Relativistic Fock Space Coupled Cluster Method for Many-Electron Systems: Non-Perturbative Account for Connected Triple Excitations

 , , and
, , and
Abstract
:1. Introduction
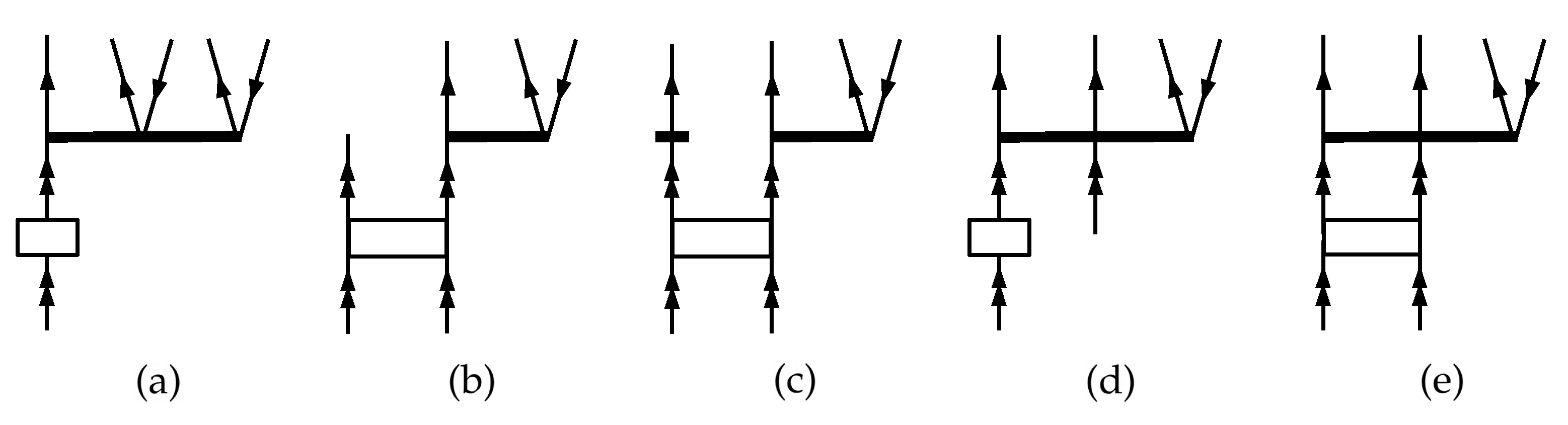
2. Theory
3. Pilot Applications
3.1. Atomic Energy Levels of Thallium and Lead
3.2. Electronic States of TlH
3.3. Static Dipole Polarizability of Lead
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Knecht, S.; Jensen, H.J.A.; Saue, T. Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond. Nat. Chem. 2019, 11, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.; Kaldor, U. Theoretical Chemistry and Physics of Heavy and Superheavy Elements—An Introduction; Springer: Cham, Switzerland, 2003; pp. 1–14. [Google Scholar] [CrossRef]
- Pyykko, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Saue, T. Relativistic Hamiltonians for chemistry: A primer. Chem. Phys. Chem. 2011, 12, 3077–3094. [Google Scholar] [CrossRef]
- Lischka, H.; Nachtigallova, D.; Aquino, A.J.; Szalay, P.G.; Plasser, F.; Machado, F.B.; Barbatti, M. Multireference approaches for excited states of molecules. Chem. Rev. 2018, 118, 7293–7361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, R.J. (Ed.) Recent Advances in Coupled-Cluster Methods; World Scientific: Singapore, 1997. [Google Scholar] [CrossRef]
- Paldus, J.; Li, X. A critical assessment of coupled cluster method in quantum chemistry. Adv. Chem. Phys. 1999, 110, 1–175. [Google Scholar]
- Pal, S.; Mukherjee, D. Use of cluster expansion methods in the open shell correlation problem. Adv. Quantum Chem. 1989, 20, 291. [Google Scholar]
- Kaldor, U. The Fock space coupled cluster method: theory and application. Theor. Chim. Acta 1991, 80, 427–439. [Google Scholar] [CrossRef]
- Kaldor, U.; Eliav, E. High-accuracy calculations for heavy and super-heavy elements. In Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1998; Volume 31, pp. 313–336. [Google Scholar]
- Jeziorski, B.; Monkhorst, H.J. Coupled-cluster method for multideterminantal reference states. Phys. Rev. A 1981, 24, 1668. [Google Scholar] [CrossRef]
- Meissner, L.; Bartlett, R.J. A general model-space coupled-cluster method using a Hilbert-space approach. J. Chem. Phys. 1990, 92, 561–567. [Google Scholar] [CrossRef]
- Balkova, A.; Kucharski, S.; Meissner, L.; Bartlett, R.J. The multireference coupled-cluster method in Hilbert space: an incomplete model space application to the LiH molecule. J. Chem. Phys. 1991, 95, 4311–4316. [Google Scholar] [CrossRef]
- Berkovic, S.; Kaldor, U. Degeneracy breaking in the Hilbert-space coupled cluster method. J. Chem. Phys. 1993, 98, 3090–3094. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Khait, Y.G. A self-consistent quasidegenerate coupled-cluster theory. Chem. Phys. Lett. 1999, 311, 372–378. [Google Scholar] [CrossRef]
- Eliav, E.; Borschevsky, A.; Shamasundar, K.R.; Pal, S.; Kaldor, U. Intermediate Hamiltonian Hilbert space coupled cluster method: Theory and pilot application. Int. J. Quantum Chem. 2009, 109, 2909–2915. [Google Scholar] [CrossRef]
- Lindgren, I. A coupled-cluster approach to the many-body perturbation theory for open-shell systems. Int. J. Quantum Chem. 1978, 14, 33–58. [Google Scholar] [CrossRef]
- Mukherjee, D.; Pal, S. Use of cluster expansion methods in the open-shell correlation problem. In Advances in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 1989; Volume 20, pp. 291–373. [Google Scholar]
- Malrieu, J.P.; Durand, P.; Daudey, J.P. Intermediate Hamiltonians as a new class of effective Hamiltonians. J. Phys. A Math. Gen. 1985, 18, 809. [Google Scholar] [CrossRef]
- Meissner, L. Fock-space coupled-cluster method in the intermediate Hamiltonian formulation: Model with singles and doubles. J. Chem. Phys. 1998, 108, 9227–9235. [Google Scholar] [CrossRef]
- Kaldor, U.; Eliav, E.; Landau, A. Study of heavy elements by relativistic Fock space and intermediate Hamiltonian coupled cluster methods. In Fundamental World of Quantum Chemistry; Springer: Cham, Switzerland, 2004; pp. 365–406. [Google Scholar]
- Eliav, E.; Vilkas, M.J.; Ishikawa, Y.; Kaldor, U. Extrapolated intermediate Hamiltonian coupled-cluster approach: Theory and pilot application to electron affinities of alkali atoms. J. Chem. Phys. 2005, 122, 224113. [Google Scholar] [CrossRef]
- Musiał, M.; Meissner, L.; Kucharski, S.A.; Bartlett, R.J. Molecular applications of the intermediate Hamiltonian Fock-space coupled-cluster method for calculation of excitation energies. J. Chem. Phys. 2005, 122, 224110. [Google Scholar] [CrossRef]
- Musiał, M.; Bartlett, R.J. Benchmark calculations of the Fock-space coupled cluster single, double, triple excitation method in the intermediate Hamiltonian formulation for electronic excitation energies. Chem. Phys. Lett. 2008, 457, 267–270. [Google Scholar] [CrossRef]
- Musiał, M.; Bartlett, R.J. Intermediate Hamiltonian Fock-space multireference coupled-cluster method with full triples for calculation of excitation energies. J. Chem. Phys. 2008, 129, 044101. [Google Scholar] [CrossRef]
- Eliav, E.; Kaldor, U. Four-component electronic structure methods. In Relativistic Methods for Chemists; Springer: Cham, Switzerland, 2010; pp. 279–349. [Google Scholar]
- Eliav, E.; Kaldor, U. Relativistic four-component multireference coupled cluster methods: towards a covariant approach. In Recent Progress in Coupled Cluster Methods; Springer: Cham, Switzerland, 2010; pp. 113–144. [Google Scholar]
- Musiał, M.; Bartlett, R.J. Multi-reference Fock space coupled-cluster method in the intermediate Hamiltonian formulation for potential energy surfaces. J. Chem. Phys. 2011, 135, 044121. [Google Scholar] [CrossRef] [PubMed]
- Eliav, E.; Borschevsky, A.; Kaldor, U. High-accuracy relativistic coupled-cluster calculations for the heaviest elements. In Handbook of Relativistic Quantum Chemistry; Liu, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 819–849. [Google Scholar]
- Mukhopadhyay, D.; Datta, B.; Mukherjee, D. The construction of a size-extensive intermediate Hamiltonian in a coupled-cluster framework. Chem. Phys. Lett. 1992, 197, 236–242. [Google Scholar] [CrossRef]
- Heully, J.L.; Evangelisti, S.; Durand, P. On some attempts to generalize the effective Hamiltonian approach. Journal de Physique II 1995, 5, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Zaitsevskii, A.; Mosyagin, N.S.; Stolyarov, A.V.; Eliav, E. Approximate relativistic coupled-cluster calculations on heavy alkali-metal diatomics: Application to the spin-orbit-coupled A1Σ+ and b3Π states of RbCs and Cs2. Phys. Rev. A 2017, 96, 022516. [Google Scholar] [CrossRef]
- Zaitsevskii, A.; Eliav, E. Padé extrapolated effective Hamiltonians in the Fock space relativistic coupled cluster method. Int. J. Quantum Chem. 2018, 118, e25772. [Google Scholar] [CrossRef]
- Visscher, L.; Eliav, E.; Kaldor, U. Formulation and implementation of the relativistic Fock-space coupled cluster method for molecules. J. Chem. Phys. 2001, 115, 9720–9726. [Google Scholar] [CrossRef]
- Oleynichenko, A.V.; Zaitsevskii, A.; Eliav, E. Towards high performance relativistic electronic structure modelling: the EXP-T program package. arXiv 2020, arXiv:2004.03682v1. [Google Scholar]
- Eliav, E.; Kaldor, U. Study of Actinides by Relativistic Coupled Cluster Methods. In Computational Methods in Lanthanide and Actinide Chemistry; Wiley: Hoboken, NJ, USA, 2015; pp. 23–54. [Google Scholar]
- Hughes, S.R.; Kaldor, U. The coupled-cluster method with full inclusion of single, double and triple excitations applied to high sectors of the Fock space. Chem. Phys. Lett. 1993, 204, 339–342. [Google Scholar] [CrossRef]
- Hughes, S.R.; Kaldor, U. The Fock-space coupled-cluster method: Electron affinities of the five halogen elements with consideration of triple excitations. J. Chem. Phys. 1993, 99, 6773–6776. [Google Scholar] [CrossRef]
- Hughes, S.R.; Kaldor, U. The coupled-cluster method in high sectors of the Fock space. Int. J. Quantum Chem. 1995, 55, 127–132. [Google Scholar] [CrossRef]
- Musiał, M.; Meissner, L.; Cembrzynska, J. The intermediate Hamiltonian Fock-space coupled-cluster method with approximate evaluation of the three-body effects. J. Chem. Phys. 2019, 151, 184102. [Google Scholar] [CrossRef] [PubMed]
- Isaev, T.A.; Berger, R. Laser cooled radium monofluoride: A molecular all-in-one probe for new physics. arXiv 2013, arXiv:1302.5682v2. [Google Scholar]
- Laatiaoui, M.; Lauth, W.; Backe, H.; Block, M.; Ackermann, D.; Cheal, B.; Chhetri, P.; Düllmann, C.E.; Duppen, P.V.; Even, J.; et al. Atom-at-a-time laser resonance ionization spectroscopy of nobelium. Nature 2016, 538, 495–498. [Google Scholar] [CrossRef]
- Garcia Ruiz, R.F.; Berger, R.; Billowes, J.; Binnersley, C.L.; Bissell, M.L.; Breier, A.A.; Brinson, A.J.; Chrysalidis, K.; Cocolios, T.E.; Cooper, B.; et al. Spectroscopy of short-lived radioactive molecules. Nature 2020, 581, 396–400. [Google Scholar] [CrossRef]
- Denis, M.; Norby, M.S.; Jensen, H.J.A.; Gomes, A.S.P.; Nayak, M.K.; Knecht, S.; Fleig, T. Theoretical study on ThF+, a prospective system in search of time-reversal violation. New J. Phys. 2015, 17, 043005. [Google Scholar] [CrossRef]
- Chubukov, D.V.; Skripnikov, L.V.; Labzowsky, L.N. P,T-odd Faraday rotation in heavy neutral atoms. Phys. Rev. A 2018, 97, 062512. [Google Scholar] [CrossRef]
- Maison, D.E.; Skripnikov, L.V.; Flambaum, V.V. Theoretical study of 173YbOH to search for the nuclear magnetic quadrupole moment. Phys. Rev. A 2019, 100, 032514. [Google Scholar] [CrossRef] [Green Version]
- Landau, A.; Eliav, E.; Ishikawa, Y.; Kaldor, U. Electronic structure of eka-lead (element 114) compared with lead. J. Chem. Phys. 2001, 114, 2977–2980. [Google Scholar] [CrossRef]
- Eliav, E.; Kaldor, U.; Hess, B.A. The relativistic Fock-space coupled-cluster method for molecules: CdH and its ions. J. Chem. Phys 1998, 108, 3409–3415. [Google Scholar] [CrossRef]
- Shavitt, I.; Bartlett, R.J. Many Body Methods in Chemistry and Physics; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar] [CrossRef] [Green Version]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef]
- Lee, Y.S.; Bartlett, R.J. A study of Be2 with many-body perturbation theory and a coupled-cluster method including triple excitations. J. Chem. Phys. 1984, 80, 4371–4377. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kucharski, S.A.; Bartlett, R.J. A coupled cluster approach with triple excitations. J. Chem. Phys. 1984, 81, 5906–5912. [Google Scholar] [CrossRef]
- Noga, J.; Bartlett, R.J.; Urban, M. Towards a full CCSDT model for electron correlation. CCSDT-n models. Chem. Phys. Lett. 1987, 134, 126–132. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Deegan, M.J.O.; Knowles, P.J. Perturbative corrections to account for triple excitations in closed and open shell coupled cluster theories. Chem. Phys. Lett. 1994, 227, 321–326. [Google Scholar] [CrossRef]
- Haque, A.; Kaldor, U. Three-electron excitation in open-shell coupled-cluster theory. Chem. Phys. Lett. 1985, 120, 261–265. [Google Scholar] [CrossRef]
- Kaldor, U.; Haque, A. Open-shell coupled-cluster method: direct calculation of excitation energies. Chem. Phys. Lett. 1986, 128, 45–48. [Google Scholar] [CrossRef]
- Bernholdt, D.E.; Bartlett, R.J. A critical assessment of multireference Fock space CCSD and perturbative third-order triples approximations for photoelectron spectra and quasidegenerate potential energy surfaces. Adv. Quantum Chem. 1999, 271–293. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Mitra, A.; Jana, D.; Ghosh, P.; Sinha, D. Full effect of triples in a valence universal multi-reference coupled cluster calculation. Chem. Phys. Lett. 2002, 361, 298–306. [Google Scholar] [CrossRef]
- Meissner, L.; Malinowski, P.; Gryniaków, J. Approximate evaluation of the effect of three-body cluster operators in the valence-universal coupled-cluster excitation energy calculations for Be and Mg. J. Phys. B At. Mol. Opt. Phys. 2004, 37, 2387–2400. [Google Scholar] [CrossRef]
- Li, X.; Paldus, J. Reduced multireference coupled cluster method with singles and doubles: perturbative corrections for triples. J. Chem. Phys. 2006, 124, 174101. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, F.A.; Prochnow, E.; Gauss, J.; Schaefer, H.F., III. Perturbative triples corrections in state-specific multireference coupled cluster theory. J. Chem. Phys. 2010, 132, 074107. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef] [Green Version]
- Vaval, N.; Pal, S.; Mukherjee, D. Fock space multireference coupled cluster theory: Noniterative inclusion of triples for excitation energies. Theor. Chem. Acc. 1998, 99, 100–105. [Google Scholar] [CrossRef]
- Evangelisti, S.; Daudey, J.P.; Malrieu, J.P. Qualitative intruder-state problems in effective Hamiltonian theory and their solution through intermediate Hamiltonians. Phys. Rev. A 1987, 35, 4930–4941. [Google Scholar] [CrossRef]
- Jana, D.; Bandyopadhyay, B.; Mukherjee, D. Development and applications of a relaxation-inducing cluster expansion theory for treating strong relaxation and differential correlation effects. Theor. Chem. Acc. 1999, 102, 317–327. [Google Scholar] [CrossRef]
- Oleynichenko, A.; Zaitsevskii, A.; Eliav, E. EXP-T, An Extensible Code for Fock Space Relativistic Coupled Cluster Calculations. 2020. Available online: http://www.qchem.pnpi.spb.ru/expt (accessed on 28 May 2020).
- Pahl, E.; Figgen, D.; Thierfelder, C.; Peterson, K.A.; Calvo, F.; Schwerdtfeger, P. A highly accurate potential energy curve for the mercury dimer. J. Chem. Phys. 2010, 132, 114301. [Google Scholar] [CrossRef]
- Skripnikov, L.V. Combined 4-component and relativistic pseudopotential study of ThO for the electron electric dipole moment search. J. Chem. Phys. 2016, 145, 214301. [Google Scholar] [CrossRef] [Green Version]
- Pašteka, L.F.; Eliav, E.; Borschevsky, A.; Kaldor, U.; Schwerdtfeger, P. Relativistic coupled cluster calculations with variational quantum electrodynamics resolve the discrepancy between experiment and theory concerning the electron affinity and ionization potential of gold. Phys. Rev. Lett. 2017, 118, 023002. [Google Scholar] [CrossRef]
- Skripnikov, L.V. Theoretical study of HfF+cation to search for the T,P-odd interactions. J. Chem. Phys. 2017, 147, 021101. [Google Scholar] [CrossRef] [Green Version]
- Fleig, T.; Skripnikov, L.V. P,T-Violating and magnetic hyperfine interactions in atomic thallium. Symmetry 2020, 12, 498. [Google Scholar] [CrossRef] [Green Version]
- Widmark, P.O.; Malmqvist, P.Å.; Roos, B.O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. Theor. Chim. Acta 1990, 77, 291–306. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Mosyagin, N.S.; Titov, A.V. Relativistic coupled-cluster calculations of spectroscopic and chemical properties for element 120. Chem. Phys. Lett. 2013, 555, 79–83. [Google Scholar] [CrossRef] [Green Version]
- CFOUR, a Quantum Chemical Program Package Written by J.F. Stanton, J. Gauss, L. Cheng, M.E. Harding, D.A. Matthews, P.G. Szalay with contributions from A.A. Auer, R.J. Bartlett, U. Benedikt, C. Berger, D.E. Bernholdt, Y.J. Bomble, O. Christiansen, F. Engel, R. Faber, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W.J. Lauderdale, F. Lipparini, T. Metzroth, L.A. Mück, D.P. O’Neill, D.R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J.D. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jorgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. For the Current Version See. Available online: http://www.cfour.de (accessed on 28 May 2020).
- All the ECPs and Basis Sets Used Throughout the Paper are Available. Available online: http://www.qchem.pnpi.spb.ru/recp (accessed on 28 May 2020).
- Mosyagin, N.S.; Zaitsevskii, A.; Titov, A.V. Shape-consistent relativistic effective potentials of small atomic cores. Int. Rev. Atomic Mol. Phys. 2010, 1, 63–72. [Google Scholar]
- Mosyagin, N.S.; Zaitsevskii, A.V.; Titov, A.V. Generalized relativistic effective core potentials for superheavy elements. Int. J. Quantum Chem. 2020, 120, e26076. [Google Scholar] [CrossRef]
- Saue, T.; Bast, R.; Gomes, A.S.P.; Jensen, H.J.A.; Visscher, L.; Aucar, I.A.; Di Remigio, R.; Dyall, K.G.; Eliav, E.; Fasshauer, E.; et al. The DIRAC code for relativistic molecular calculations. J. Chem. Phys. 2020, 152, 204104. [Google Scholar] [CrossRef] [PubMed]
- Eliav, E.; Kaldor, U.; Ishikawa, Y.; Seth, M.; Pyykkö, P. Calculated energy levels of thallium and eka-thallium (element 113). Phys. Rev. A 1996, 53, 3926–3933. [Google Scholar] [CrossRef]
- Dzuba, V.A.; Flambaum, V.V. Electron structure of superheavy elements Uut, Fl and Uup (Z=113 to 115). Hyperfine Interact. 2016, 237, 160. [Google Scholar] [CrossRef] [Green Version]
- Malli, G.L.; Da Silva, A.B.F.; Ishikawa, Y. Universal Gaussian basis set for accurate ab initio relativistic Dirac-Fock calculations. Phys. Rev. A 1993, 47, 143–146. [Google Scholar] [CrossRef]
- Shabaev, V.M.; Tupitsyn, I.I.; Yerokhin, V.A. QEDMOD: Fortran program for calculating the model Lamb-shift operator. Comput. Phys. Commun. 2015, 189, 175–181. [Google Scholar] [CrossRef]
- Sansonetti, J.E.; Martin, W.C. Handbook of Basic Atomic Spectroscopic Data. J. Phys. Chem. Ref. Data 2005, 34, 1559–2259. [Google Scholar] [CrossRef] [Green Version]
- Titov, A.V.; Mosyagin, N.S.; Alekseyev, A.B.; Buenker, R.J. GRECP/MRD-CI calculations of spin-orbit splitting in ground state of Tl and of spectroscopic properties of TlH. Int. J. Quantum Chem. 2001, 81, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Grundström, B.; Valberg, P. Das Bandenspektrum des Thalliumhydrids. I. Z. Phys. 1938, 108, 326–337. [Google Scholar] [CrossRef]
- Neuhaus, H.; Muld, V. Das Bandenspektrum des Thalliumdeutrids. Z. Phys. 1959, 153, 412–422. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Sundholm, D. VIBROT. Available online: http://www.chem.helsinki.fi/$\sim$sundholm/software/GPL/ (accessed on 28 May 2020).
- Urban, R.D.; Bahnmaier, A.H.; Magg, U.; Jones, H. The diode laser spectrum of thallium hydride (205TlH and 203TlH) in its ground electronic state. Chem. Phys. Lett. 1989, 158, 443–446. [Google Scholar] [CrossRef]
- Le Roy, R.J. RKR1: A computer program implementing the first-order RKR method for determining diatomic molecule potential energy functions. J. Quant. Spectrosc. Ra. 2017, 186, 158–166. [Google Scholar] [CrossRef]
- Skripnikov, L.V.; Titov, A.V.; Petrov, A.N.; Mosyagin, N.S.; Sushkov, O.P. Enhancement of the electron electric dipole moment in Eu2+. Phys. Rev. A 2011, 84, 022505. [Google Scholar] [CrossRef] [Green Version]
- Skripnikov, L.V.; Maison, D.E.; Mosyagin, N.S. Scalar-pseudoscalar interaction in the francium atom. Phys. Rev. A 2017, 95, 022507. [Google Scholar] [CrossRef] [Green Version]
- Hanni, M.E.; Keele, J.A.; Lundeen, S.R.; Fehrenbach, C.W.; Sturrus, W.G. Polarizabilities of Pb2+ and Pb4+ and ionization energies of Pb+ and Pb3+ from spectroscopy of high-L Rydberg states of Pb+ and Pb3+. Phys. Rev. A 2010, 81, 042512. [Google Scholar] [CrossRef]
- Ross, C.B.; Wood, D.R.; Scholl, P.S. Series limit and hydrogenlike series in PbII. J. Opt. Soc. Am. 1976, 66, 36–39. [Google Scholar] [CrossRef]
- Thierfelder, C.; Assadollahzadeh, B.; Schwerdtfeger, P.; Schäfer, S.; Schäfer, R. Relativistic and electron correlation effects in static dipole polarizabilities for the group-14 elements from carbon to element Z = 114: Theory and experiment. Phys. Rev. A 2008, 78, 052506. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Nagle, J.K. 2018 Table of static dipole polarizabilities of the neutral elements in the periodic table. Mol. Phys. 2019, 117, 1200–1225. [Google Scholar] [CrossRef] [Green Version]
- Porsev, S.G.; Kozlov, M.G.; Safronova, M.S.; Tupitsyn, I.I. Development of the configuration-interaction + all-order method and application to the parity-nonconserving amplitude and other properties of Pb. Phys. Rev. A 2016, 93, 012501. [Google Scholar] [CrossRef] [Green Version]
- Pershina, V.; Borschevsky, A.; Eliav, E.; Kaldor, U. Prediction of the adsorption behavior of elements 112 and 114 on inert surfaces from ab initio Dirac-Coulomb atomic calculations. J. Chem. Phys. 2008, 128, 024707. [Google Scholar] [CrossRef]
- Gould, T.; Bučko, T. C6 coefficients and dipole polarizabilities for all atoms and many ions in rows 1–6 of the periodic table. J. Chem. Theory Comput. 2016, 12, 3603–3613. [Google Scholar] [CrossRef]
- Maltsev, D.A.; Lomachuk, Y.V.; Shakhova, V.M.; Mosyagin, N.S.; Skripnikov, L.V.; Titov, A.V. Compound-tunable embedding potential method and its application to fersmite crystal. arXiv 2019, arXiv:1907.06947. [Google Scholar]
- Hughes, S.R.; Kaldor, U. Fock-space coupled-cluster method: the (1,2) sector. Phys. Rev. A 1993, 47, 4705–4712. [Google Scholar] [CrossRef]
- Musiał, M.; Olszówka, M.; Lyakh, D.I.; Bartlett, R.J. The equation-of-motion coupled cluster method for triple electron attached states. J. Chem. Phys. 2012, 137, 174102. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Exptl | IH-FS- | FS- | FS-RCCSD/LB + T/SB | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| [84] | RCCSD [47] | RCCSD/LB | SDT-1 | SDT-1’ | SDT-2 | SDT-3 | SDT | |||
| Tl, ground state | ||||||||||
| IP | 49,266 | −56 | −38 | −38 | −204 | −151 | −32 | |||
| EE | 7793 | −112 | 23 | 23 | 1 | 9 | −31 | |||
| Pb, ground state | ||||||||||
| IP | 121,245 | −168 | −143 | −28 | −28 | −190 | −158 | −59 | ||
| EE | 14,081 | −196 | −136 | 25 | 25 | 12 | 14 | −42 | ||
| Pb, ground state | ||||||||||
| IP | 59,819 | −543 | 364 | −44 | −285 | −347 | −336 | 7 | ||
| EE | 7819 | −288 | −302 | 76 | 5 | −4 | −3 | −28 | ||
| 10,650 | −343 | −235 | 130 | 129 | 97 | 102 | 13 | |||
| 21,458 | −605 | −394 | 215 | 203 | 158 | 167 | 5 | |||
| 29,467 | −208 | 414 | 170 | 248 | 293 | 302 | 173 | |||
| , Å | , cm | , cm | , Å | , cm | , cm | |
| FS-RCCSD/LB | 1.775 | 1800 | 0 | 1.749 | 1572 | 15,914 |
| FS-RCCSD/LB + SDT-1/SB | 1.862 | 1378 | 0 | 1.812 | 1222 | 17,899 |
| FS-RCCSD/LB + SDT/SB | 1.840 | 1500 | 0 | 1.801 | 1302 | 17,501 |
| RKR() + FS-RCCSD/LB | 1.909 | 1018 | 15,948 | |||
| RKR() + FS-RCCSD/LB + SDT-1/SB | 1.829 | 1218 | 17,931 | |||
| RKR() + FS-RCCSD/LB + SDT/SB | 1.846 | 1138 | 17,567 | |||
| Exptl. [86,87,90] | 1.873 | 1391 | 0 | 1.840 | 1043 | 17,723 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oleynichenko, A.V.; Zaitsevskii, A.; Skripnikov, L.V.; Eliav, E. Relativistic Fock Space Coupled Cluster Method for Many-Electron Systems: Non-Perturbative Account for Connected Triple Excitations. Symmetry 2020, 12, 1101. https://doi.org/10.3390/sym12071101
Oleynichenko AV, Zaitsevskii A, Skripnikov LV, Eliav E. Relativistic Fock Space Coupled Cluster Method for Many-Electron Systems: Non-Perturbative Account for Connected Triple Excitations. Symmetry. 2020; 12(7):1101. https://doi.org/10.3390/sym12071101
Chicago/Turabian StyleOleynichenko, Alexander V., Andréi Zaitsevskii, Leonid V. Skripnikov, and Ephraim Eliav. 2020. "Relativistic Fock Space Coupled Cluster Method for Many-Electron Systems: Non-Perturbative Account for Connected Triple Excitations" Symmetry 12, no. 7: 1101. https://doi.org/10.3390/sym12071101
APA StyleOleynichenko, A. V., Zaitsevskii, A., Skripnikov, L. V., & Eliav, E. (2020). Relativistic Fock Space Coupled Cluster Method for Many-Electron Systems: Non-Perturbative Account for Connected Triple Excitations. Symmetry, 12(7), 1101. https://doi.org/10.3390/sym12071101