Computational and Spectral Means for Characterizing the Intermolecular Interactions in Solutions and for Estimating Excited State Dipole Moment of Solute

 ,
,
Abstract
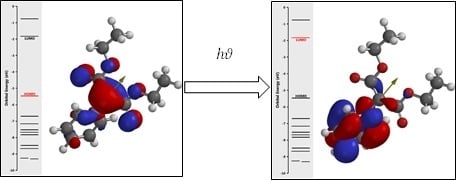
1. Introduction
2. Experimental and Computational Details
3. Theoretical Notions
4. Results and Discussion
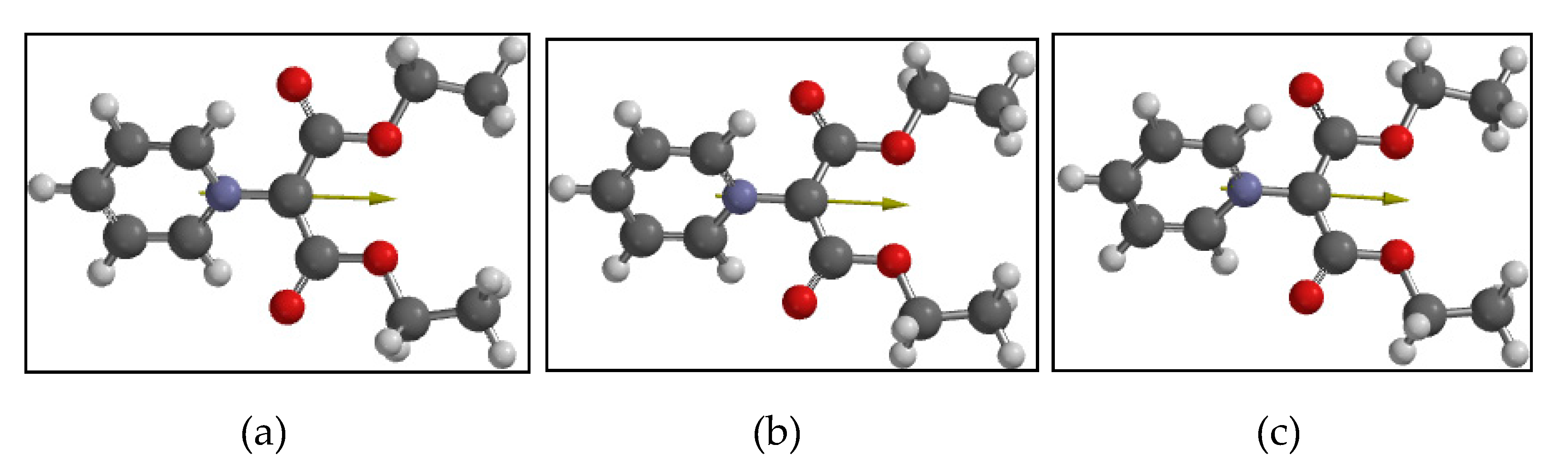
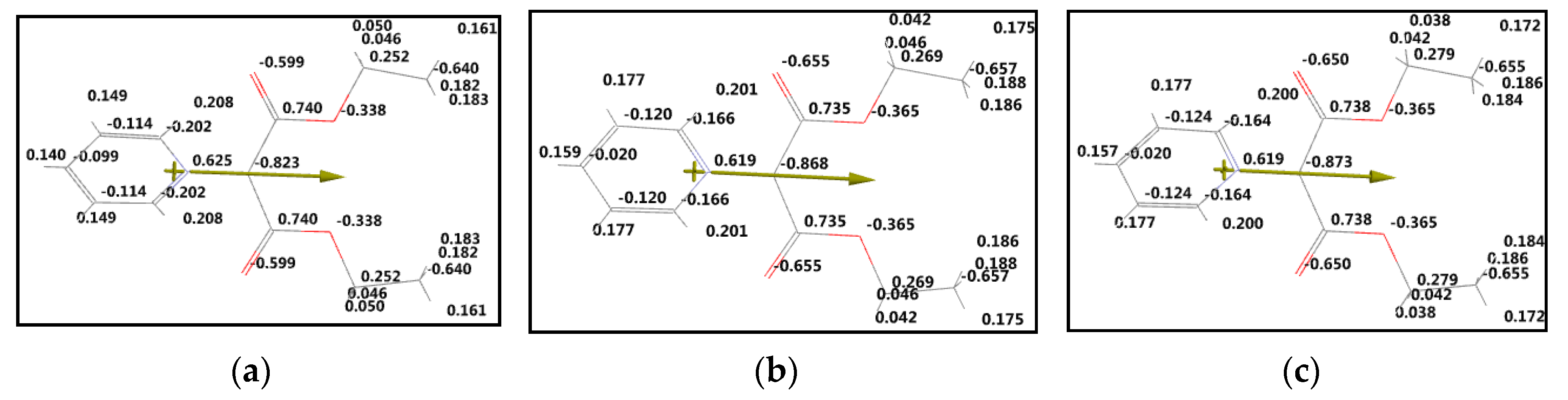
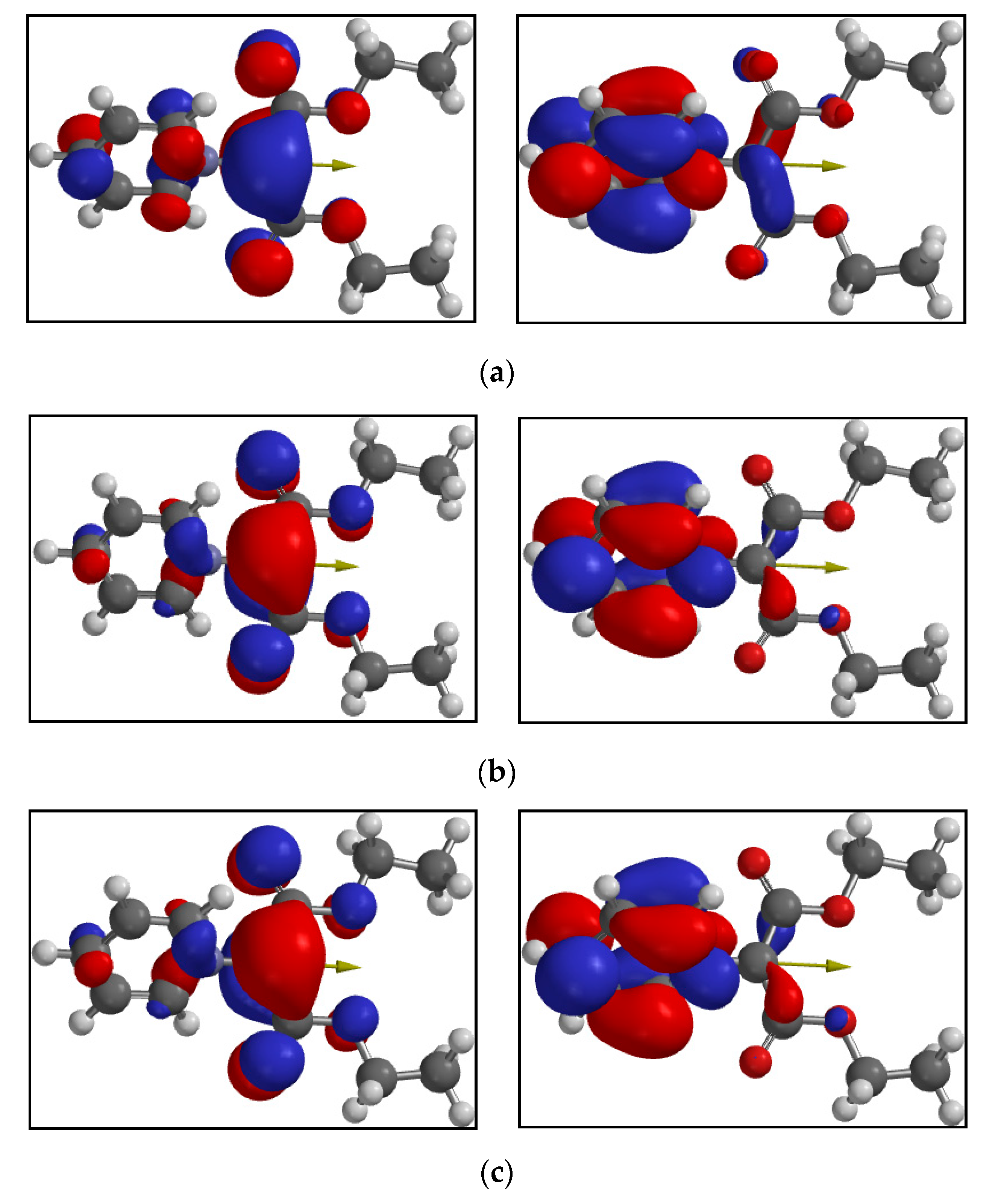
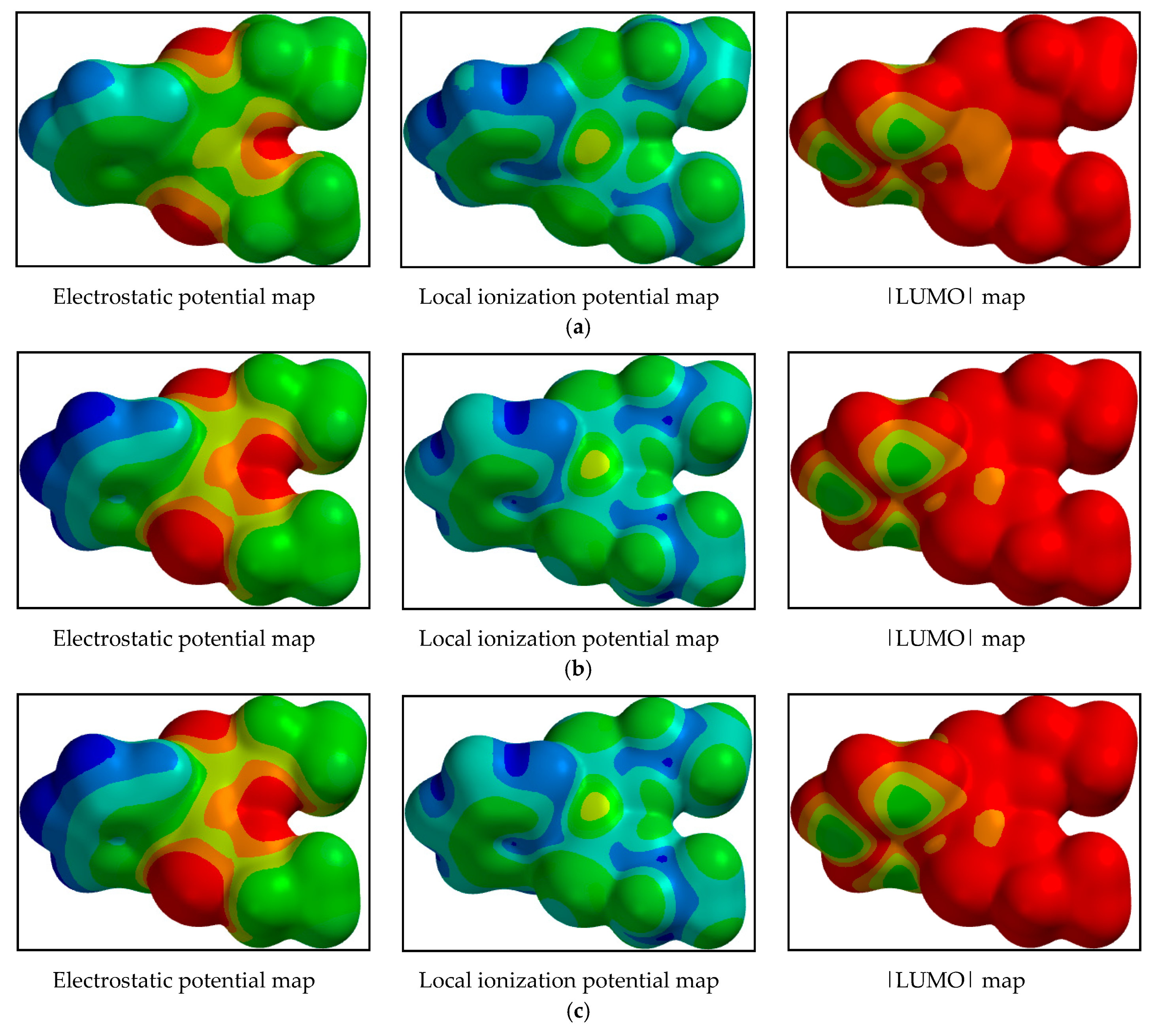
4.1. Computational Results
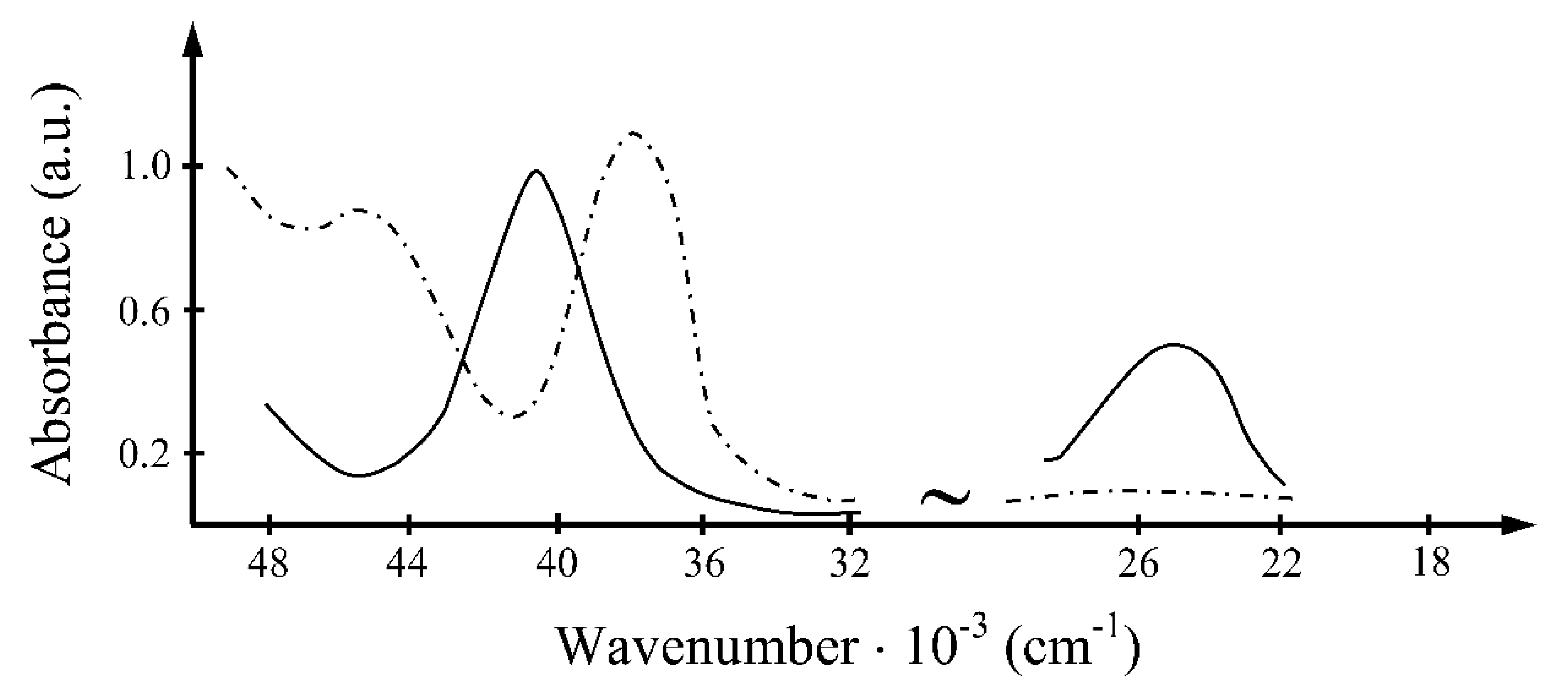
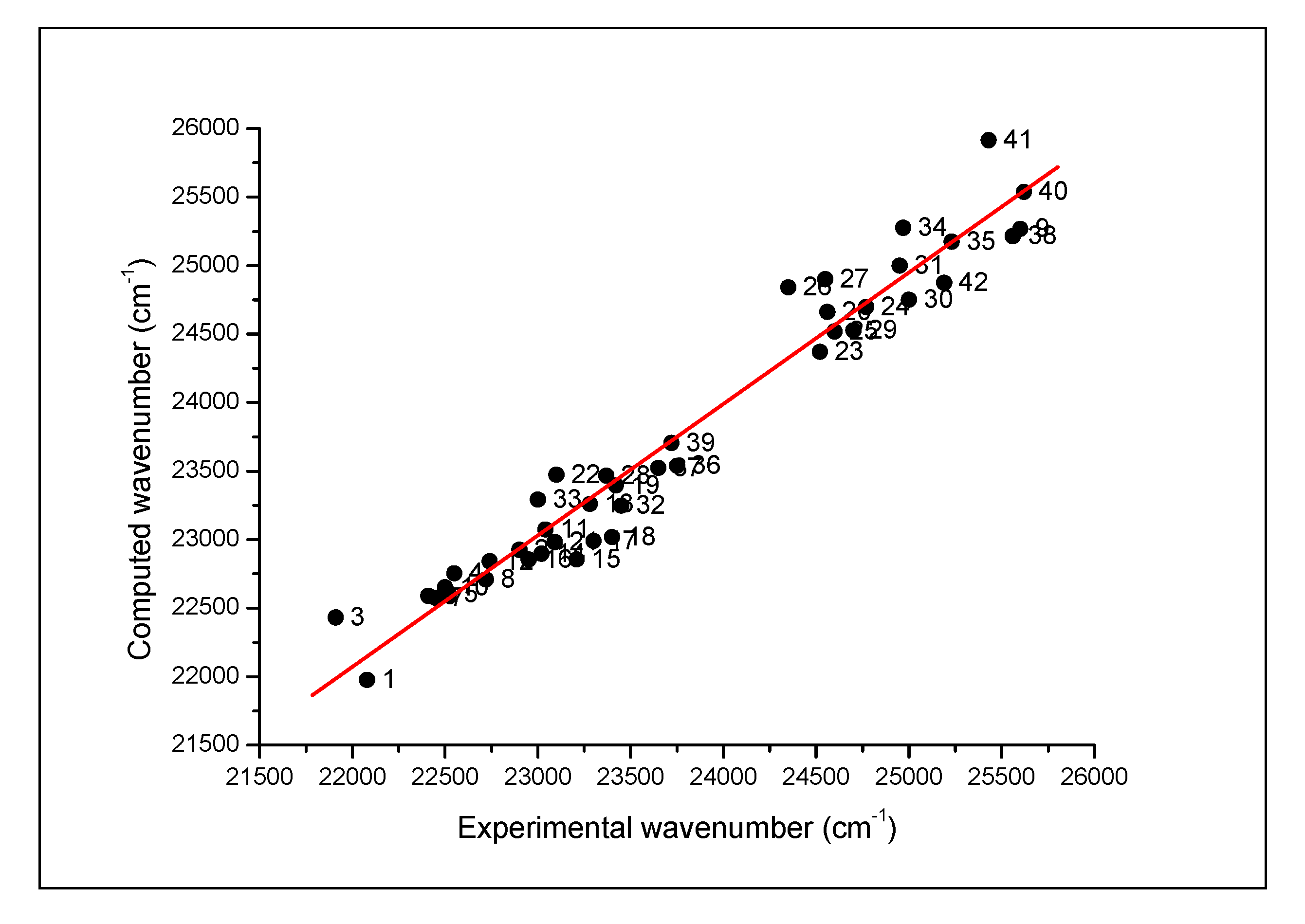
4.2. Spectral Results
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Onsager, L. Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- McRae, E.G. Theory of solvent influence on the molecular electronic spectra. Frequency shifts. J. Phys. Chem. 1957, 61, 562–572. [Google Scholar] [CrossRef]
- Bakhshiev, N.G. Spectroscopy of Intermolecular Interactions; Nauka: St. Petersburg, Russian, 1972. (In Russian) [Google Scholar]
- Abe, T. Theory of solvent influence on the molecular electronic spectra. Frequency shifts. Bull. Chem. Soc. Jpn. 1965, 38, 1314–1318. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Dorohoi, D.O.; Holban, V. Intermolecular interactions in some pyridazinium ylids solutions. J. Mol. Struct. 1993, 293, 133–136. [Google Scholar] [CrossRef]
- Kawski, A. Ground and excited state dipole moments of 6-propionyl-2-(dimethyl amino)-naphthalene determined by solvatochromic shifts. Z. Naturforsch 1999, 54, 379–381. [Google Scholar] [CrossRef]
- Zakerhamidi, M.S.; Moghadam, M.; Ghanadzadeh, A.; Hosseini, S. Anisotropic and isotropic solvent effects on the dipole moment and photophysical properties of rhodamine dyes. J. Lum. 2012, 132, 931–937. [Google Scholar] [CrossRef]
- Stanculescu, E.R.; Stroia, L.; Dorohoi, D.O. Solvatochromism Used in Determining Some Electro-Optical Parameters. Proceedings SPIE nr. 8001, 2011, nr. Art. 80012S, Published in 2011. Available online: https://www.spiedigitallibrary.org/conference-proceedings-of-spie/8001/80012S/Solvatochromism-used-in-determining-some-molecular-electro-optical-parameters/10.1117/12.891949.short (accessed on 20 June 2020).
- Ivan, L.M.; Dimitriu, D.G.; Gritco-Todirascu, A.; Morosanu, A.C.; Dorohoi, D.O.; Cheptea, C. Excited state dipole moments of two pyridazinium p-nitro-phenacylids estimated from solvatochromic study. Spectrosc. Lett. 2020, 53, 1–11. [Google Scholar] [CrossRef]
- Zugravescu, I.; Petrovanu, M. N-Ylid Chemistry; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Beak, P.; Reitz, D.B. Dipole stabilized carbanions: Novel and useful intermediates. Chem. Rev. 1978, 78, 275–316. [Google Scholar] [CrossRef]
- Radzhabov, M.R.; Tsyganov, D.V.; Pivina, T.S.; Krayushkin, M.M.; Seeboth, N.; Ivanov, S.; Frederique-Salit, A. Syntheses of pyridinium ylids and simulation of their 1, 3 dipolar Cycloaddition mechanism. Mendeleev Commun. 2017, 27, 503–505. [Google Scholar] [CrossRef]
- Jacobs, J.; van Hende, E.; Claessens, S.; De Kimpe, N. Pyridinium ylids in heterocyclic synthesis. Curr. Org. Chem. 2011, 15, 1340–1362. [Google Scholar] [CrossRef]
- Pawda, A.; Austin, D.J.; Percedo, L.; Lin, Z. Cycloaddition reactions of pyridinium and related azomethine ylides. J. Org. Chem. 1993, 58, 1144–1150. [Google Scholar] [CrossRef]
- Allgäuer, D.S.; Mayer, P.; Mayr, H. Nucleophilicity parameters of pyridinium ylides and their use in mechanistic analyses. J. Am. Chem. Soc. 2013, 135, 15216–15224. [Google Scholar] [CrossRef] [PubMed]
- Bonte, S.; Ghenea, I.O.; Dinica, R.; Baussanne, I.; Demeunynck, M. Investigation of the pyridinium ylide-alkene Cycloaddition as a fluorogenic coupling reaction. Molecules 2016, 21, 332. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Yulikov, M.; Kohn, T.; Jeschke, G. Pyridinium salts and ylides as partial structures of photoresponsive Merrifield resins. J. Mat. Chem. 2010, 20, 3025–3034. [Google Scholar] [CrossRef]
- Schwalm, R.; Bottcher, A.; Koch, H. MID UV resistant materials containing pyridinium ylides. Proc. SPIE 1988. [Google Scholar] [CrossRef]
- Streith, J. The photochemistry of N-iminopyridinium ylides in retrospect. From a simple concept to same applications. CHIMIA 1991, 45, 65–76. [Google Scholar]
- Taylor, L.D.; Karl, M.; Haubs, J. Polymeric Pyridinium Ylide and Products from Same; European Patent Office: Munich, Germany, 1985.
- Leontie, L.; Olariu, I.; Rusu, I.G. On the charge transport in some new carbanion disubstituted in thin films. Mater. Chem. Phys. 2003, 80, 506–511. [Google Scholar] [CrossRef]
- Leontie, L.; Roman, M.; Branza, F.; Podaru, C.; Rusu, I.G. Electrical and optical properties of some new synthetized ylids in thin films. Synth. Mat. 2003, 138, 157–163. [Google Scholar] [CrossRef]
- Dorohoi, D.O. Electronic spectroscopy of N-ylids. J. Mol. Struct. 2004, 704, 31–43. [Google Scholar] [CrossRef]
- Pop, V.; Dorohoi, D.O.; Holban, V. Molecular interactions in binary solutions of 4-aminophthalimide and 3p-cumyl-pyridazinium acetyl benzoyl methylid. Spectrochim. Acta A 1994, 50, 2281–2289. [Google Scholar] [CrossRef]
- Dorohoi, D.O.; Partenie, H.D. The Spectroscopy of N-ylids. J. Mol. Struct. 1993, 293, 129–132. [Google Scholar] [CrossRef]
- Gheorghies, C.; Gheorghies, L.V.; Dorohoi, D.O. Solvent influence on some complexes realized by Hydrogen bond. J. Mol. Struct. 2008, 887, 122–127. [Google Scholar] [CrossRef]
- Morosanu, A.C.; Gritco Todirascu, A.; Creanga, D.E.; Dorohoi, D.O. Computational and solvatochromic study of pyridinium acetyl benzoyl methylid (PABM). Spectrochim. Acta A 2018, 189, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, C.K. Symbiotic ligands, hard and soft central atoms. Inorg. Chem. 1964, 3, 1201–1202. [Google Scholar] [CrossRef]
- Pearson, R.G.; Songstad, J. Application of the principle of Hard and Soft acids and bases to Organic Chemistry. J. Am. Chem. Soc. 1967, 89, 1827–1836. [Google Scholar] [CrossRef]
- Surpateanu, G.; Dorohoi, D.O. Determination of the group electronegativity. An. Univ. Al. I. Cuza Sect. Ib Fizica 1977, 23, 99–102. [Google Scholar]
- Dorohoi, D.O.; Partenie, D.H.; Calugaru, A.C. Specific and universal interactions in Benzo-[f]-Quinolinium Acetyl-Benzoyl Methylid (BQABM) solutions; excited state dipole moment of BQABM. Spectrochim. Acta A 2019, 213, 184–191. [Google Scholar] [CrossRef]
- Spartan’14 for Windows, Macintosh and Linux, Tutorial and User’s Guide, January 10, 2014 Modeling. 2014. Available online: http://downloads.wavefun.com/Spartan14Manual.pdf (accessed on 3 June 2020).
- Kawschi, A. On the estimation of excited state dipole moments from solvatochromic shifts of absorption and fluorescence spectra. Z. Nat. 2002, 57, 255–262. [Google Scholar]
- Kamlet, M.J.; Abboud, J.I.; Abraham, M.; Taft, R.W. Linear solvation energy relationships 23. A comprehensive collection of the solvatochromic parameters π*, α,β and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Nadejde, C.; Creanga, D.E.; Humelnicu, I.; Dorohoi, D.O. Study of intermolecular interactions in rifampicin ternary solutions. J. Mol. Liq. 2009, 150, 51–55. [Google Scholar] [CrossRef]
- Morosanu, A.C.; Benchea, A.C.; Babusca, D.; Dimitriu, D.G.; Dorohoi, D.O. Quantum mechanical and solvatochromic characterization of Quercetin. Anal. Lett. 2017, 50, 2725–2739. [Google Scholar] [CrossRef]
- Babusca, D.; Morosanu, A.C.; Dorohoi, D.O. Spectral study of specific interactions between zwitterionic compounds and protic solvents. Spectrochim. Acta A 2017, 172, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Dorohoi, D.O.; Surpateanu, G.; Gheorghies, L.V. A spectral method for determination some molecular parameters of cycloimmonium ylids. Balk. Phys. Lett. 1998, 6, 198–203. [Google Scholar]
- Dorohoi, D.O. Excited state molecular parameters determined by spectral means. Ukr. J. Phys. 2018, 63, 701–707. [Google Scholar] [CrossRef]
- Pantil, S.K.; Wari, M.N.; Yohannan Panicker, C.; Inamdar, S.R. Solvatochromic study of coumarin 545 in alcohols for the determination of ground and excited state dipole moments. Int. J. Adv. Res. 2013, 1, 616–626. [Google Scholar]
- Thipperudrappa, J. Analysis of solvatochromism of a biologically active ketocyanine due using different solvent polarity scales and estimation of dipole moments. Int. J. Life Sci. Pharma Res. 2014, 4, 1–11. [Google Scholar]
- Zakerhamidi, M.S.; Golghasemi Sorkhabi, S.H.; Shamkhali, S. Polar and low polar solvents media effect on dipole moments of some diazo-Sudan dyes. Spectrochim. Acta A 2014, 127, 340–348. [Google Scholar] [CrossRef]
- Gahlaut, R.; Tewari, N.; Bridhkoti, J.P.; Koshi, H.C.; Pant, S. Determination of ground and excited state dipole moments of some naphthols using solvatochromic shift method. J. Mol. Liq. 2011, 163, 141–146. [Google Scholar] [CrossRef]
- Pop, V.; Dorohoi, D.O.; Delibas, M. Considerations on the statistic model of the intermolecular interactions in ternary solutions. An. Stiin. Univ. Al. I. Cuza Iasi 1987, 33, 78–86. [Google Scholar]
- Dorohoi, D.O.; Pop, V. Spectral shifts of the electronic absorption spectra of some cycloimmonium ylids in ternary solutions. An. Stiint. Univ. Al. I. Cuza Iasi Ib Fizica 1987, 33, 78–85. [Google Scholar]
- Puica-Melniciuc, N.; Avadanei, M.I.; Caprosu, M.; Dorohoi, D.O. Interaction energy in pairs of phthalazinium-dibenzoyl methylid (PDBM)–protic solvent molecules estimated in the limits of ternary solution model. Spectrochim. Acta A 2012, 96, 271–277. [Google Scholar]
- Dorohoi, D.O.; Avadanei, M.; Postolache, M. Characterization of the solvation spheres of some dipolar spectrally active molecules in binary solvents. Optoelectron. Adv. Mater. 2008, 2, 51–55. [Google Scholar]
- Avadanei, M.; Dorohoi, D.O. Interaction energy in pairs of pyridazinium ylid-solvent molecules estimated by spectral means within the cell ternary solution model. Ukr. J. Phys. 2012, 57, 118–122. [Google Scholar]
- Sasirekha, V.; Vanelle, P.; Terme, T.; Ramakrishnan, V. Solvatochromism and preferential solvation of 1, 4-dihydroxy-2, 3-dimethyl-9, 10-anthraquinone by UV-vis absorption and laser-induced fluorescence measurements. Spectrochim. Acta A 2008, 71, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Buhvestov, U.; Rived, F.; Ràfols, C.; Bosch, E.; Rosés, M. Solute-solvent and solvent-solvent interactions in binary solvent mixtures. Part 7. Comparison of the enhancement of the water structure in alcohol-water mixtures measured by solvatochromic indicators. J. Phys. Org. Chem. 1998, 11, 185–192. [Google Scholar] [CrossRef]
- Rosés, M.; Buhvestov, U.; Ráfols, C.; Rived, F.; Bosch, E. Solute-solvent and solvent-solvent interactions in binary solvent mixtures. Part.6. A quantitative measurement of the enhancement of water structure in 2-methylpropan-2-ol-water and propan-2-ol-water mixtures by solvatochromic indicators. J. Chem. Soc. Perkin Trans. 1997, 2, 1341–1348. [Google Scholar] [CrossRef]
- Dulcescu, M.M.; Stan, C.; Dorohoi, D.O. Spectral investigations of the influence of the hydroxyl solvents on the intermolecular interactions in some pyridinium ylid ternary solution. Rev. Chim. Buchar. 2010, 61, 1219–1222. [Google Scholar]
- Dulcescu, M.M.; Stan, C.; Dorohoi, D.O. Spectral study of intermolecular interactions in water-ethanol solutions of some carbanion di-substituted pyridinium ylids. Rev. Roum. Chim. 2010, 55, 403–408. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule Properties | |||
|---|---|---|---|
| Density Functional EDF2, 6–31 G*, in Vacuum | Density Functional EDF2, 6–31 G*, in Water | Density Functional EDF2, 6–31 G*, in Ethanol | |
| Formula | C12H15NO4 | C12H15NO4 | C12H15NO4 |
| Weight | 237.255 amu | 237.255 amu | 237.255 amu |
| Energy | −821.401842 au | −821.422833 au | −821.428045 au |
| Energy (aq) | −821.414893 au | - | - |
| Solvation E | −34.27 kJ/mol | - | - |
| EHOMO | −5.12 eV | −5.58 eV | −5.47 eV |
| ELUMO | −1.93 eV | −1.86 eV | −1.83 eV |
| Dipole Moment | 2.87 Debye | 6.28 Debye | 6.15 Debye |
| Tautomers | 0 | 0 | 0 |
| Conformers | 36 | 36 | 36 |
| QSAR (Quantitative Structure–Activity Relationship) | |||
| Area | 270.44 Å2 | 274.40 Å2 | 274.30 Å2 |
| Volume | 243.96 Å3 | 245.24 Å3 | 245.15 Å3 |
| PSA | 35.050 Å2 | 37.812 Å2 | 37.737 Å2 |
| Ovality | 1.43 | 1.45 | 1.45 |
| Polarizability | 60.41 Å3 | 60.39 Å3 | 60.40 Å3 |
| HBD Count | 0 | 0 | 0 |
| HBA Count | 3 | 3 | 3 |
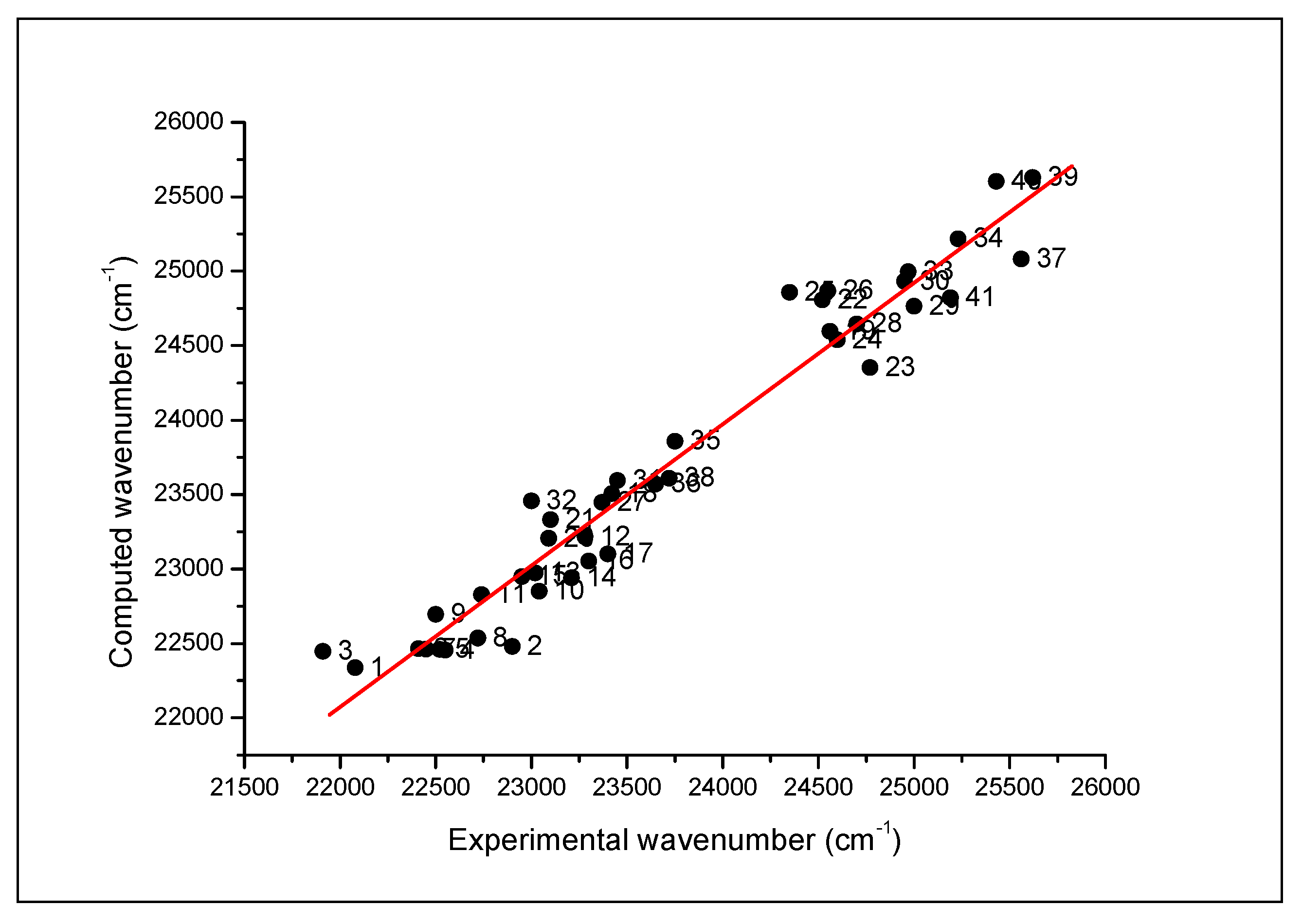
| No. | Solvent | f(ε) | f(n) | β | α | (cm−1) | |||
|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc. (1′) | Calc. (2′′) | |||||||
| 1 | n-Heptane | 0.227 | 0.240 | −0.08 | 0.00 | 0.00 | 22,080 | 21,975 | 22,337 |
| 2 | Dioxane | 0.286 | 0.251 | 0.55 | 0.37 | 0.00 | 22,900 | 22,925 | 22,480 |
| 3 | Carbon tetrachloride | 0.292 | 0.274 | 0.28 | 0.10 | 0.00 | 21,910 | 22,430 | 22,448 |
| 4 | Benzene | 0.299 | 0.295 | 0.59 | 0.10 | 0.00 | 22,550 | 22,753 | 22,454 |
| 5 | p-Xylene | 0.299 | 0.291 | 0.40 | 0.13 | 0.00 | 22,520 | 22,602 | 22,459 |
| 6 | 1,3,5 Trimethylbenzene | 0.302 | 0.293 | 0.41 | 0.13 | 0.00 | 22,410 | 22,589 | 22,463 |
| 7 | o-Xylene | 0.302 | 0.292 | 0.41 | 0.11 | 0.00 | 22,450 | 22,574 | 22,461 |
| 8 | Toluene | 0.348 | 0.297 | 0.54 | 0.11 | 0.00 | 22,720 | 22,709 | 22,535 |
| 9 | Trichloroethylene | 0.448 | 0.282 | 0.53 | 0.05 | 0.00 | 22,500 | 22,651 | 22,695 |
| 10 | Anisole | 0.524 | 0.302 | 0.73 | 0.32 | 0.00 | 23,040 | 23,074 | 22,851 |
| 11 | 1,2-Dibromoethane | 0.538 | 0.313 | 0.75 | 0.00 | 0.00 | 22,740 | 22,841 | 22,828 |
| 12 | Chloroform | 0.552 | 0.267 | 0.69 | 0.10 | 0.20 | 23,280 | 23,259 | 23,216 |
| 13 | n-Butyl acetate | 0.577 | 0.240 | 0.46 | 0.45 | 0.00 | 23,020 | 22,895 | 22,972 |
| 14 | Iso-amyl acetate | 0.589 | 0.241 | 0.71 | 0.07 | 0.00 | 23,210 | 22,855 | 22,940 |
| 15 | Chlorobenzene | 0.605 | 0.307 | 0.71 | 0.07 | 0.00 | 22,950 | 22,855 | 22,949 |
| 16 | Ethyl acetate | 0.625 | 0.228 | 0.55 | 0.45 | 0.00 | 23,300 | 22,989 | 23,054 |
| 17 | Methyl acetate | 0.655 | 0.221 | 0.60 | 0.42 | 0.00 | 23,400 | 23,017 | 22,101 |
| 18 | Dichloromethane | 0.727 | 0.256 | 0.82 | 0.10 | 0.20 | 23,420 | 23,395 | 22,506 |
| 19 | N-Octyl alcohol | 0.745 | 0.258 | 0.40 | 0.81 | 0.77 | 24,560 | 24,662 | 24,597 |
| 20 | 1,2-Dichloroethane | 0.752 | 0.264 | 0.81 | 0.10 | 0.00 | 23,090 | 22,983 | 23,206 |
| 21 | Pyridine | 0.790 | 0.299 | 0.87 | 0.64 | 0.00 | 23,100 | 23,473 | 23,332 |
| 22 | N-hexyl alcohol | 0.839 | 0.252 | 0.04 | 0.84 | 0.80 | 24,520 | 24,370 | 24,807 |
| 23 | Benzyl alcohol | 0.804 | 0.311 | 0.98 | 0.52 | 0.60 | 24,770 | 24,697 | 24,352 |
| 24 | Cyclohexanol | 0.824 | 0.276 | 0.45 | 0.84 | 0.66 | 24,600 | 24,517 | 24,340 |
| 25 | N-Amyl alcohol | 0.826 | 0.241 | 0.40 | 0.86 | 0.84 | 24,350 | 24,842 | 24,860 |
| 26 | N-Butyl alcohol | 0.833 | 0.242 | 0.47 | 0.84 | 0.84 | 24,550 | 24,899 | 24,868 |
| 27 | Acetophenone | 0.833 | 0.312 | 0.90 | 0.49 | 0.04 | 23,370 | 23,466 | 23,446 |
| 28 | Iso-Butyl alcohol | 0.852 | 0.239 | 0.40 | 0.84 | 0.69 | 24,700 | 24,525 | 24,646 |
| 29 | Iso propyl alcohol | 0.852 | 0.234 | 0.48 | 0.84 | 0.76 | 25,000 | 24,749 | 24,646 |
| 30 | N-Propyl alcohol | 0.866 | 0.239 | 0.52 | 0.90 | 0.84 | 24,950 | 24,999 | 24,931 |
| 31 | Acetone | 0.868 | 0.222 | 0.62 | 0.48 | 0.08 | 23,450 | 23,246 | 23,594 |
| 32 | Benzo nitrile | 0.889 | 0.308 | 0.90 | 0.37 | 0.00 | 23,000 | 23,291 | 23,456 |
| 33 | Ethanol | 0.895 | 0.221 | 0.86 | 0.75 | 0.86 | 24,970 | 25,274 | 24,997 |
| 34 | Methanol | 0.909 | 0.203 | 0.60 | 0.66 | 0.98 | 25,230 | 25,173 | 25,216 |
| 35 | Acetonitrile | 0.921 | 0.219 | 0.75 | 0.40 | 0.19 | 23,750 | 25,539 | 23,857 |
| 36 | DMF | 0.924 | 0.258 | 0.88 | 0.69 | 0.00 | 23,650 | 23,523 | 23,560 |
| 37 | Ethylene glycol | 0.930 | 0.259 | 0.90 | 0.52 | 0.90 | 25,560 | 23,515 | 25,081 |
| 38 | DMSO | 0.946 | 0.281 | 1.00 | 0.76 | 0.00 | 23,720 | 23,704 | 23,609 |
| 39 | 1,2,3-Propanetriol | 0.948 | 0.280 | 0.62 | 0.51 | 1.21 | 25,620 | 25,536 | 25,630 |
| 40 | Water | 0.964 | 0.206 | 1.09 | 0.47 | 1.17 | 25,430 | 25,915 | 25,600 |
| 41 | Formamide | 0.973 | 0.271 | 0.97 | 0.48 | 0.71 | 24,190 | 24,875 | 24,821 |
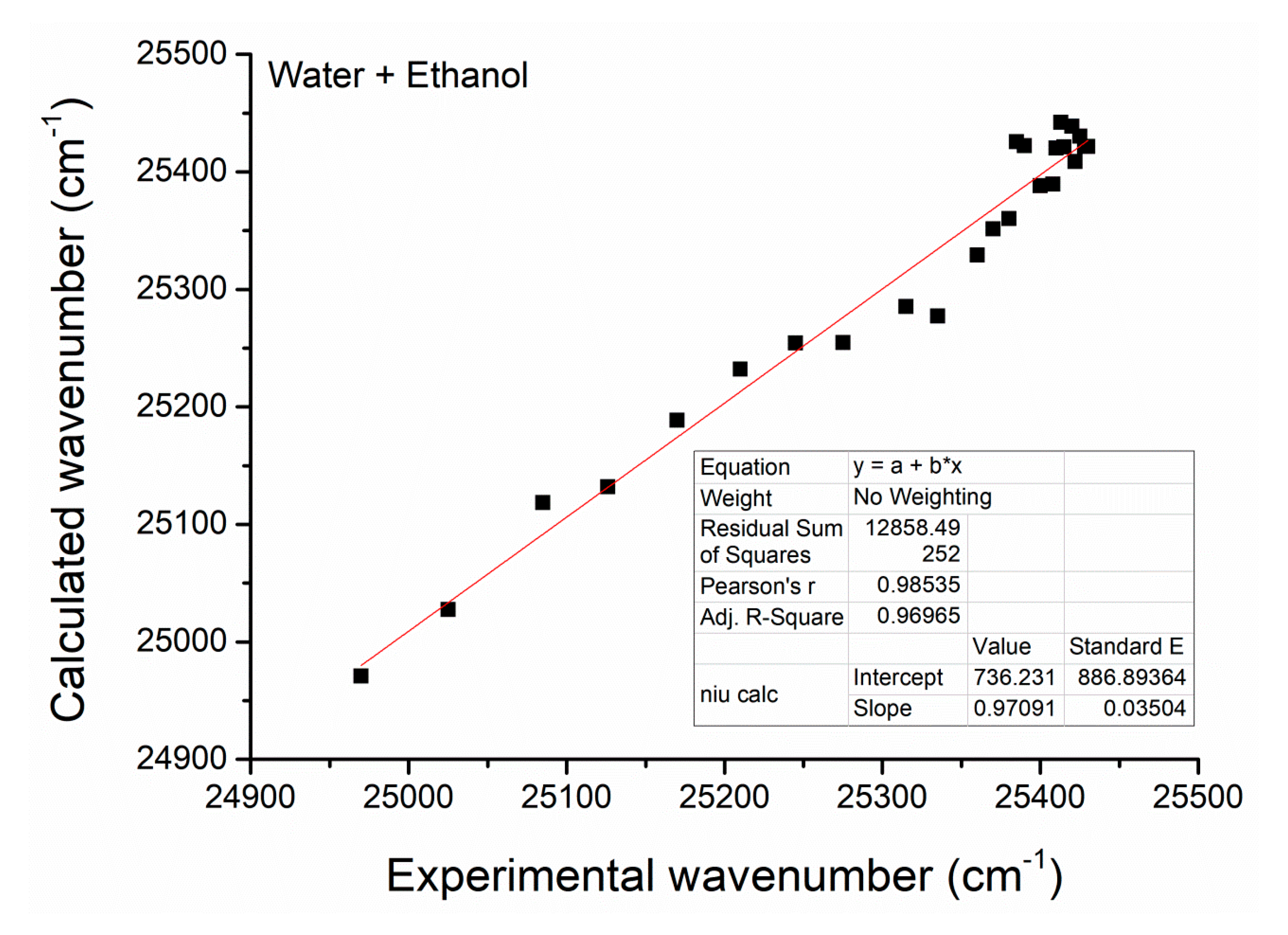
| No. | α | Β | PCCM | |||||
|---|---|---|---|---|---|---|---|---|
| (cm−1) | ||||||||
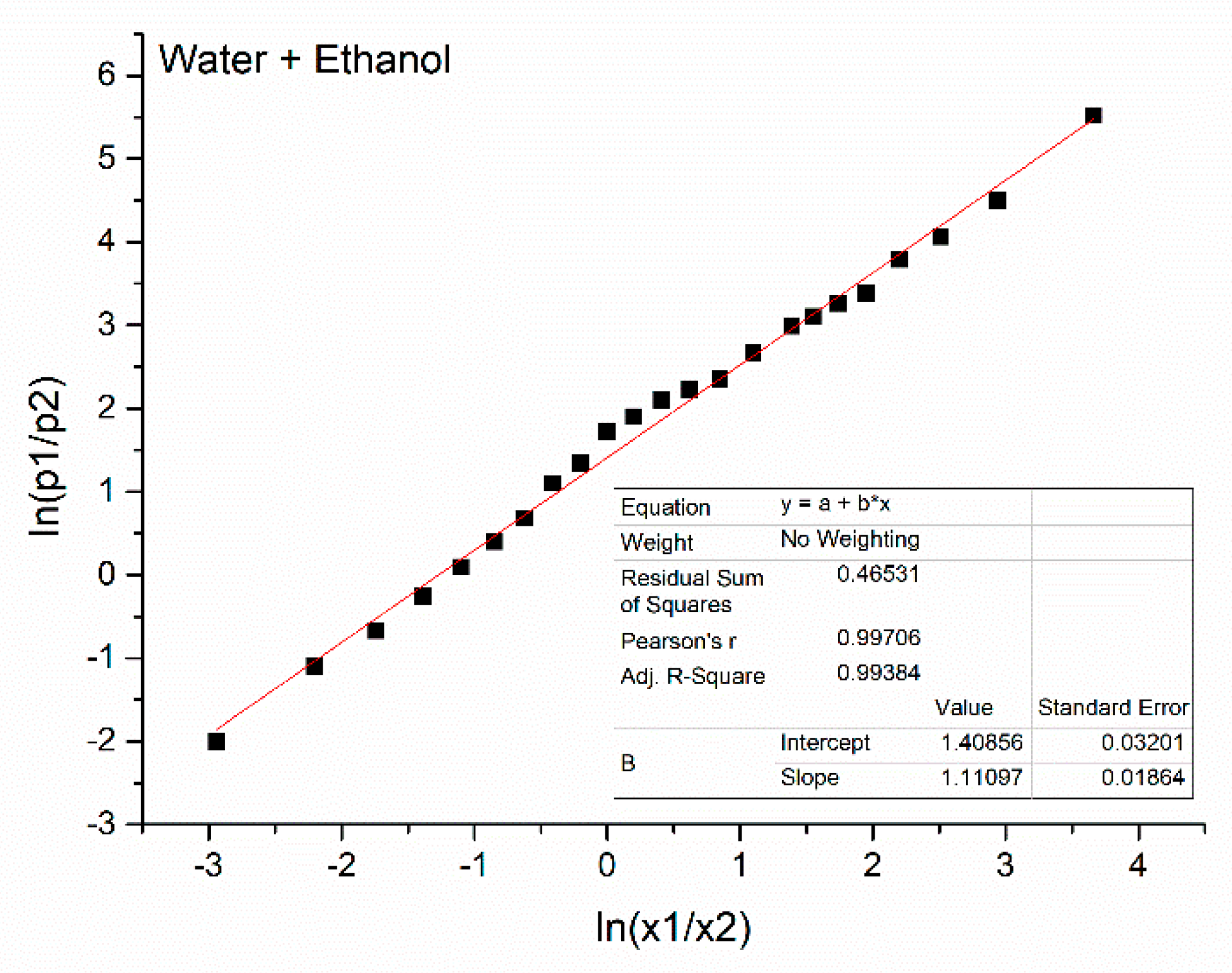
| 1 | 0.000 | 0.51 | 0.83 | 0.98 | - | 24,970 | - | - |
| 2 | 0.050 | 0.54 | 0.83 | 0.97 | −2.94 | 25,025 | 0.119 | −2.00 |
| 3 | 0.100 | 0.57 | 0.84 | 0.96 | −2.20 | 25,085 | 0.249 | −1.10 |
| 4 | 0.150 | 0.60 | 0.83 | 0.94 | −1.74 | 25,126 | 0.338 | −0.67 |
| 5 | 0.200 | 0.63 | 0.83 | 0.93 | −1.39 | 25,170 | 0.435 | −0.26 |
| 6 | 0.250 | 0.65 | 0.83 | 0.93 | −1.10 | 25,210 | 0.522 | 0.09 |
| 7 | 0.300 | 0.68 | 0.82 | 0.92 | −0.85 | 25,245 | 0.599 | 0.40 |
| 8 | 0.350 | 0.70 | 0.81 | 0.91 | −0.62 | 25,275 | 0.663 | 0.68 |
| 9 | 0.400 | 0.73 | 0.80 | 0.91 | −0.41 | 25,315 | 0.750 | 1.10 |
| 10 | 0.450 | 0.75 | 0.79 | 0.89 | −0.20 | 25,335 | 0.792 | 1.34 |
| 11 | 0.500 | 0.77 | 0.79 | 0.90 | 0.00 | 25,360 | 0.848 | 1.72 |
| 12 | 0550 | 0.80 | 0.78 | 0.89 | 0.20 | 25,370 | 0.870 | 1.90 |
| 13 | 0.600 | 0.82 | 0.77 | 0.89 | 0.41 | 25,380 | 0.891 | 2.10 |
| 14 | 0.650 | 0.85 | 0.77 | 0.89 | 0.62 | 25,385 | 0.902 | 2.22 |
| 15 | 0.700 | 0.90 | 0.74 | 0.88 | 0.85 | 25,390 | 0.913 | 2.35 |
| 16 | 0.750 | 0.94 | 0.71 | 0.86 | 1.10 | 25,400 | 0.935 | 2.67 |
| 17 | 0.800 | 1.00 | 0.67 | 0.87 | 1.39 | 25,408 | 0.952 | 2.99 |
| 18 | 0.825 | 1.03 | 0.66 | 0.87 | 1.55 | 25,410 | 0.957 | 3.10 |
| 19 | 0.850 | 1.06 | 0.64 | 0.90 | 1.74 | 25,413 | 0.963 | 3.26 |
| 20 | 0.875 | 1.09 | 0.61 | 0.92 | 1.95 | 25,415 | 0.967 | 3.38 |
| 21 | 0.900 | 1.11 | 0.59 | 0.97 | 2.20 | 25,420 | 0.978 | 3.79 |
| 22 | 0.925 | 1.12 | 0.56 | 1.03 | 2.51 | 25,422 | 0.983 | 4.06 |
| 23 | 0.950 | 1.13 | 0.54 | 1.11 | 2.94 | 25,425 | 0.989 | 4.50 |
| 24 | 0.975 | 1.13 | 0.52 | 1.18 | 3.66 | 25,428 | 0.996 | 5.52 |
| 25 | 1.000 | 1.13 | 0.50 | 1.26 | - | 25,430 | - | - |
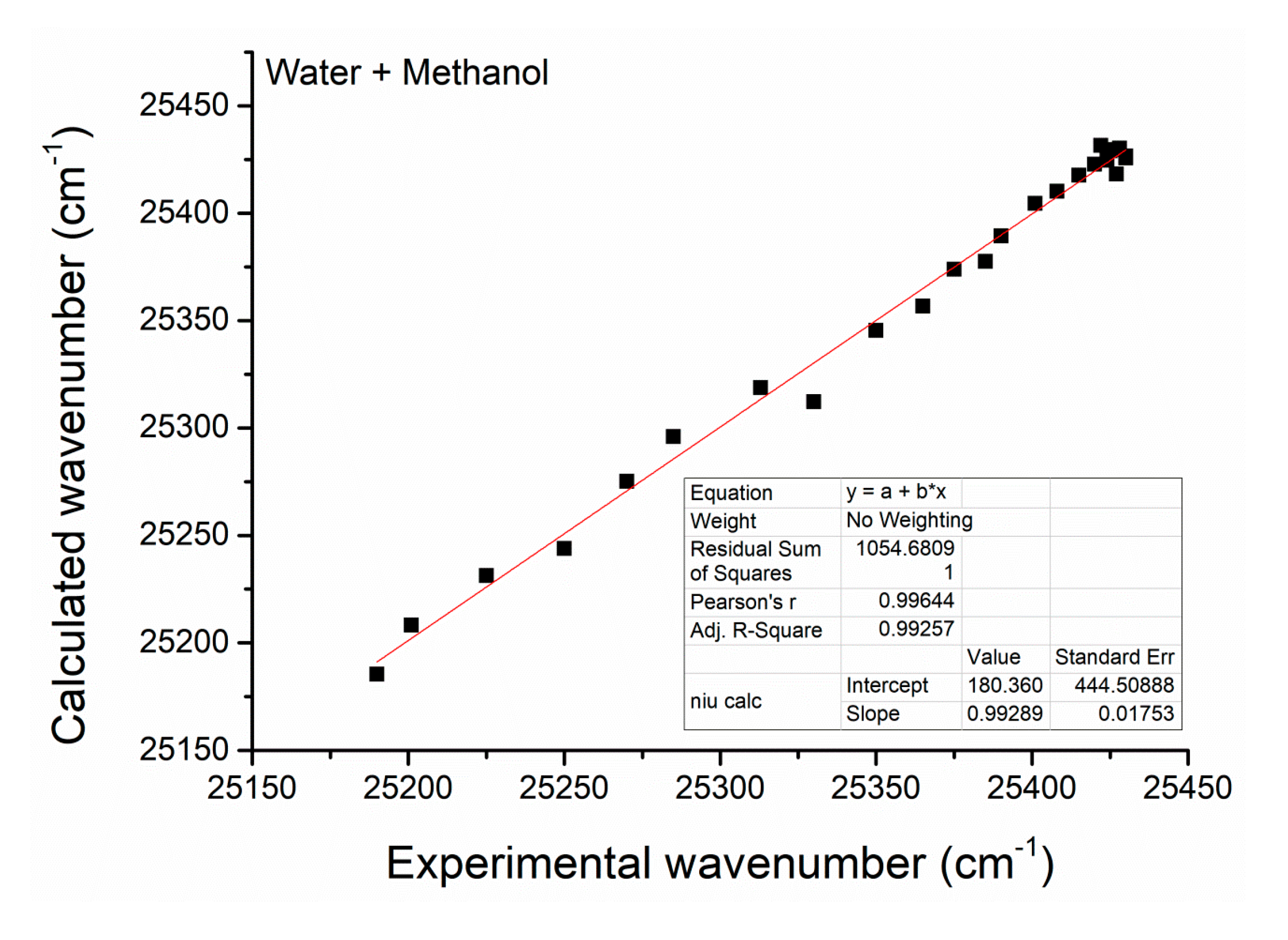
| No. | α | Β | PCCM | |||||
|---|---|---|---|---|---|---|---|---|
| (cm−1) | ||||||||
| 1 | 0.000 | 0.58 | 0.74 | 1.14 | - | 25,190 | - | - |
| 2 | 0.050 | 0.61 | 0.74 | 1.13 | −2.94 | 25,201 | 0.046 | −3.03 |
| 3 | 0.100 | 0.64 | 0.74 | 1.12 | −2.20 | 25,225 | 0.146 | −1.77 |
| 4 | 0.150 | 0.66 | 0.74 | 1.10 | −1.74 | 25,250 | 0.249 | −1.10 |
| 5 | 0.200 | 0.70 | 0.74 | 1.09 | −1.39 | 25,270 | 0.334 | −0.69 |
| 6 | 0.250 | 0.73 | 0.74 | 1.07 | −1.10 | 25,285 | 0.397 | −0.42 |
| 7 | 0.300 | 0.76 | 0.74 | 1.06 | −0.85 | 25,313 | 0.512 | 0.05 |
| 8 | 0.350 | 0.78 | 0.72 | 1.04 | −0.62 | 25,330 | 0.584 | 0.34 |
| 9 | 0.400 | 0.82 | 0.72 | 1.04 | −0.41 | 25,350 | 0.666 | 0.69 |
| 10 | 0.450 | 0.85 | 0.71 | 1.02 | −0.20 | 25,365 | 0.729 | 0.99 |
| 11 | 0.500 | 0.88 | 0.70 | 1.03 | 0.00 | 25,375 | 0.770 | 1.21 |
| 12 | 0550 | 0.91 | 0.68 | 1.02 | 0.20 | 25,385 | 0.813 | 1.47 |
| 13 | 0.600 | 0.95 | 0.66 | 1.01 | 0.41 | 25,390 | 0.833 | 1.61 |
| 14 | 0.650 | 0.98 | 0.65 | 1.01 | 0.62 | 25,401 | 0.879 | 1.98 |
| 15 | 0.700 | 1.01 | 0.63 | 1.01 | 0.85 | 25,408 | 0.908 | 2.29 |
| 16 | 0.750 | 1.04 | 0.61 | 1.02 | 1.10 | 25,415 | 0.938 | 2.72 |
| 17 | 0.800 | 1.06 | 0.59 | 1.06 | 1.39 | 25,420 | 0.958 | 3.13 |
| 18 | 0.825 | 1.08 | 0.58 | 1.07 | 1.55 | 25,422 | 0.967 | 3.38 |
| 19 | 0.850 | 1.09 | 0.56 | 1.09 | 1.74 | 25,424 | 0.975 | 3.66 |
| 20 | 0.875 | 1.10 | 0.55 | 1.12 | 1.95 | 25,425 | 0.979 | 3.84 |
| 21 | 0.900 | 1.11 | 0.54 | 1.13 | 2.20 | 25,426 | 0.983 | 4.06 |
| 22 | 0.925 | 1.11 | 0.52 | 1.17 | 2.51 | 25,427 | 0.988 | 4.41 |
| 23 | 0.950 | 1.12 | 0.52 | 1.19 | 2.94 | 25,428 | 0.992 | 4.82 |
| 24 | 0.975 | 1.12 | 0.51 | 1.22 | 3.66 | 25,430 | - | - |
| 25 | 1.000 | 1.14 | 0.49 | 1.23 | - | 25,430 | - | - |
| No. | Solvent | (cm−1) | (%) | (cm−1) | (%) | (cm−1) | (%) | (cm−1) | (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | n-Heptane | 372.7 | 85.6 | 62.6 | 14.4 | 0.0 | 0.0 | 0.0 | 0.0 |
| 2 | Dioxane | 469.6 | 80.3 | 65.5 | 11.2 | 0.0 | 0.0 | 49.8 | 8.5 |
| 3 | Carbon tetrachloride | 479.4 | 85.0 | 71.5 | 12.7 | 0.0 | 0.0 | 13.5 | 2.4 |
| 4 | Benzene | 490.9 | 84.5 | 76.9 | 13.2 | 0.0 | 0.0 | 13.5 | 2.3 |
| 5 | p-Xylene | 490.9 | 84.0 | 75.9 | 13.0 | 0.0 | 0.0 | 17.5 | 3.0 |
| 6 | 1,3,5-Trimethylbenzene | 495.9 | 84.1 | 76.4 | 13.0 | 0.0 | 0.0 | 17.5 | 3.0 |
| 7 | o-Xylene | 495.9 | 84.5 | 76.1 | 13.0 | 0.0 | 0.0 | 14.8 | 2.5 |
| 8 | Toluene | 571.4 | 86.1 | 77.5 | 11.7 | 0.0 | 0.0 | 14.8 | 2.2 |
| 9 | Trichloroethylene | 735.6 | 90.2 | 73.5 | 9.0 | 0.0 | 0.0 | 6.7 | 0.8 |
| 10 | Anisole | 860.4 | 87.6 | 78.8 | 8.0 | 0.0 | 0.0 | 43.1 | 4.4 |
| 11 | 1,2-Dibromoethane | 883.4 | 91.5 | 81.6 | 8.5 | 0.0 | 0.0 | 0.0 | 0.0 |
| 12 | Chloroform | 906.3 | 68.2 | 69.6 | 5.2 | 339.0 | 25.5 | 13.5 | 1.0 |
| 13 | n-Butyl acetate | 947.4 | 88.5 | 62.6 | 5.8 | 0.0 | 0.0 | 60.6 | 5.7 |
| 14 | Iso-amyl acetate | 967.1 | 93.0 | 62.8 | 6.0 | 0.0 | 0.0 | 9.4 | 0.9 |
| 15 | Chlorobenzene | 993.4 | 91.7 | 80.1 | 7.4 | 0.0 | 0.0 | 9.4 | 0.9 |
| 16 | Ethyl acetate | 1026.2 | 89.5 | 59.5 | 5.2 | 0.0 | 0.0 | 60.6 | 5.3 |
| 17 | Methyl acetate | 1075.5 | 90.4 | 57.6 | 4.8 | 0.0 | 0.0 | 56.5 | 4.8 |
| 18 | Dichloromethane | 1193.7 | 74.0 | 66.8 | 4.1 | 339.0 | 21.0 | 13.5 | 0.8 |
| 19 | N-Octyl alcohol | 1223.2 | 45.2 | 67.3 | 2.5 | 1305.2 | 48.3 | 109.0 | 4.0 |
| 20 | 1,2-Dichloroethane | 1234.7 | 93.8 | 68.8 | 5.2 | 0.0 | 0.0 | 13.5 | 1.0 |
| 21 | Pyridine | 1297.1 | 88.8 | 78.0 | 5.3 | 0.0 | 0.0 | 86.1 | 5.9 |
| 22 | N-hexyl alcohol | 1377.6 | 47.3 | 65.7 | 2.3 | 1356.0 | 46.6 | 113.0 | 3.9 |
| 23 | Benzyl alcohol | 1320.1 | 53.1 | 81.1 | 3.3 | 1017.0 | 40.9 | 70.0 | 2.8 |
| 24 | Cyclohexanol | 1353.0 | 50.9 | 72.0 | 2.7 | 1118.7 | 42.1 | 113.0 | 4.3 |
| 25 | N-Amyl alcohol | 1356.2 | 45.8 | 62.8 | 2.1 | 1423.9 | 48.1 | 115.7 | 3.9 |
| 26 | N-Butyl alcohol | 1367.7 | 46.1 | 63.1 | 2.1 | 1423.9 | 48.0 | 113.0 | 3.8 |
| 27 | Acetophenone | 1367.7 | 86.4 | 81.4 | 5.1 | 67.8 | 4.3 | 65.9 | 4.2 |
| 28 | Iso-Butyl alcohol | 1398.9 | 51.0 | 62.3 | 2.3 | 1169.6 | 42.6 | 113.0 | 4.1 |
| 29 | Iso propyl alcohol | 1398.9 | 48.9 | 61.0 | 2.1 | 1288.2 | 45.0 | 113.0 | 4.0 |
| 30 | N-Propyl alcohol | 1421.9 | 46.9 | 62.3 | 2.1 | 1423.9 | 47.0 | 121.1 | 4.0 |
| 31 | Acetone | 1425.2 | 84.7 | 57.9 | 3.4 | 135.6 | 8.1 | 64.6 | 3.8 |
| 32 | Benzo nitrile | 1459.7 | 91.8 | 80.3 | 5.1 | 0.0 | 0.0 | 49.8 | 3.1 |
| 33 | Ethanol | 1469.5 | 47.6 | 57.6 | 1.9 | 1457.8 | 47.2 | 100.9 | 3.3 |
| 34 | Methanol | 1492.5 | 45.3 | 52.9 | 1.6 | 1661.2 | 50.4 | 88.8 | 2.7 |
| 35 | Acetonitrile | 1512.2 | 77.7 | 57.1 | 2.9 | 322.1 | 16.6 | 53.8 | 2.8 |
| 36 | DMF | 1517.1 | 90.5 | 67.3 | 4.0 | 0.0 | 0.0 | 92.9 | 5.5 |
| 37 | Ethylene glycol | 1527.0 | 47.9 | 67.5 | 2.1 | 1525.6 | 47.8 | 70.0 | 2.2 |
| 38 | DMSO | 1553.3 | 89.8 | 73.3 | 4.2 | 0.0 | 0.0 | 102.3 | 5.9 |
| 39 | 1,2,3-Propanetriol | 1556.5 | 41.5 | 73.0 | 1.9 | 2051.0 | 54.7 | 68.6 | 1.8 |
| 40 | Water | 1582.8 | 43.0 | 53.7 | 1.5 | 1983.2 | 53.8 | 63.2 | 1.7 |
| 41 | Formamide | 1597.6 | 54.4 | 70.7 | 2.4 | 1203.5 | 41.0 | 64.6 | 2.2 |
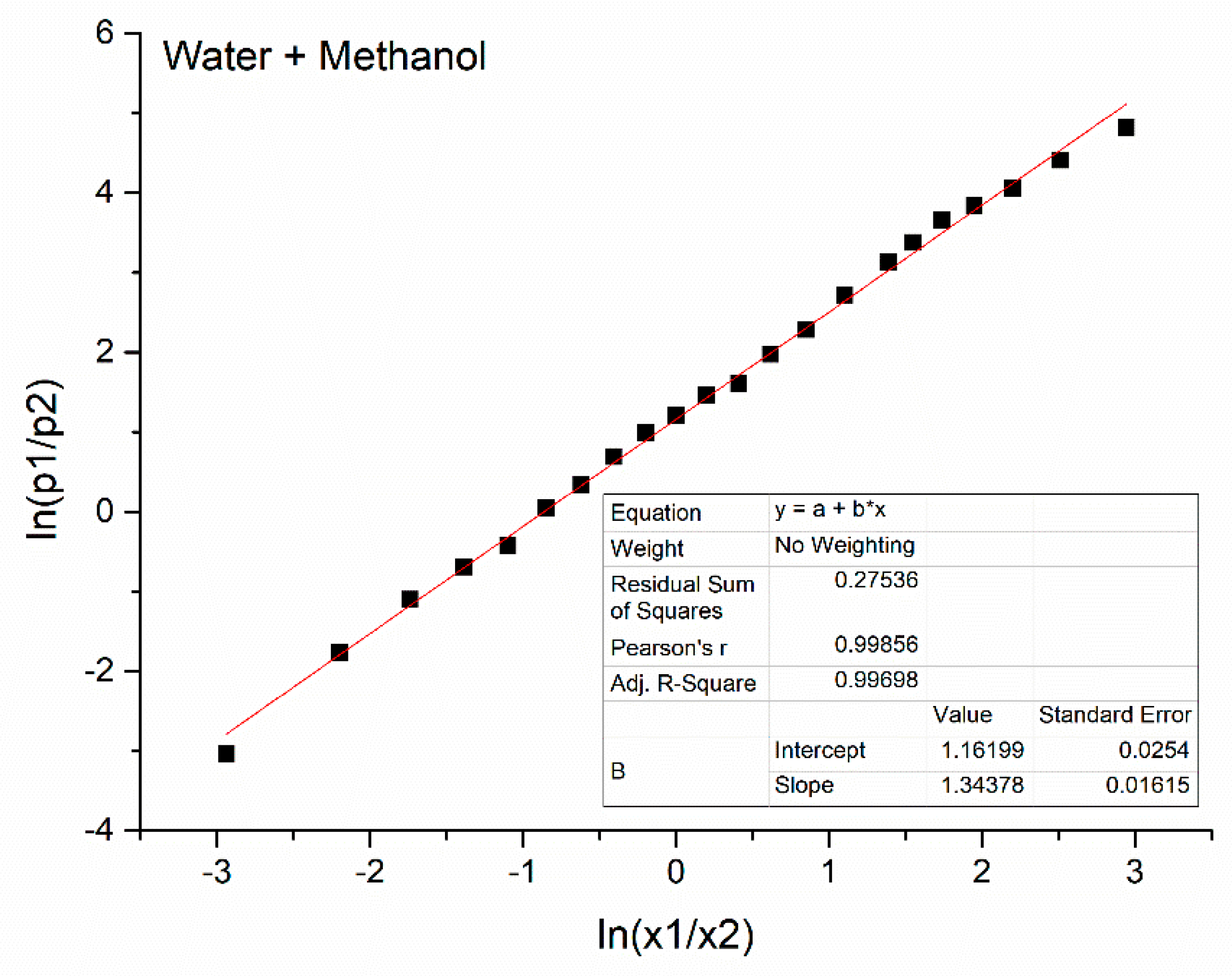
| No. | x1 | Water + Ethanol | Water + Methanol | ||||||
|---|---|---|---|---|---|---|---|---|---|
(cm−1) | C1π* % | C2α % | C3β % | (cm−1) | C1π* % | C2α % | C3β % | ||
| 1 | 0.000 | 4817 | 23.0 | 59.5 | 17.5 | 1417 | 33.9 | 50.3 | 15.8 |
| 2 | 0.050 | 4872 | 24.0 | 58.8 | 17.2 | 1428 | 35.5 | 49.9 | 15.6 |
| 3 | 0.100 | 4932 | 25.0 | 58.5 | 16.5 | 1452 | 35.5 | 49.2 | 15.3 |
| 4 | 0.150 | 4973 | 26.1 | 57.6 | 16.3 | 1477 | 37.0 | 48.3 | 14.7 |
| 5 | 0.200 | 5017 | 27.0 | 57.0 | 16.0 | 1497 | 38.7 | 47.3 | 14.0 |
| 6 | 0.250 | 5057 | 28.0 | 56.6 | 15.4 | 1512 | 39.9 | 47.2 | 13.9 |
| 7 | 0.300 | 5092 | 27.8 | 55.6 | 15.6 | 1540 | 40.8 | 46.2 | 13.0 |
| 8 | 0.350 | 5122 | 29.7 | 55.0 | 15.3 | 1557 | 41.4 | 45.5 | 13.1 |
| 9 | 0.400 | 5162 | 30.8 | 54.0 | 15.2 | 1577 | 43.0 | 43.9 | 13.1 |
| 10 | 0.450 | 5182 | 31.6 | 53.6 | 14.8 | 1592 | 44.2 | 42.8 | 13.0 |
| 11 | 0.500 | 5207 | 32.3 | 52.8 | 14.9 | 1602 | 45.4 | 42.0 | 12.6 |
| 12 | 0550 | 5217 | 33.7 | 51.6 | 14.7 | 1612 | 46.7 | 40.8 | 12.5 |
| 13 | 0.600 | 5227 | 34.5 | 50.8 | 14.7 | 1617 | 48.6 | 39.2 | 12.2 |
| 14 | 0.650 | 5232 | 35.4 | 50.8 | 14.8 | 1628 | 49.8 | 38.2 | 12.0 |
| 15 | 0.700 | 5237 | 37.4 | 47.8 | 14.8 | 1635 | 51.0 | 37.0 | 12.00 |
| 16 | 0.750 | 5247 | 39.2 | 46.7 | 14.1 | 1642 | 52.4 | 35.6 | 12.0 |
| 17 | 0.800 | 5255 | 41.5 | 44.1 | 14.4 | 1647 | 53.1 | 34.4 | 12.5 |
| 18 | 0.825 | 5257 | 42.6 | 43.2 | 14.2 | 1649 | 54.0 | 33.8 | 12.2 |
| 19 | 0.850 | 5260 | 43.8 | 42.0 | 14.2 | 1651 | 54.6 | 32.6 | 12.8 |
| 20 | 0.875 | 5262 | 45.0 | 40.0 | 15.0 | 1652 | 55.0 | 32.0 | 13.0 |
| 21 | 0.900 | 5267 | 45.8 | 38.7 | 15.5 | 1653 | 55.5 | 31.4 | 13.1 |
| 22 | 0.925 | 5269 | 46.4 | 36.7 | 16.9 | 1654 | 55.5 | 30.6 | 13.9 |
| 23 | 0.950 | 5272 | 46.6 | 35.4 | 18.0 | 1655 | 56.0 | 30.0 | 14.0 |
| 24 | 0.975 | 5275 | 46.6 | 34.0 | 19.4 | 1657 | 55.9 | 29.7 | 14.4 |
| 25 | 1.000 | 5277 | 23.0 | 59.5 | 17.53 | 46.6 | 32.7 | 20.7 | |
| No. | Φ (Degree) | µe (D) | αe (cm3) |
|---|---|---|---|
| 1 | 0 | 5.778 | 60.48 |
| 2 | 5 | 5.801 | 60.45 |
| 3 | 8 | 5.837 | 60.40 |
| 4 | 8.5 | 5.842 | 60.39 |
| 5 | 8.6 | 5.845 | 60.388 |
| 6 | 9 | 5.853 | 70.37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorohoi, D.O.; Creanga, D.E.; Dimitriu, D.G.; Morosanu, A.C.; Gritco-Todirascu, A.; Mariciuc, G.G.; Melniciuc, N.P.; Ardelean, E.; Cheptea, C. Computational and Spectral Means for Characterizing the Intermolecular Interactions in Solutions and for Estimating Excited State Dipole Moment of Solute. Symmetry 2020, 12, 1299. https://doi.org/10.3390/sym12081299
Dorohoi DO, Creanga DE, Dimitriu DG, Morosanu AC, Gritco-Todirascu A, Mariciuc GG, Melniciuc NP, Ardelean E, Cheptea C. Computational and Spectral Means for Characterizing the Intermolecular Interactions in Solutions and for Estimating Excited State Dipole Moment of Solute. Symmetry. 2020; 12(8):1299. https://doi.org/10.3390/sym12081299
Chicago/Turabian StyleDorohoi, Dana Ortansa, Dorina Emilia Creanga, Dan Gheorghe Dimitriu, Ana Cezarina Morosanu, Antonina Gritco-Todirascu, Gabriel Grigore Mariciuc, Nicoleta Puica Melniciuc, Elena Ardelean, and Corina Cheptea. 2020. "Computational and Spectral Means for Characterizing the Intermolecular Interactions in Solutions and for Estimating Excited State Dipole Moment of Solute" Symmetry 12, no. 8: 1299. https://doi.org/10.3390/sym12081299
APA StyleDorohoi, D. O., Creanga, D. E., Dimitriu, D. G., Morosanu, A. C., Gritco-Todirascu, A., Mariciuc, G. G., Melniciuc, N. P., Ardelean, E., & Cheptea, C. (2020). Computational and Spectral Means for Characterizing the Intermolecular Interactions in Solutions and for Estimating Excited State Dipole Moment of Solute. Symmetry, 12(8), 1299. https://doi.org/10.3390/sym12081299