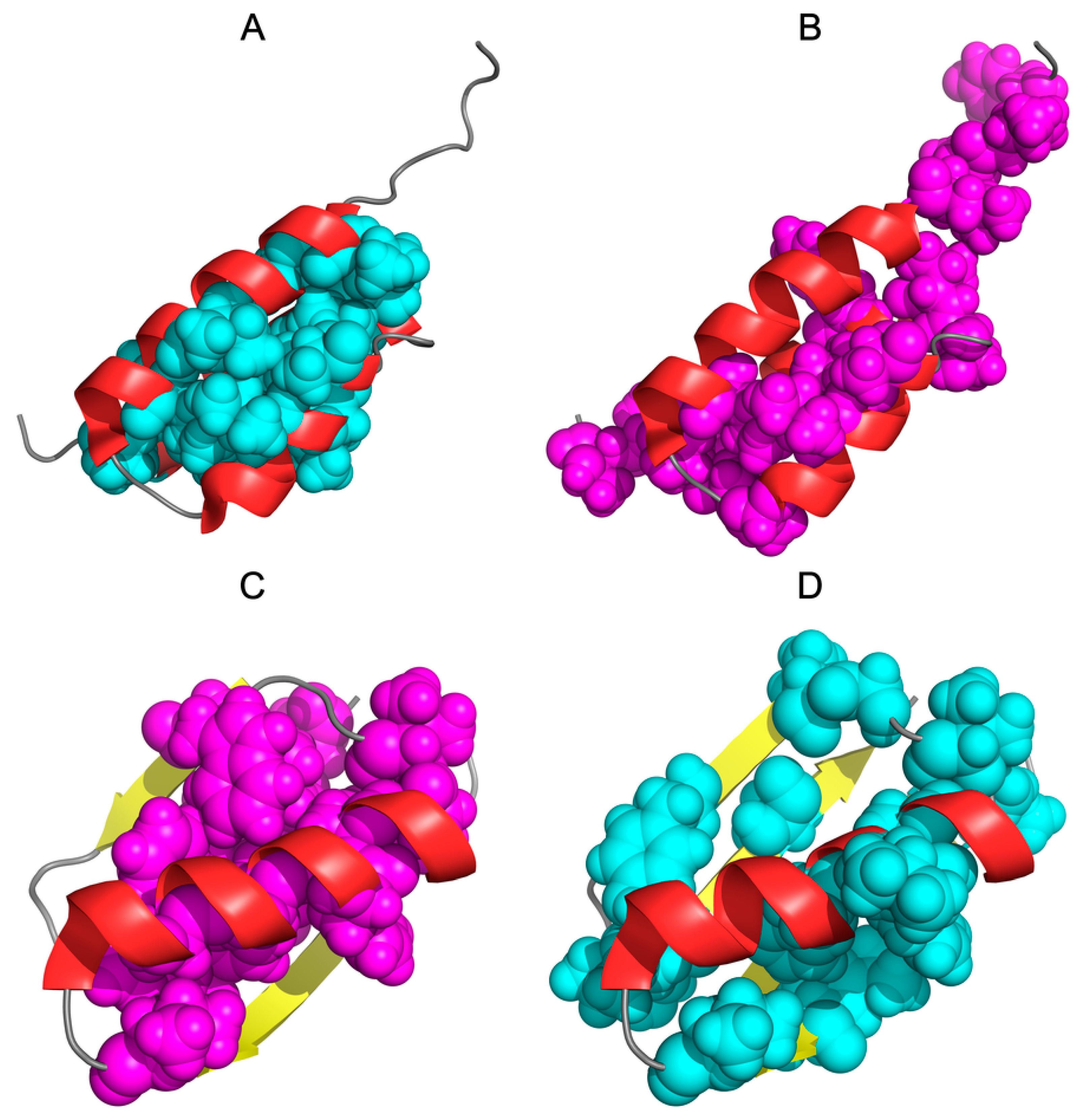
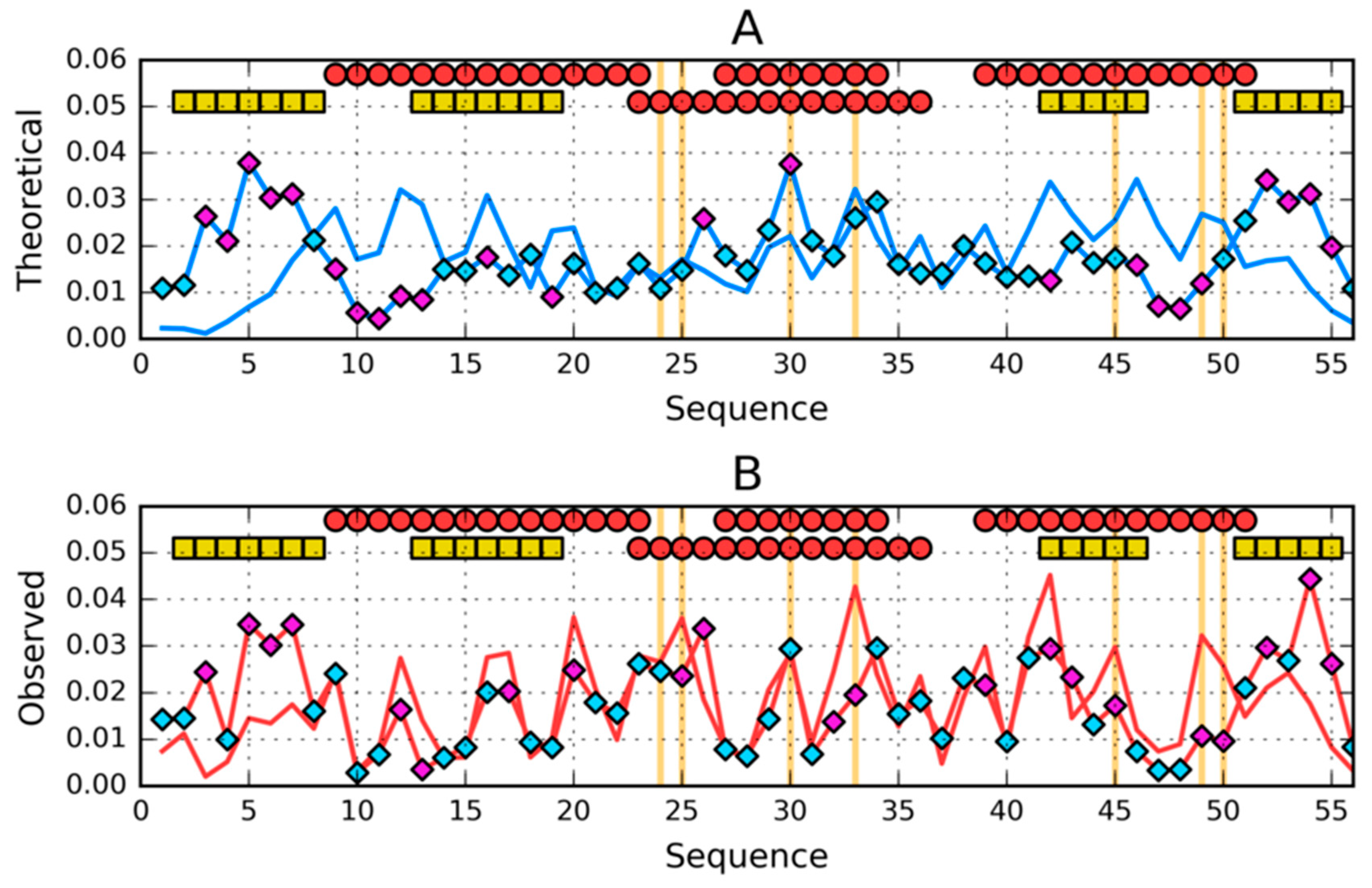
2.3. The Structure of the Hydrophobic Core in the Native Structure
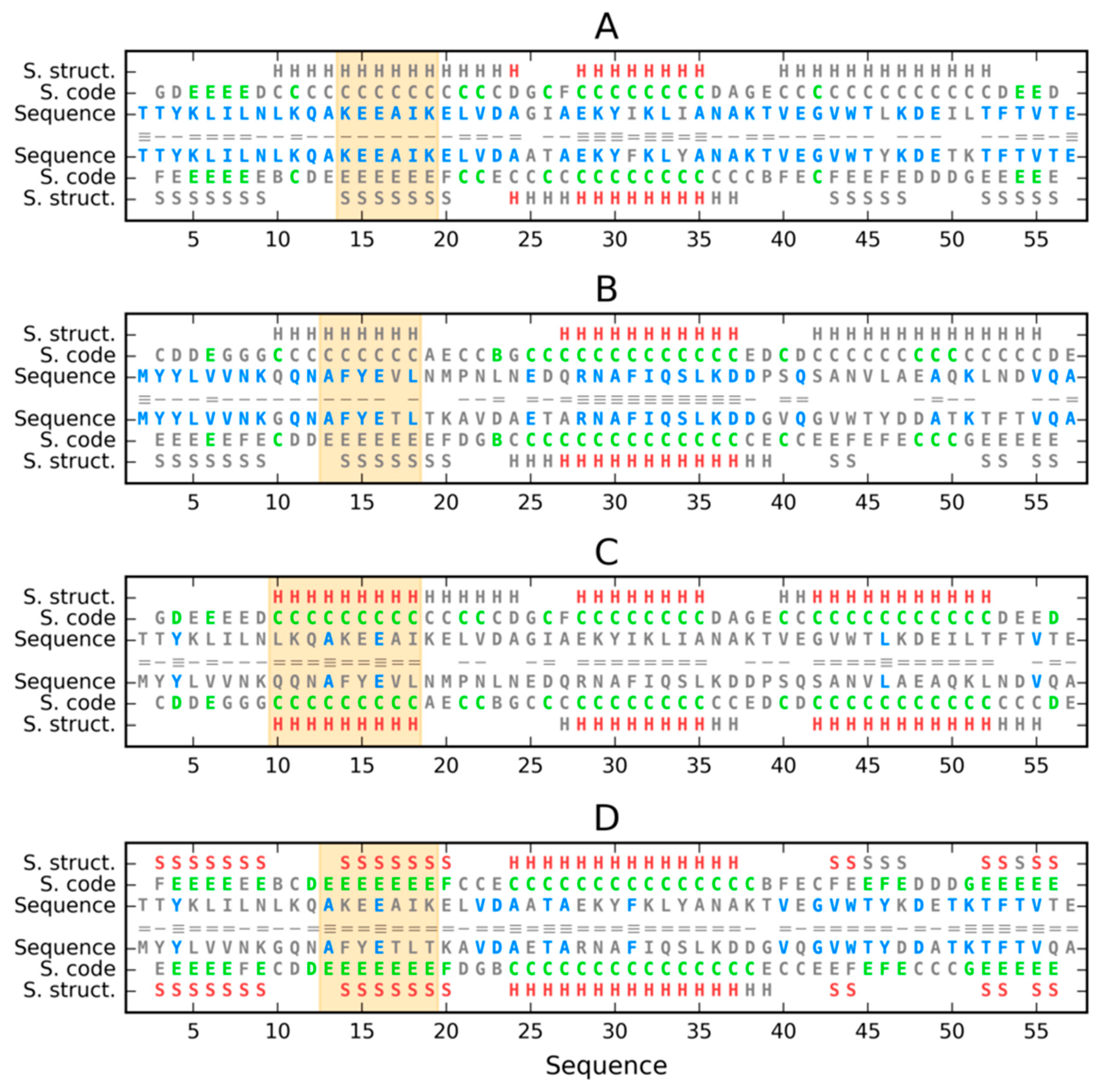
3. Results
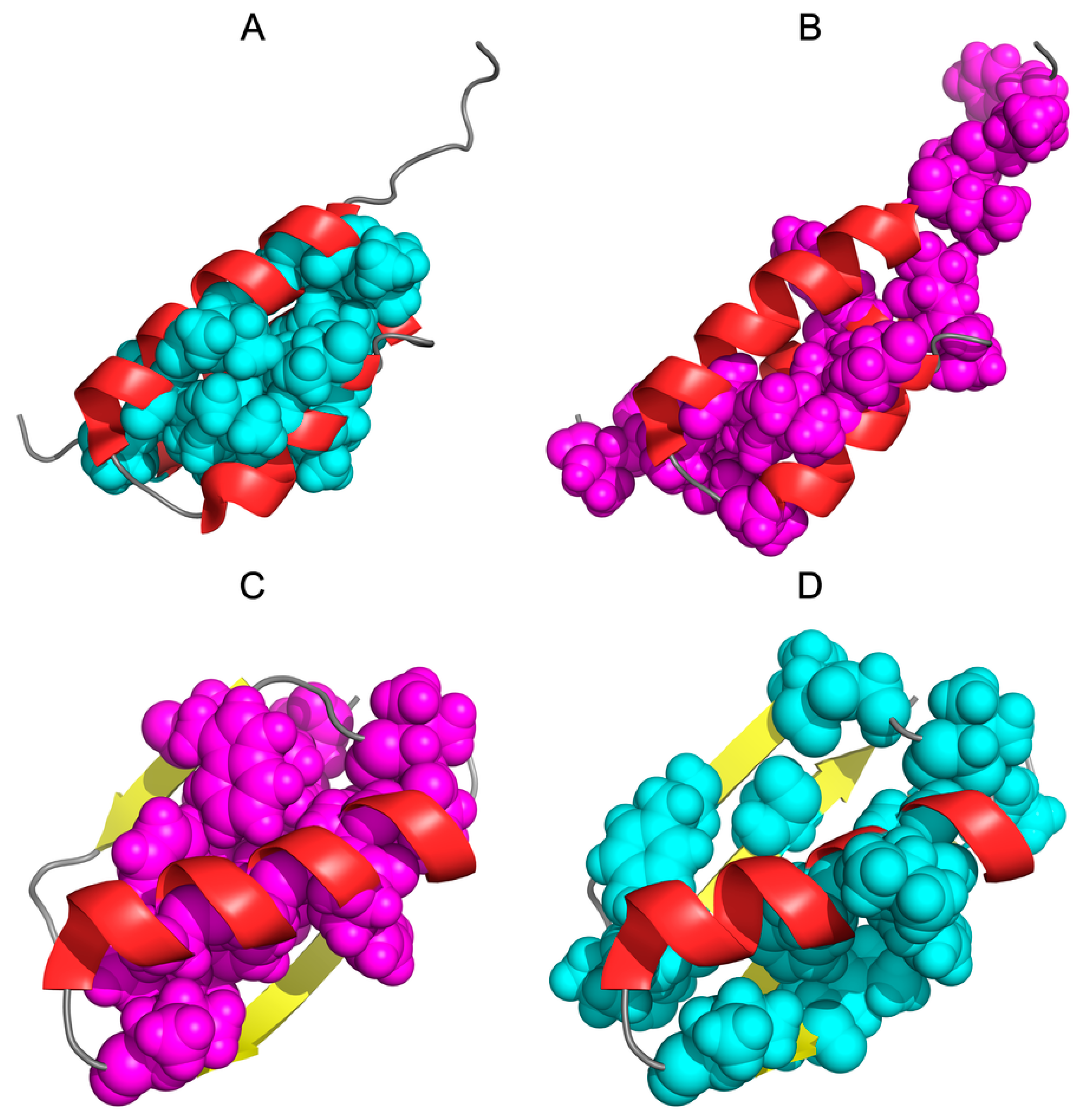
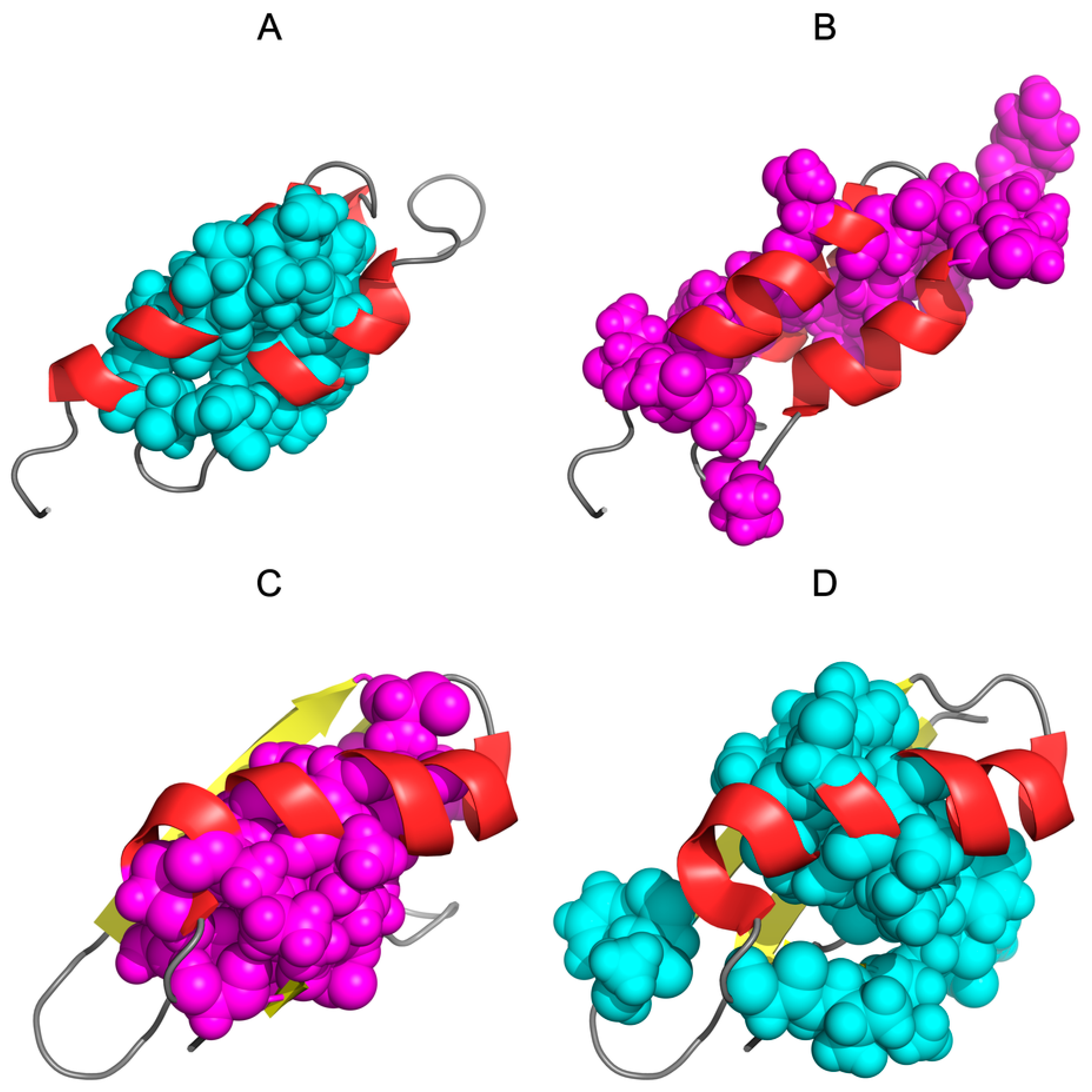
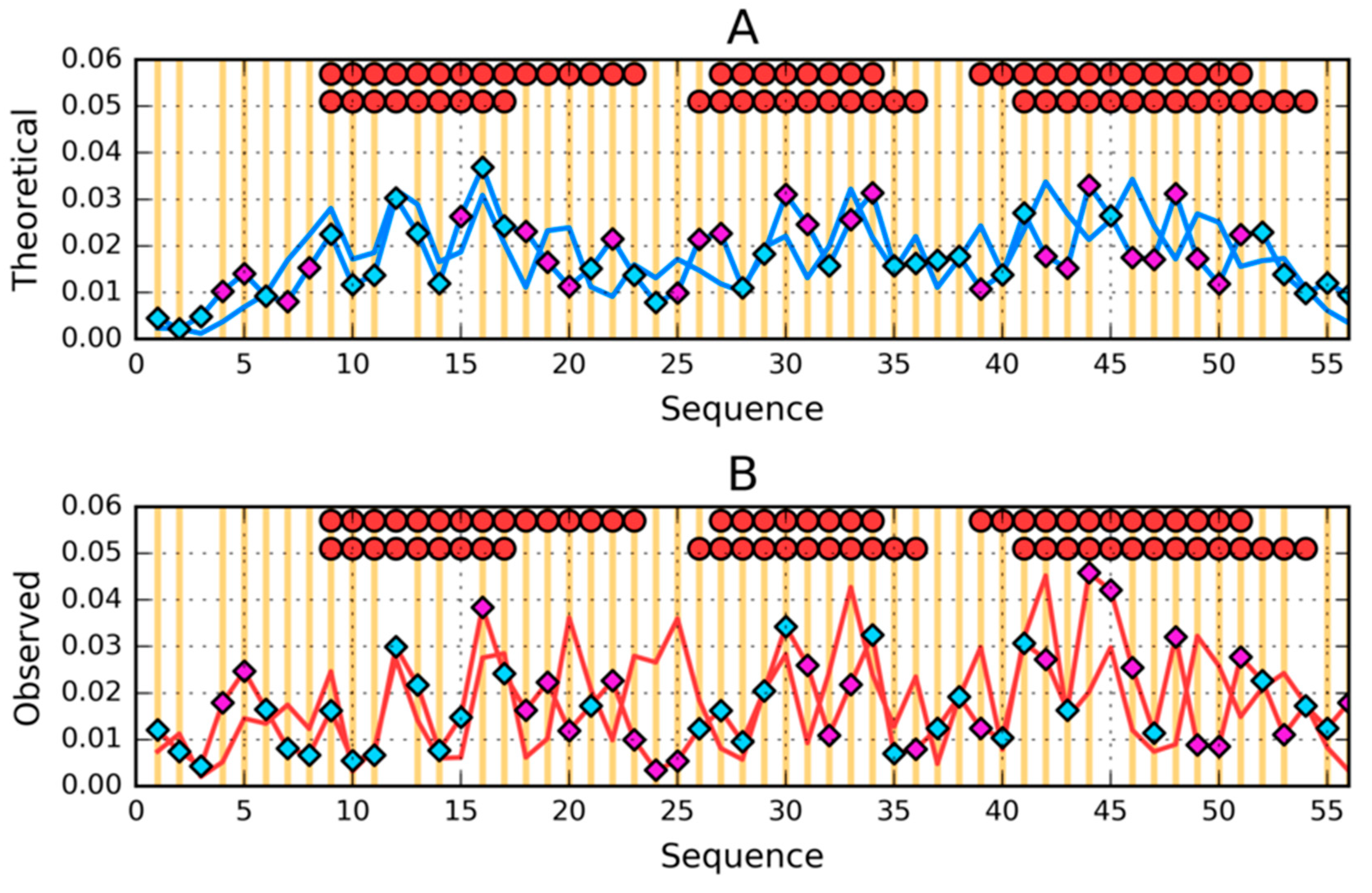
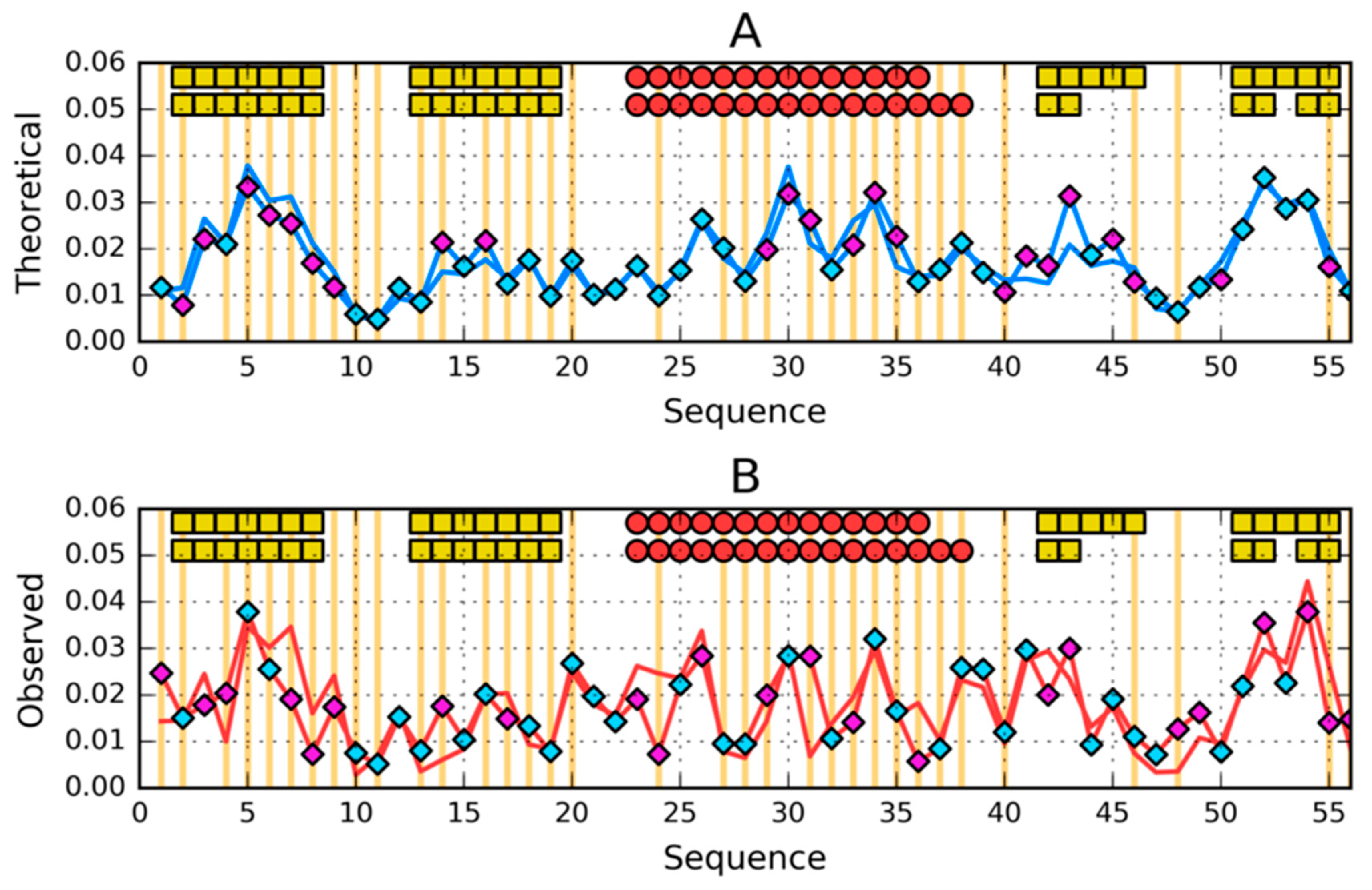
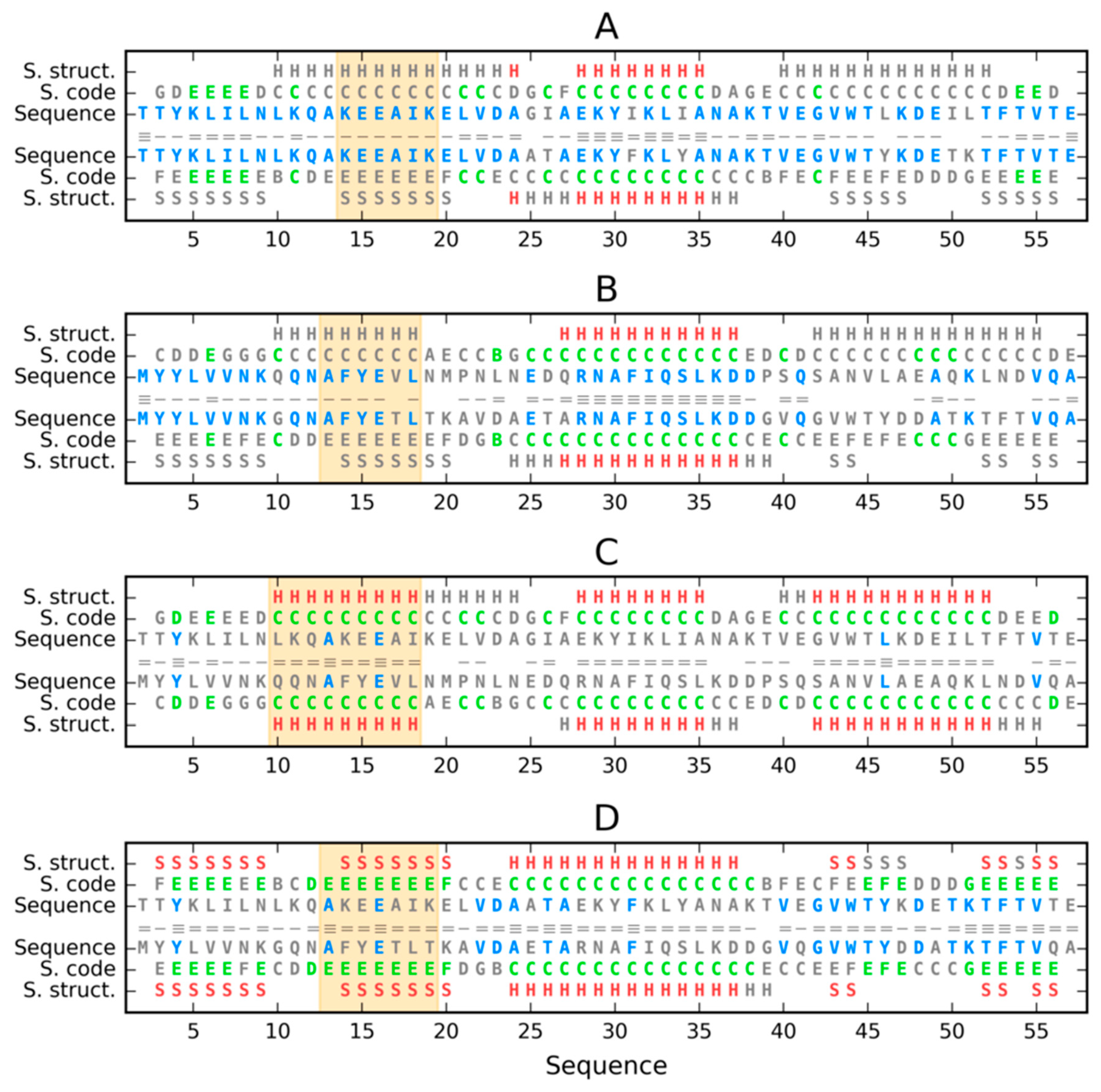
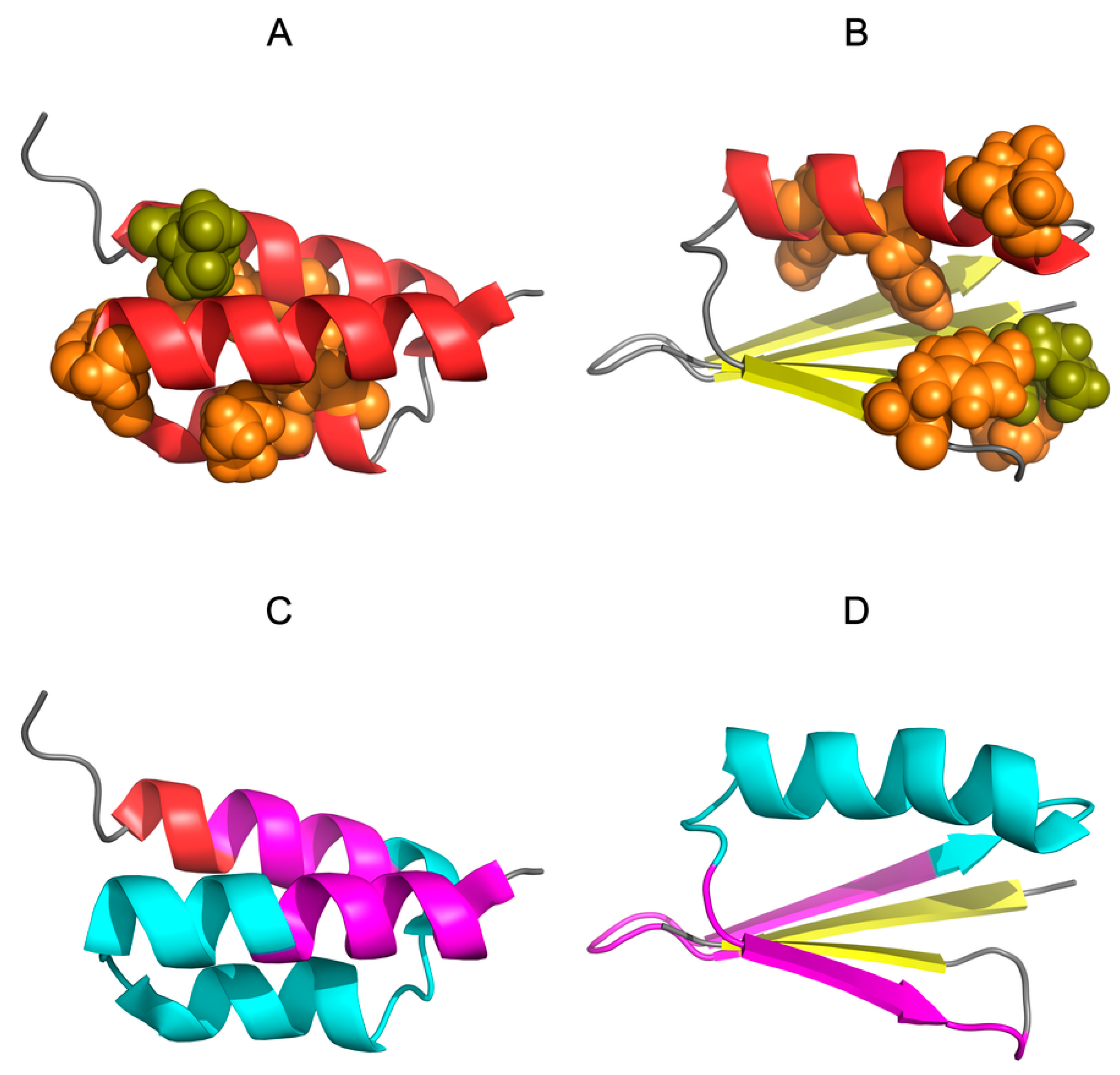
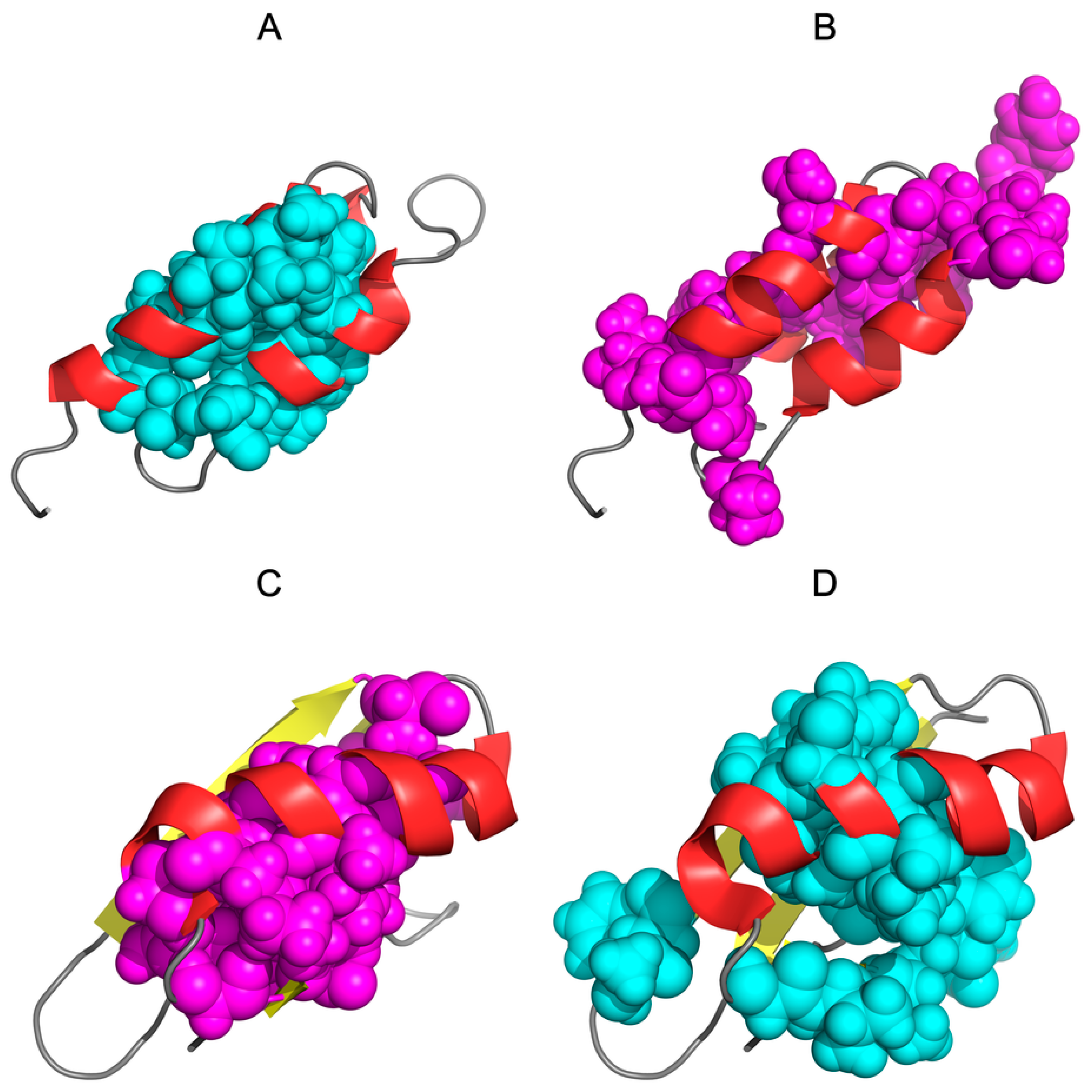
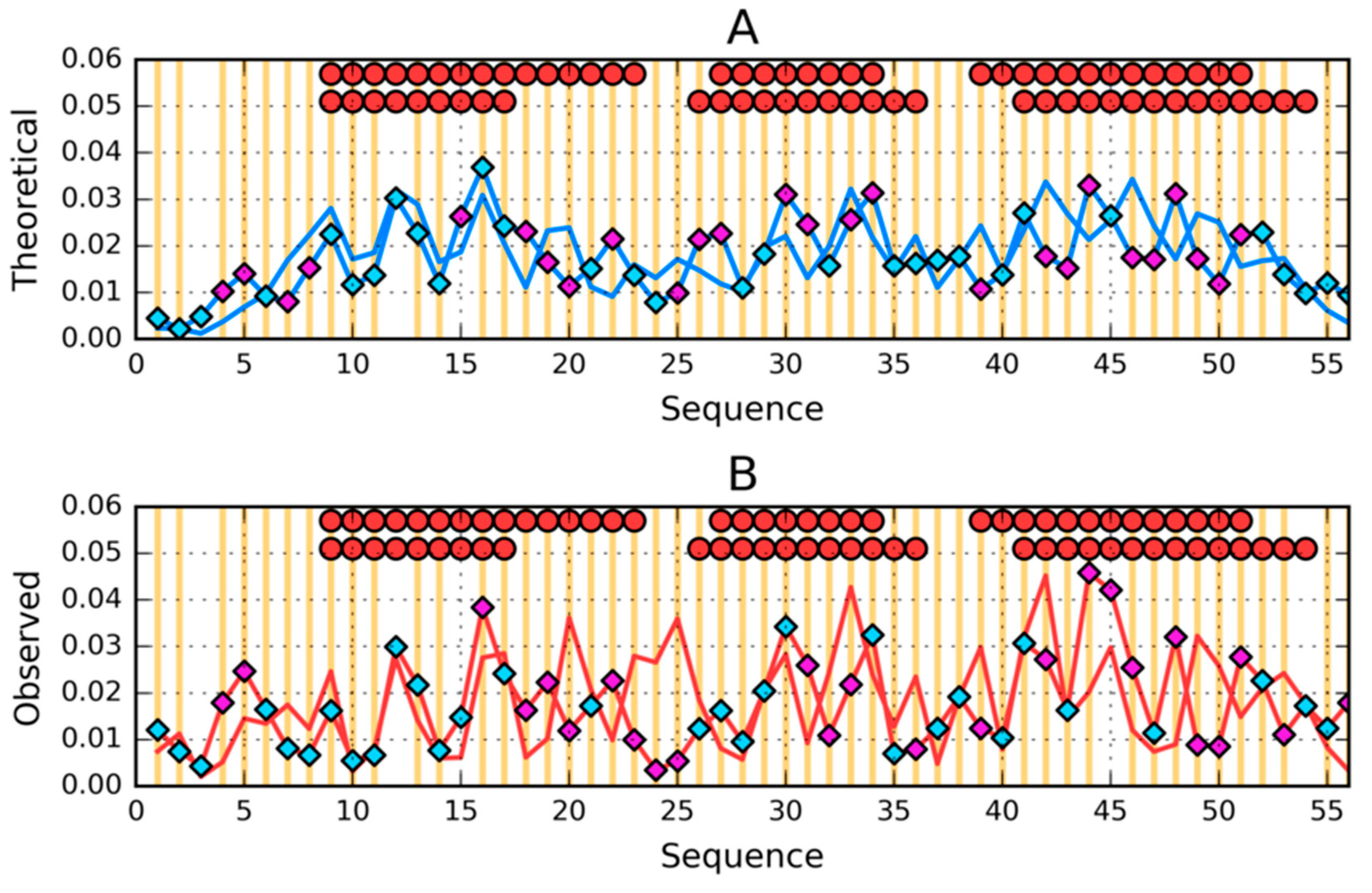
3.1. Similar Sequence—Different Structure 2JWU-2JWS
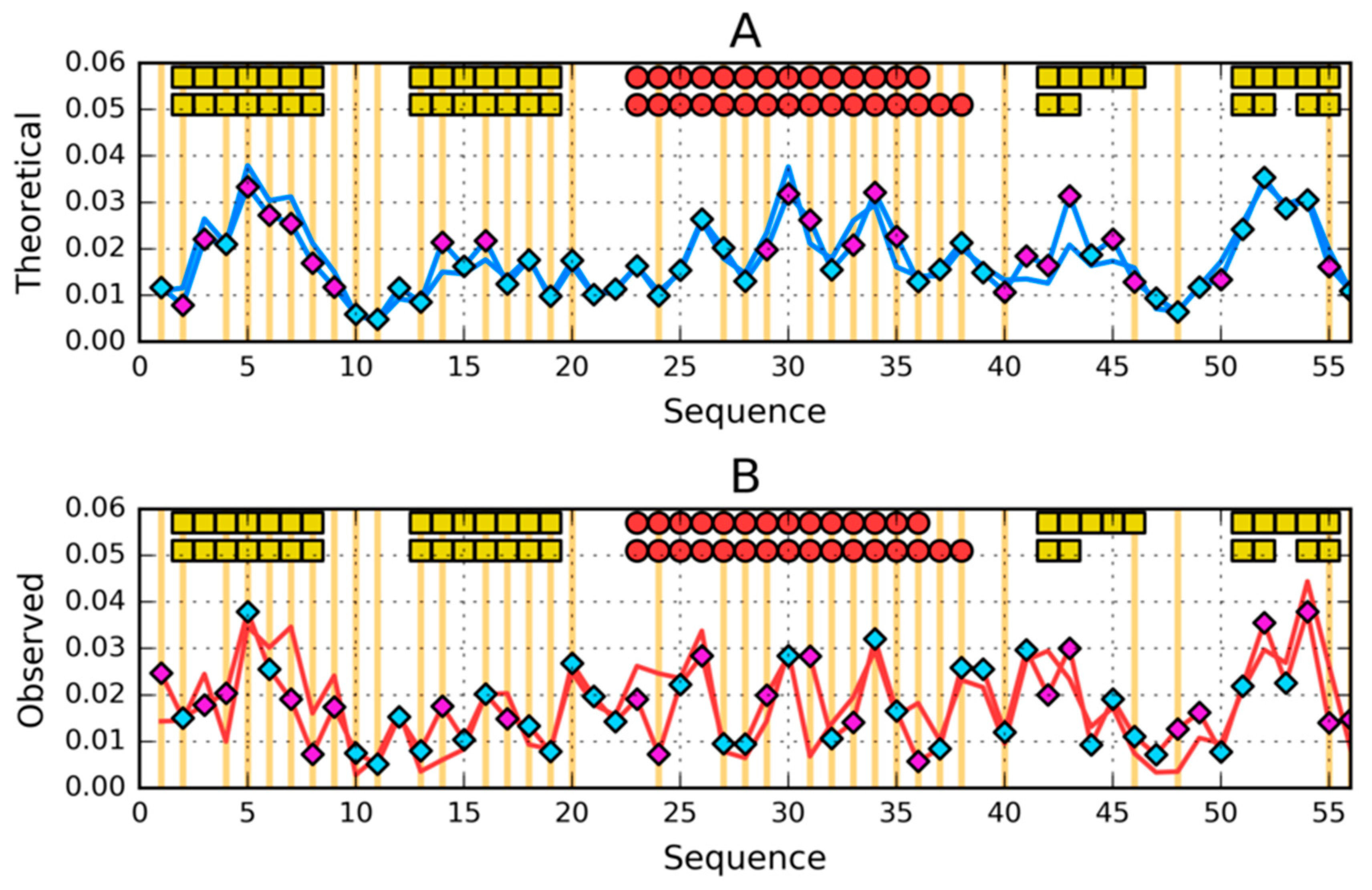
3.2. Highly Homologous Sequences—Different Three-Dimensional Structures
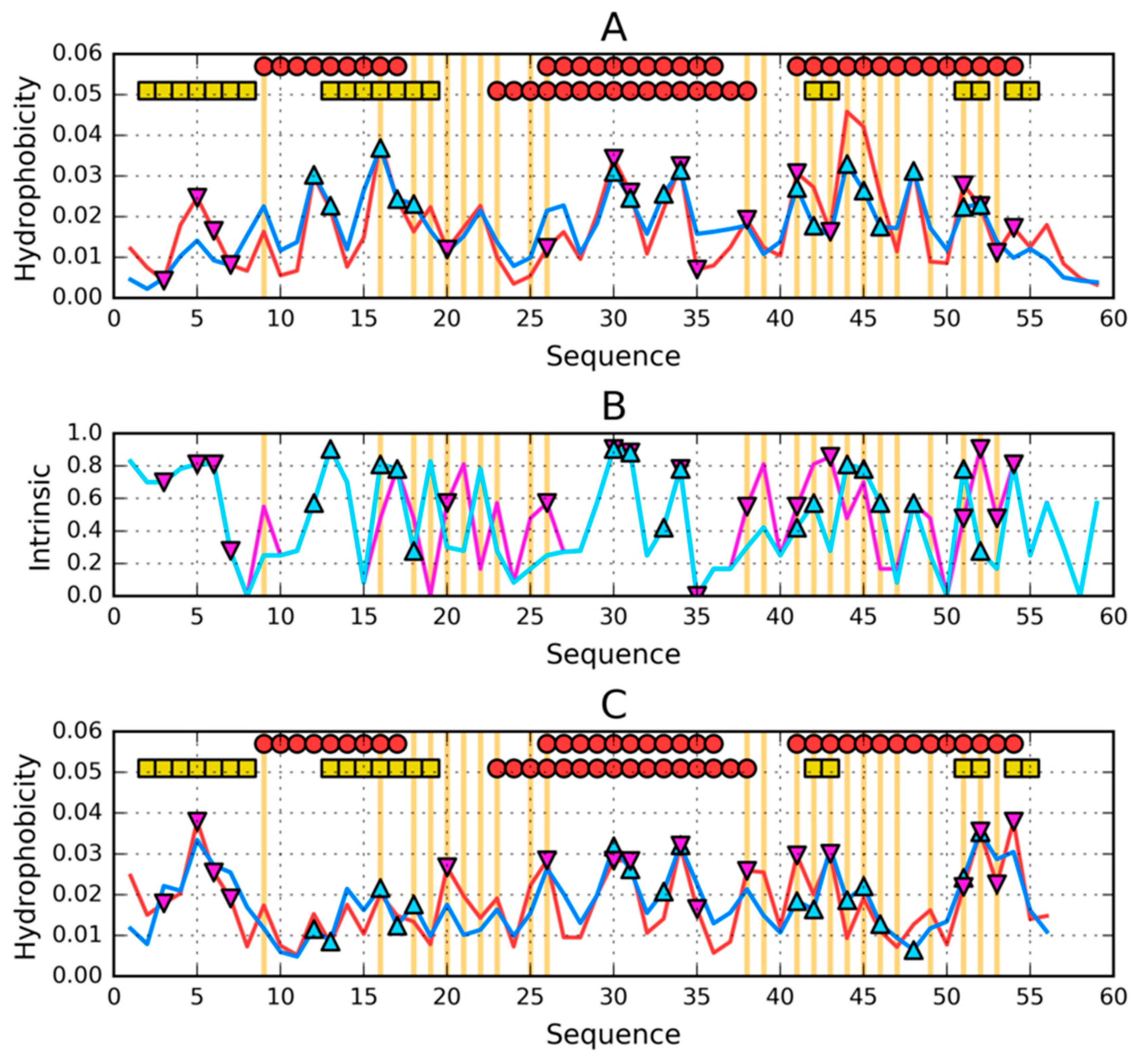
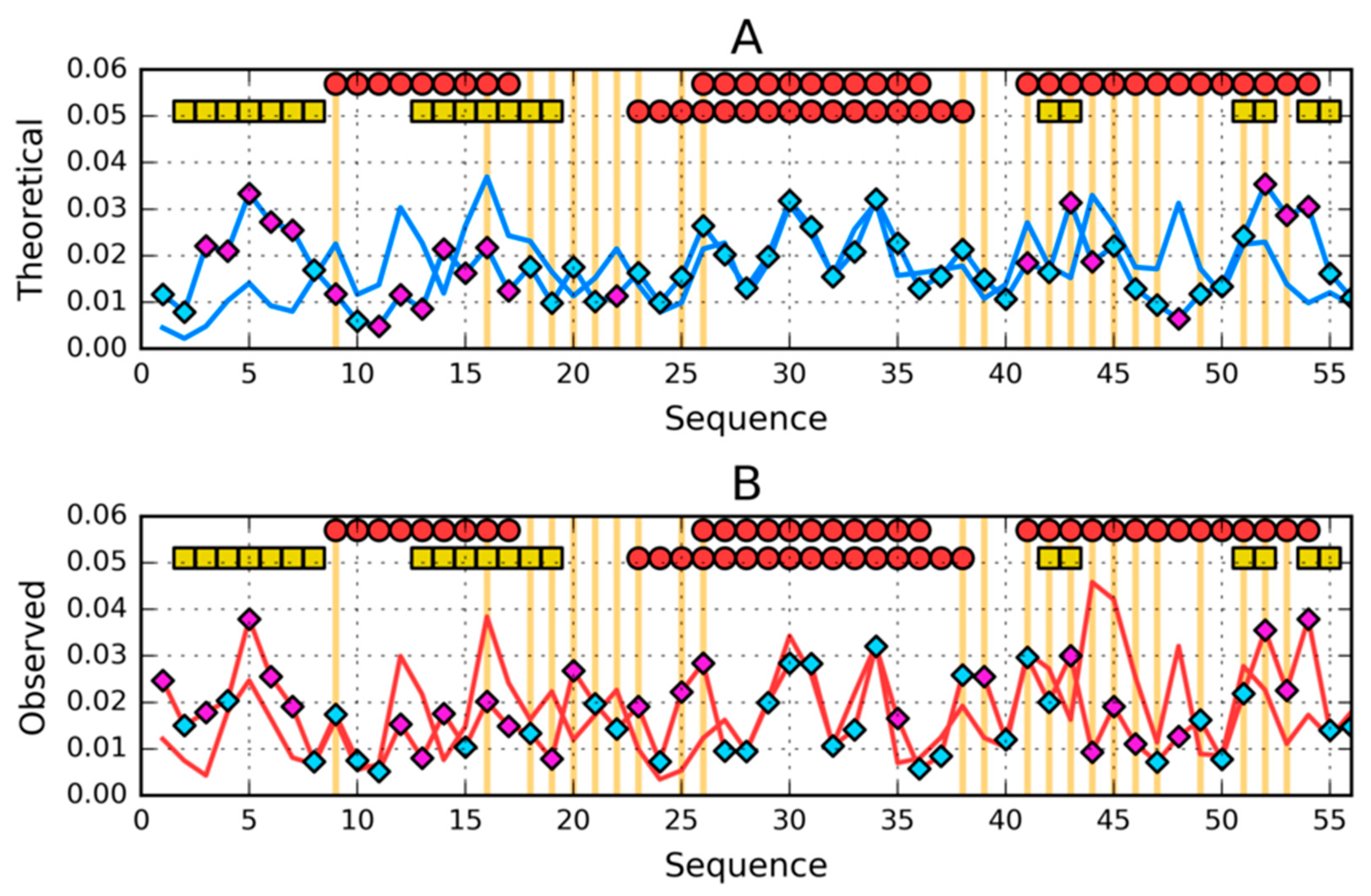
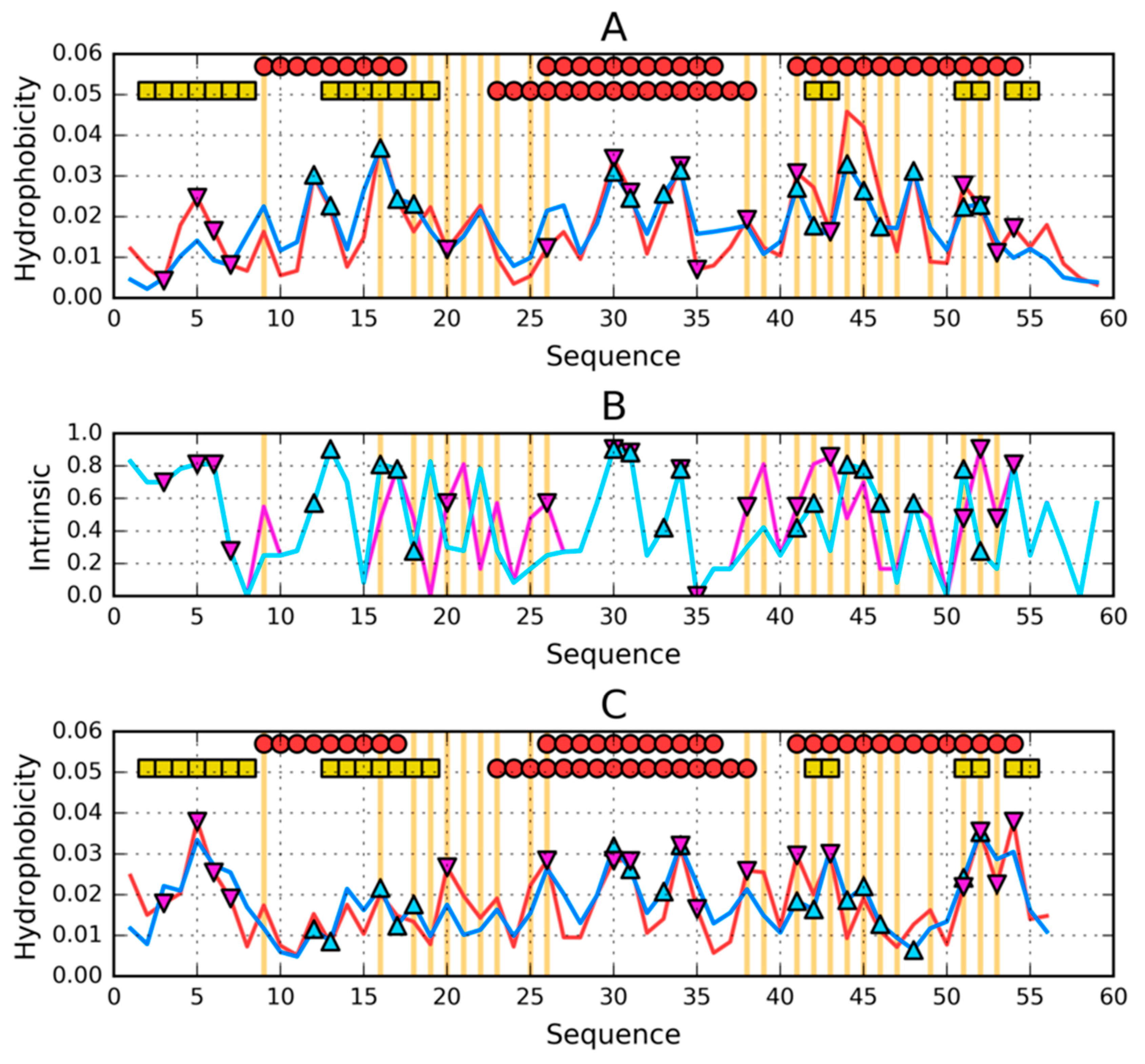
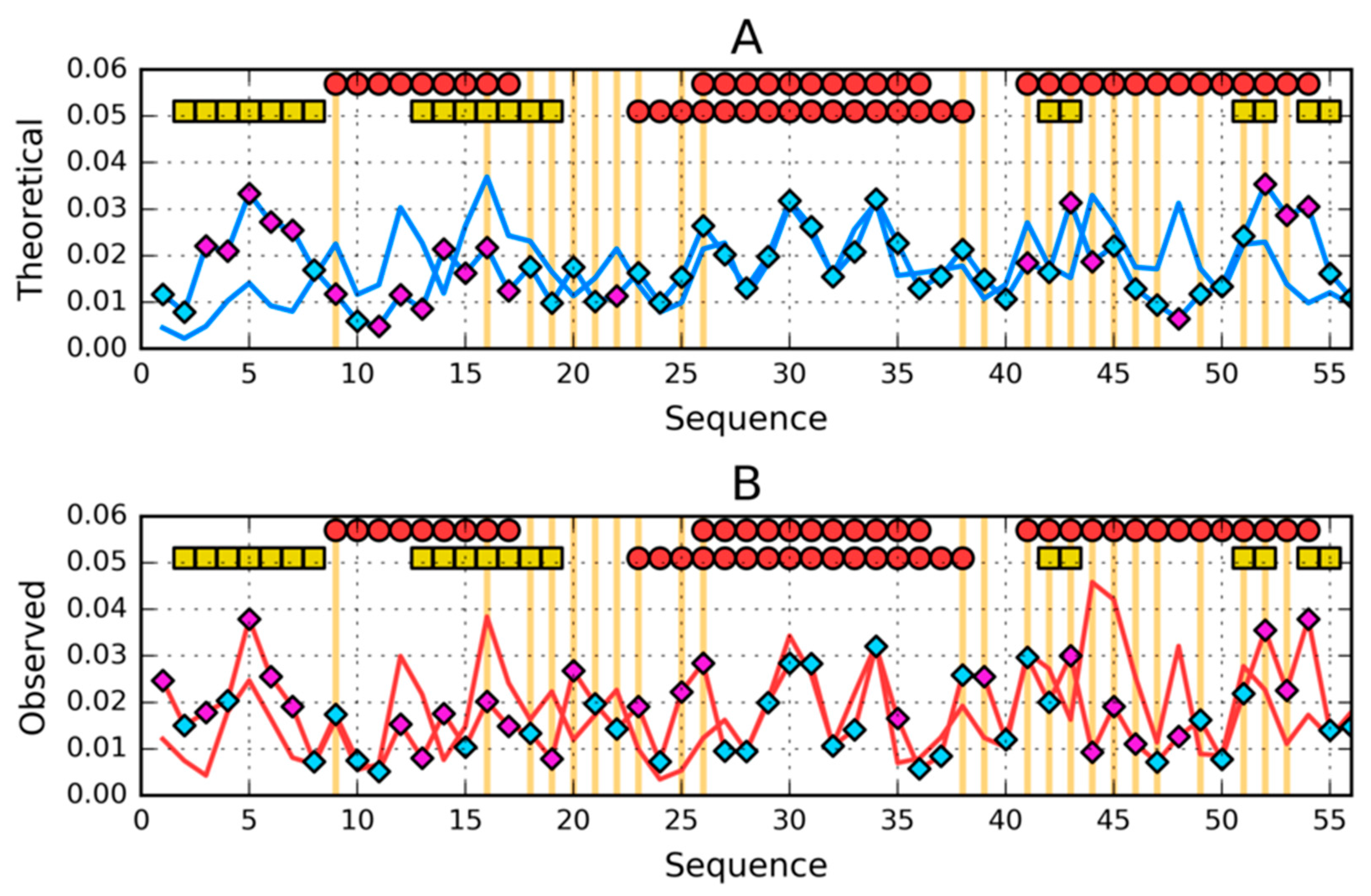
3.3. Comparative Cross-Analysis
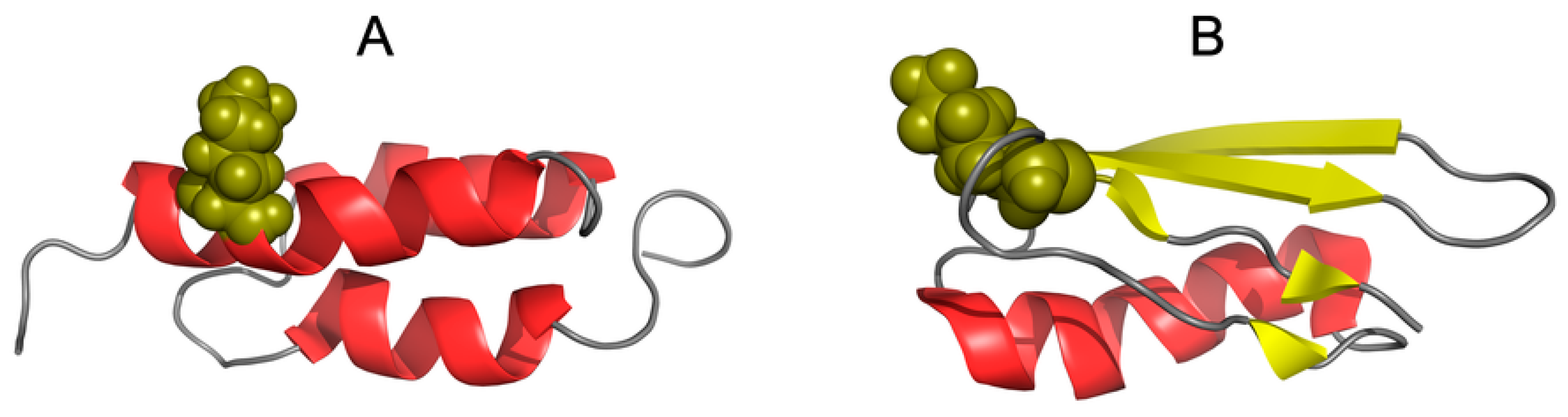
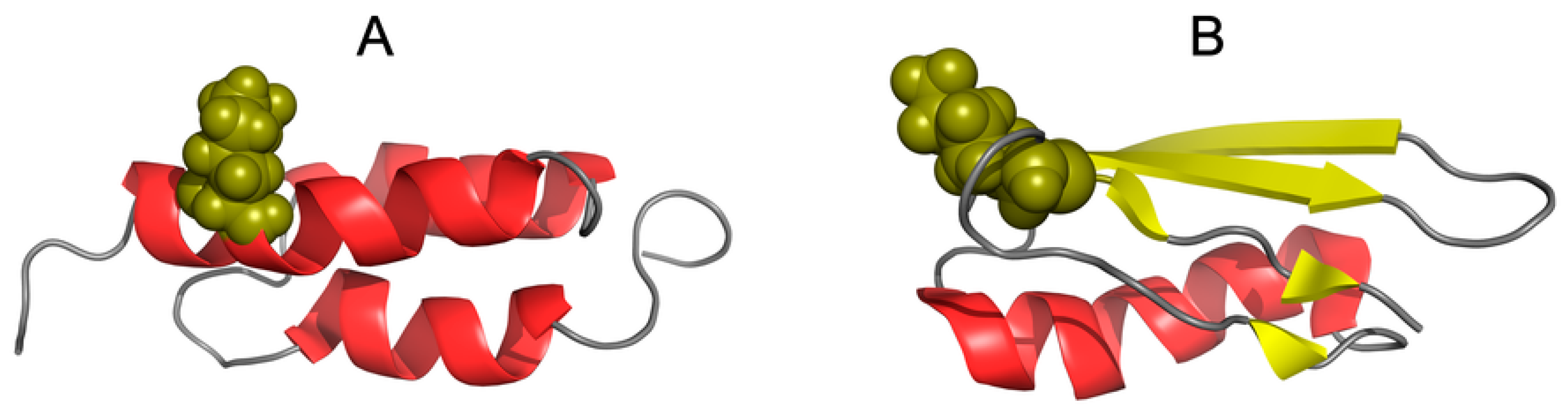
3.4. Status of Sections with a Specific Secondary Structure
3.5. Comparison of the Structural Code List
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dill, K.A.; MacCallum, J.L. The Protein-Folding Problem, 50 Years On. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Levinthal, C. Are there pathways for protein folding? J. Chim. Phys. 1968, 65, 44–45. [Google Scholar] [CrossRef]
- Levinthal, C. How to Fold Graciously. Mossbauer Spectroscopy in Biological Systems. In Proceedings of the Meeting Held at Allerton House, Monticello, IL, USA, 17–18 March 1969; University of Illinois Press: Urbana, IL, USA, 1969; Volume 67, pp. 22–24. [Google Scholar]
- Zwanzig, R.; Szabo, A.; Bagchi, B. Levinthal’s paradox. Proc. Natl. Acad. Sci. USA 1992, 89, 20–22. [Google Scholar] [CrossRef] [Green Version]
- Dill, K.A.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Struct. Mol. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef]
- Karplus, M. The Levinthal paradox: Yesterday and today. Fold. Des. 1997, 2, S69–S75. [Google Scholar] [CrossRef] [Green Version]
- Durup, J. On “Levinthal paradox” and the theory of protein folding. J. Mol. Struct. THEOCHEM 1998, 424, 157–169. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfinsen, C.B.; Scheraga, H.A. Experimental and Theoretical Aspects of Protein Folding. Adv. Protein Chem. 1975, 29, 205–300. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y. Hidden intermediates and levinthal paradox in the folding of small proteins. Biochem. Biophys. Res. Commun. 2003, 305, 785–788. [Google Scholar] [CrossRef]
- Chothia, C. Hydrophobic bonding and accessible surface area in proteins. Nature 1974, 248, 338–339. [Google Scholar] [CrossRef]
- Tanford, C. The hydrophobic effect and the organization of living matter. Science 1978, 200, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Tanford, C. Interfacial free energy and the hydrophobic effect. Proc. Natl. Acad. Sci. USA 1979, 76, 4175–4176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauzmann, W. Structural factors in protein denaturation. J. Cell. Physiol. Suppl. 1956, 47 (Suppl. 1), 113–131. [Google Scholar] [CrossRef] [PubMed]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Tanford, C. My debt to Walter Kauzmann. Biophys. Chem. 2003, 105, 159–160. [Google Scholar] [CrossRef]
- Ripoll, D.R.; Piela, L.; Váasquez, M.; Scheraga, H.A. On the multiple-minima problem in the conformational analysis of polypeptides. V. Application of the self-consistent electrostatic field and the electrostatically driven monte carlo methods to bovine pancreatic trypsin inhibitor. Proteins Struct. Funct. Genet. 1991, 10, 188–198. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K.; Moult, J.; Schwede, T.; Tramontano, A. Evaluation of the template-based modeling in CASP12. Proteins Struct. Funct. Bioinform. 2017, 86, 321–334. [Google Scholar] [CrossRef]
- Osguthorpe, D. Ab initio protein folding. Curr. Opin. Struct. Biol. 2000, 10, 146–152. [Google Scholar] [CrossRef]
- Available online: http://predictioncenter.org (accessed on 25 January 2020).
- Elofsson, A.; Joo, K.; Keasar, C.; Lee, J.; Maghrabi, A.H.A.; Manavalan, B.; McGuffin, L.J.; Hurtado, D.M.; Mirabello, C.; Pilstål, R.; et al. Methods for estimation of model accuracy in CASP12. Proteins Struct. Funct. Bioinform. 2017, 86, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K.; Schwede, T.; Tramontano, A. Assessment of model accuracy estimations in CASP12. Proteins Struct. Funct. Bioinform. 2017, 86, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Keasar, C.; McGuffin, L.J.; Wallner, B.; Chopra, G.; Adhikari, B.; Bhattacharya, D.; Blake, L.; Bortot, L.O.; Cao, R.; Dhanasekaran, B.K.; et al. An analysis and evaluation of the WeFold collaborative for protein structure prediction and its pipelines in CASP11 and CASP12. Sci. Rep. 2018, 8, 9939. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Liwo, A.; Khatib, F.; Zhou, H.; Chopra, G.; Bacardit, J.; Bortot, L.O.; Faccioli, R.A.; Deng, X.; He, Y.; et al. WeFold: A coopetition for protein structure prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 1850–1868. [Google Scholar] [CrossRef]
- Gregersen, N.; Bross, P.; Vang, S.; Christensen, J.H. Protein Misfolding and Human Disease. Annu. Rev. Genom. Hum. Genet. 2006, 7, 103–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, P.; Best, R.B. Structural Determinants of Misfolding in Multidomain Proteins. PLoS Comput. Biol. 2016, 12, e1004933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, F.; Iturria-Medina, Y.; Evans, A.C. Mathematical Modeling of Protein Misfolding Mechanisms in Neurological Diseases: A Historical Overview. Front. Neurol. 2018, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L. Amyloid structure. Essays Biochem. 2014, 56, 1–10. [Google Scholar] [CrossRef]
- Al-Garawi, Z.S.; Morris, K.L.; Marshall, K.E.; Eichler, J.; Serpell, L.C. The diversity and utility of amyloid fibrils formed by short amyloidogenic peptides. Interface Focus 2017, 7, 20170027. [Google Scholar] [CrossRef]
- Chothia, C. Principles that Determine the Structure of Proteins. Annu. Rev. Biochem. 1984, 53, 537–572. [Google Scholar] [CrossRef]
- Creighton, T.E.; Chothia, C. Selecting buried residues. Nature 1989, 339, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987, 196, 641–656. [Google Scholar] [CrossRef]
- Dill, K.A.; Flory, P.J. Molecular organization in micelles and vesicles. Proc. Natl. Acad. Sci. USA 1981, 78, 676–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sałapa, K.; Kalinowska, B.; Jadczyk, T.; Roterman, I. Measurement of Hydrophobicity Distribution in Proteins—Non-redundant Protein Data Bank. Bio-Algorithms Med.-Syst. 2012, 8, 327–338. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Is the hydrophobic core a universal structural element in proteins? J. Mol. Mod. 2017, 23, 205. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman-Konieczna, I. Ligand-binding-site recognition. In Protein Folding in Silico; Woodhead Publishing: Cambridge, UK, 2012; pp. 79–93. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman-Konieczna, I. Can the Structure of the Hydrophobic Core Determine the Complexation Site. In Identification of Ligand Binding Site and Protein-Protein Interaction Area; Roterman-Konieczna, I., Ed.; Springer—Focus on Structural Biology: Berlin/Heidelberg, Germany, 2012; pp. 41–54. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman-Konieczna, I. Use of the “fuzzy oil drop” model to identify the complexation area in protein homodimers. In Protein Folding in Silico; Roterman-Konieczna, I., Ed.; Woodhead Publishing: Cambridge, UK, 2012; pp. 95–122. [Google Scholar] [CrossRef]
- Lesk, A.M.; Chothia, C. Solvent accessibility, protein surfaces, and protein folding. Biophys. J. 1980, 32, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, M.; Chothia, C. Packing at the protein-water interface. Proc. Natl. Acad. Sci. USA 1996, 93, 10167–10172. [Google Scholar] [CrossRef] [Green Version]
- Chiche, L.; Gregoret, L.M.; Cohen, F.E.; Kollman, P.A. Protein model structure evaluation using the solvation free energy of folding. Proc. Natl. Acad. Sci. USA 1990, 87, 3240–3243. [Google Scholar] [CrossRef] [Green Version]
- Fisher, H.F. A limiting law relating the size and shape of protein molecules to their composition. Proc. Natl. Acad. Sci. USA 1964, 51, 1285–1291. [Google Scholar] [CrossRef] [Green Version]
- Tanford, C. How protein chemists learned about the hydrophobic factor. Protein Sci. 1997, 6, 1358–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, L.; Brylinski, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. Silico Biol. 2006, 6, 15–22. [Google Scholar]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Fabian, P.; Stapor, K.; Banach, M.; Ptak-Kaczor, M.; Konieczny, L.; Roterman, I. Different Synergy in Amyloids and Biologically Active Forms of Proteins. Int. J. Mol. Sci. 2019, 20, 4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dułak, D.; Gadzała, M.; Banach, M.; Ptak, M.; Wisniowski, Z.; Konieczny, L.; Roterman, I. Filamentous Aggregates of Tau Proteins Fulfil Standard Amyloid Criteria. Int. J. Mol. Sci. 2018, 19, 2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roterman, I.; Dułak, D.; Gadzała, M.; Banach, M.; Konieczny, L. Structural analysis of the Aβ(11–42) amyloid fibril based on hydrophobicity distribution. J. Comput. Aided Mol. Des. 2019, 33, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dułak, D.; Banach, M.; Gadzała, M.; Konieczny, L.; Roterman, I. Structural analysis of the Aβ(15-40) amyloid fibril based on hydrophobicity distribution. Acta Biochim. Pol. 2018, 65, 595–604. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Chen, Y.; Alexander, P.; Bryan, P.; Orban, J. NMR structures of two designed proteins with 88% sequence identity but different fold and function. Proc. Natl. Acad. Sci. USA 2008, 105, 14412–14417. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Yeh, D.C.; Alexander, P.; Bryan, P.N.; Orban, J. Solution NMR Structures of IgG Binding Domains with Artificially Evolved High Levels of Sequence Identity but Different Folds. Biochemistry 2005, 44, 14055–14061. [Google Scholar] [CrossRef]
- Dawson, N.L.; Lewis, T.E.; Das, S.; Lees, J.G.; Lee, D.; Ashford, P.; Orengo, C.A.; Sillitoe, I. CATH: An expanded resource to predict protein function through structure and sequence. Nucleic Acids Res. 2016, 45, D289–D295. [Google Scholar] [CrossRef]
- Banach, M.; Kalinowska, B.; Konieczny, L.; Roterman, I. Structural Role of Hydrophobic Core in Proteins-Selected Examples. J. Proteom. Bioinform. 2016, 9, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Jurkowski, W.; Baster, Z.; Dułak, D.; Roterman-Konieczna, I. The early-stage intermediate. In Protein Folding in Silico; Woodhead Publishing: Cambridge, UK, 2012; pp. 1–20. [Google Scholar] [CrossRef]
- Kalinowska, B.; Fabian, P.; Stąpor, K.; Roterman, I. Statistical dictionaries for hypothetical in silico model of the early-stage intermediate in protein folding. J. Comput. Aided Mol. Des. 2015, 29, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roterman, I. The geometrical analysis of peptide backbone structure and its local deformations. Biochimie 1995, 77, 204–216. [Google Scholar] [CrossRef]
- Roterman, I. Modelling the Optimal Simulation Path in the Peptide Chain Folding–Studies Based on Geometry of Alanine Heptapeptide. J. Theor. Biol. 1995, 177, 283–288. [Google Scholar] [CrossRef]
- Roterman, I.; Konieczny, L.; Jurkowski, W.; Prymula, K.; Banach, M. Two-intermediate model to characterize the structure of fast-folding proteins. J. Theor. Biol. 2011, 283, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Alexander, P.A.; He, Y.; Chen, Y.; Orban, J.; Bryan, P.N. The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc. Natl. Acad. Sci. USA 2007, 104, 11963–11968. [Google Scholar] [CrossRef] [Green Version]
- De Beer, T.A.P.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2013, 42, D292–D296. [Google Scholar] [CrossRef] [Green Version]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- He, Y.; Chen, Y.; Alexander, P.A.; Bryan, P.N.; Orban, J. Mutational tipping points for switching protein folds and functions. Structure 2012, 20, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN | PDB ID [Ref] | SEQUENCE IDENTITY | Length | Secondary Structure | Description |
|---|---|---|---|---|---|
| Highly similar sequences—different 3D structures | |||||
| De novo designed | 2JWS [52] | 87.5% | 56 | α + RC | 1.10.8.40 Alpha Orthogonal Bundle |
| De novo designed | 2JWU [52] | 56 | β + α | 3.10.20.10 Alpha-Beta Roll | |
| Highly homologous sequences—different three-dimensional structures | |||||
| G311 | 1ZXH [53] | 59% | 56 | α + β | 3.10.20.10 Alpha Beta Roll |
| A219 | 1ZXG [53] | 59 | α | 1.20.5.420 Mainly Alpha Up-down bundle | |
| 2JWU | 2JWS | 1ZXH | 1ZXG | ||||
|---|---|---|---|---|---|---|---|
| Fragment | RD | Fragment | RD | Fragment | RD | Fragment | RD |
| 1–N | 0.335 | 1–N | 0.370 | 1–N | 0.310 | 1–N | 0.248 |
| 1–9 β | 0.249 | 1–7 RC | 0.493 | 1–9 β | 0.471 | 1–7 RC | 0.308 |
| 12–20 β | 0.441 | 8–24 H | 0.449 | 12–20 β | 0.305 | 8–18 H | 0.205 |
| 22–37 H | 0.418 | 26–35 H | 0.180 | 22–39 H | 0.313 | 25–37 H | 0.211 |
| 42–47 β | 0.558 | 38–52 H | 0.418 | 42–44 β | 0.266 | 40–55 H | 0.241 |
| 50–56 β | 0.268 | 53–56 RC | 0.054 | 50–53 β | 0.179 | 56–59 RC | 0.168 |
| 1–25 | 0.345 | 1-25 | 0.432 | 1–25 | 0.348 | 1–25 | 0.296 |
| 26–35 | 0.280 | 26–35 | 0.180 | 26–35 | 0.229 | 26–35 | 0.205 |
| 36–N | 0.318 | 36–N | 0.342 | 36–N | 0.306 | 36–N | 0.215 |
| DISTRIBUTION | CORRELATION COEFFICIENT | |||||
|---|---|---|---|---|---|---|
| 2JWS–2JWU | 2JWS–1ZXG | 2JWS–1ZXH | 2JWU–1ZXG | 2JWU–1ZXH | 1ZXG–1ZXH | |
| Residues 1–25 | ||||||
| HvH | 0.969 | −0.056 | 0.337 | −0.003 | 0.351 | 0.554 |
| OvO | 0.524 | 0.213 | 0.270 | 0.009 | 0.656 | 0.225 |
| TvT | −0.315 | 0.707 | −0.226 | −0.196 | 0.943 | −0.066 |
| Residues 26–35 | ||||||
| HvH | 0.987 | 0.189 | 0.226 | 0.217 | 0.261 | 0.945 |
| OvO | 0.545 | 0.415 | 0.154 | 0.401 | 0.673 | 0.681 |
| TvT | 0.573 | 0.503 | 0.241 | 0.846 | 0.801 | 0.870 |
| Residues 36–56 | ||||||
| HvH | 0.823 | 0.041 | 0.560 | 0.262 | 0.790 | 0.486 |
| OvO | 0.417 | 0.189 | 0.299 | 0.026 | 0.810 | 0.123 |
| TvT | −0.224 | 0.243 | −0.077 | −0.159 | 0.905 | 0.003 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fabian, P.; Stapor, K.; Banach, M.; Ptak-Kaczor, M.; Konieczny, L.; Roterman, I. Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy. Symmetry 2020, 12, 273. https://doi.org/10.3390/sym12020273
Fabian P, Stapor K, Banach M, Ptak-Kaczor M, Konieczny L, Roterman I. Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy. Symmetry. 2020; 12(2):273. https://doi.org/10.3390/sym12020273
Chicago/Turabian StyleFabian, Piotr, Katarzyna Stapor, Mateusz Banach, Magdalena Ptak-Kaczor, Leszek Konieczny, and Irena Roterman. 2020. "Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy" Symmetry 12, no. 2: 273. https://doi.org/10.3390/sym12020273
APA StyleFabian, P., Stapor, K., Banach, M., Ptak-Kaczor, M., Konieczny, L., & Roterman, I. (2020). Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy. Symmetry, 12(2), 273. https://doi.org/10.3390/sym12020273