
Unsymmetrical Hexafluorocyclopentene-Linked Twisted ?-Conjugated Molecules as Dual-State Emissive Luminophores †

 ,
, 
 , and
, and
Abstract
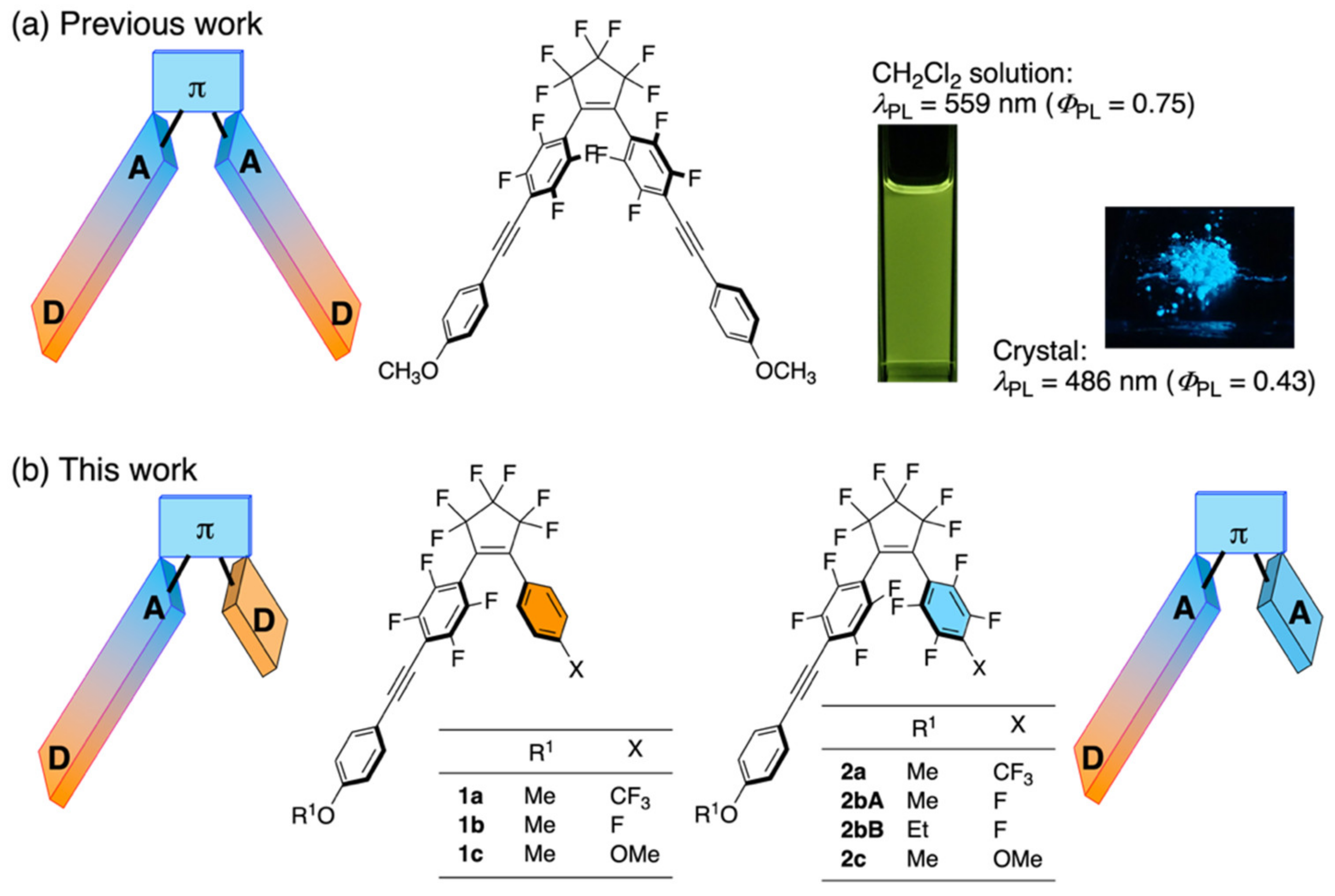
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Materials
2.3. Single Crystal X-ray Diffraction
2.4. DFT
2.5. Photophysical Property
3. Results and Discussion
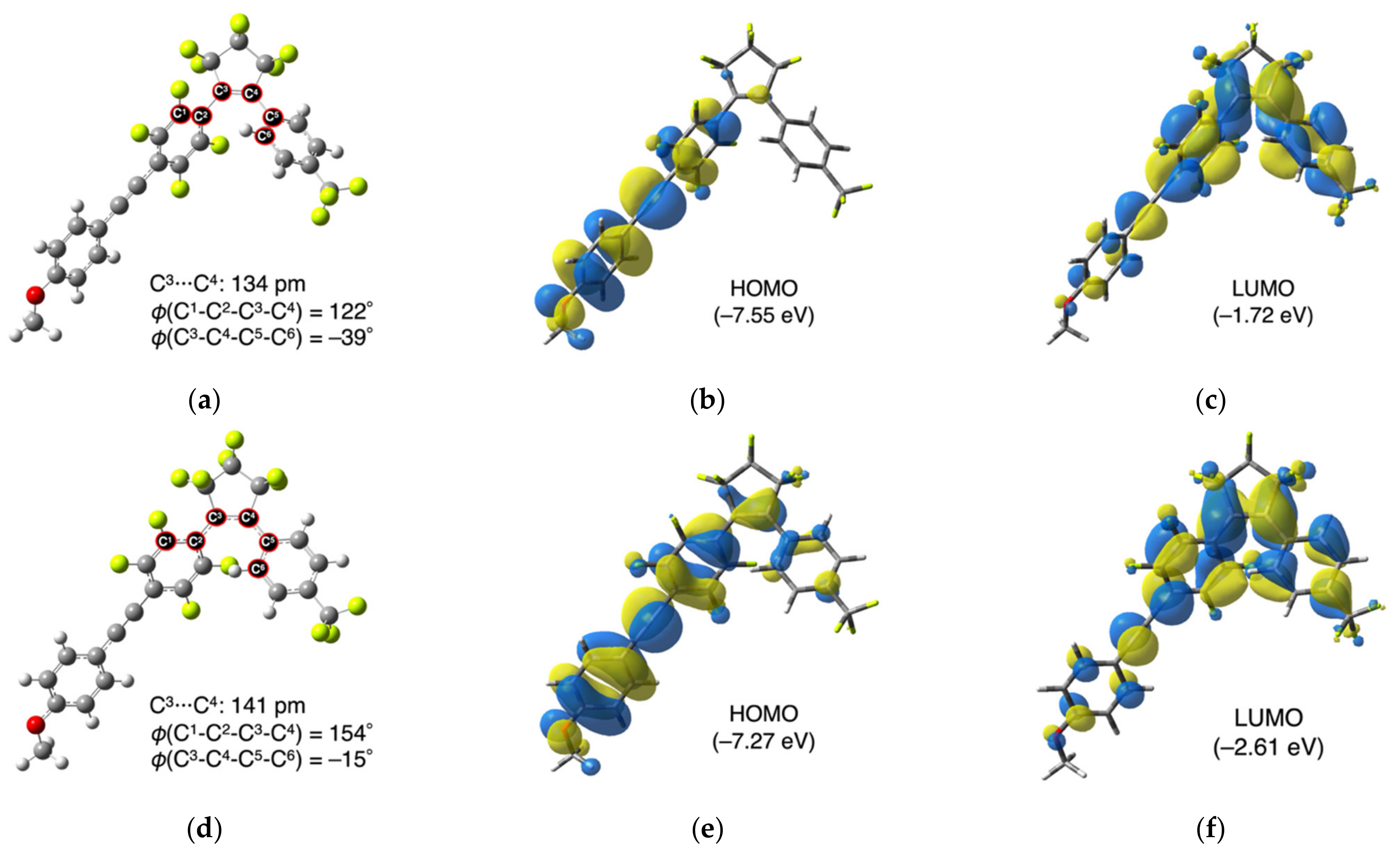
3.1. Theoretical Assessment
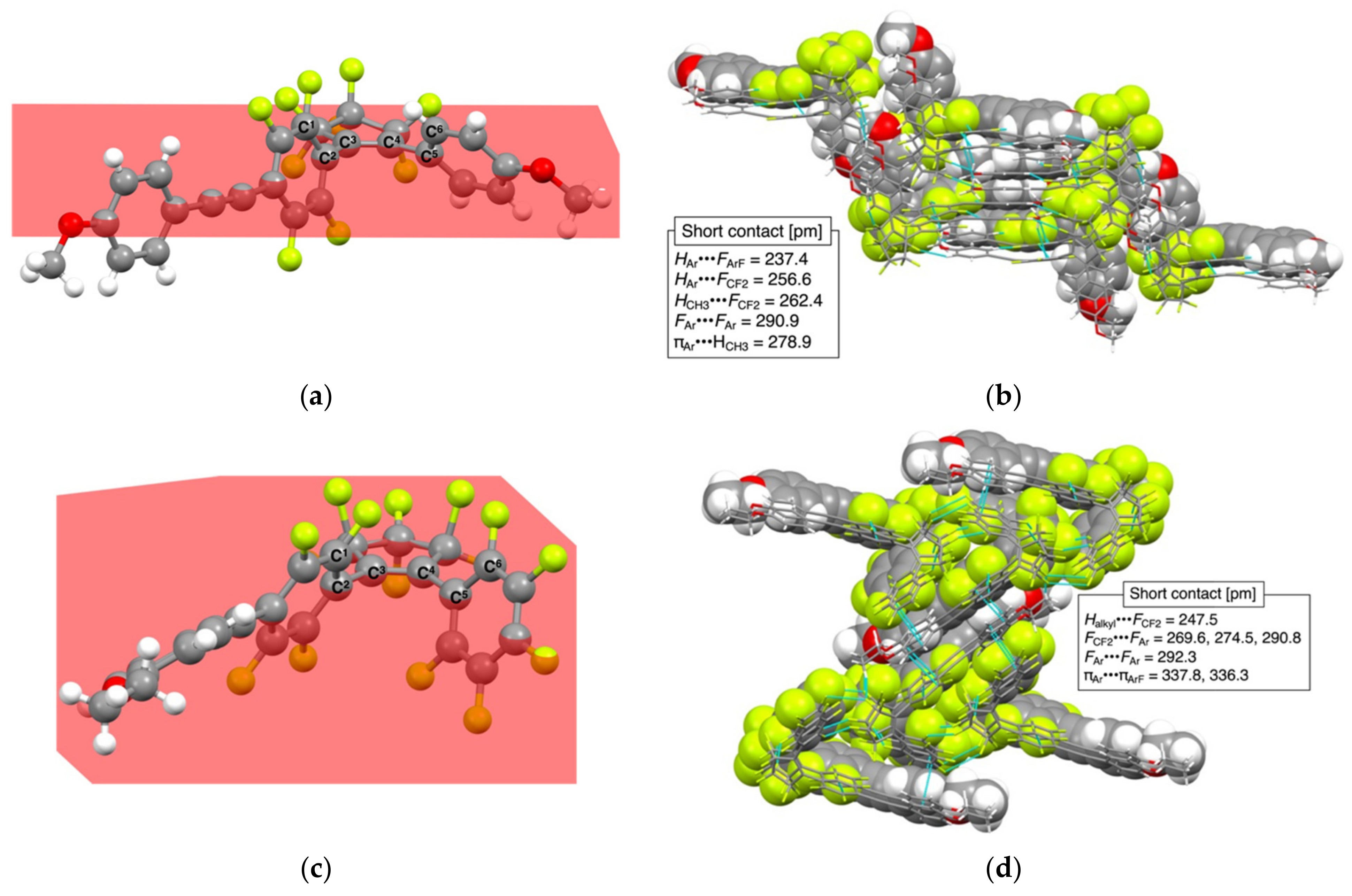
3.2. Crystal Structures of 1c and 2bB
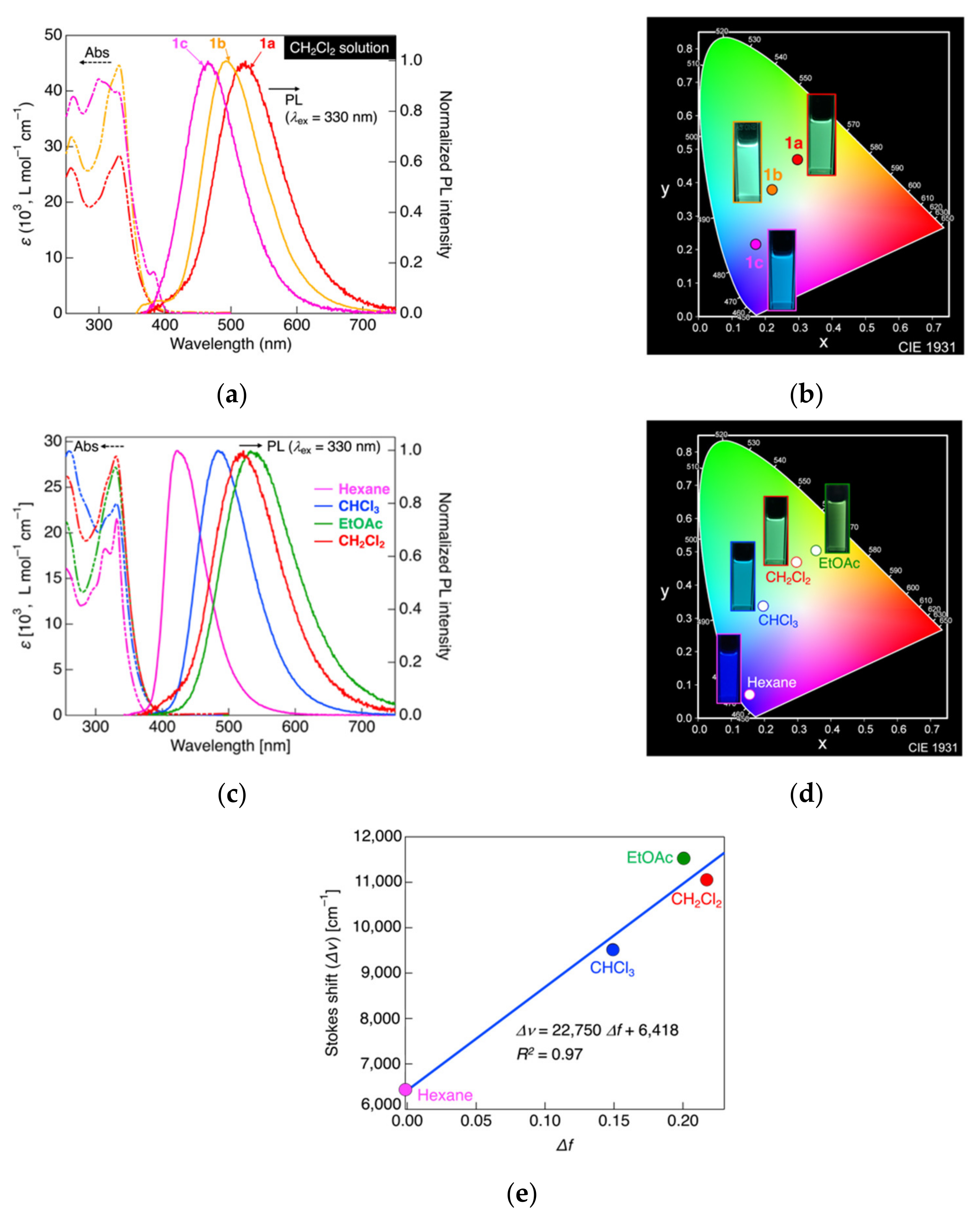
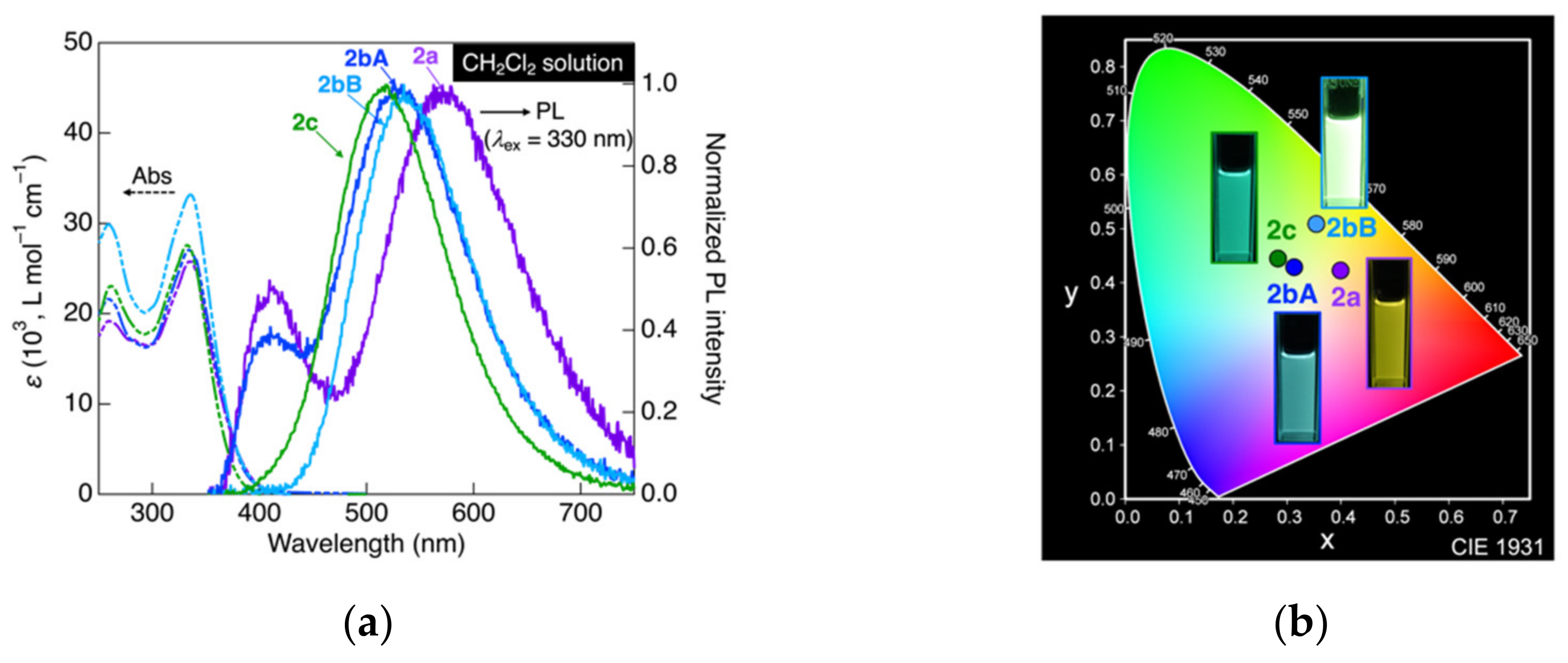
3.3. Photophysical Measurements in Solution State
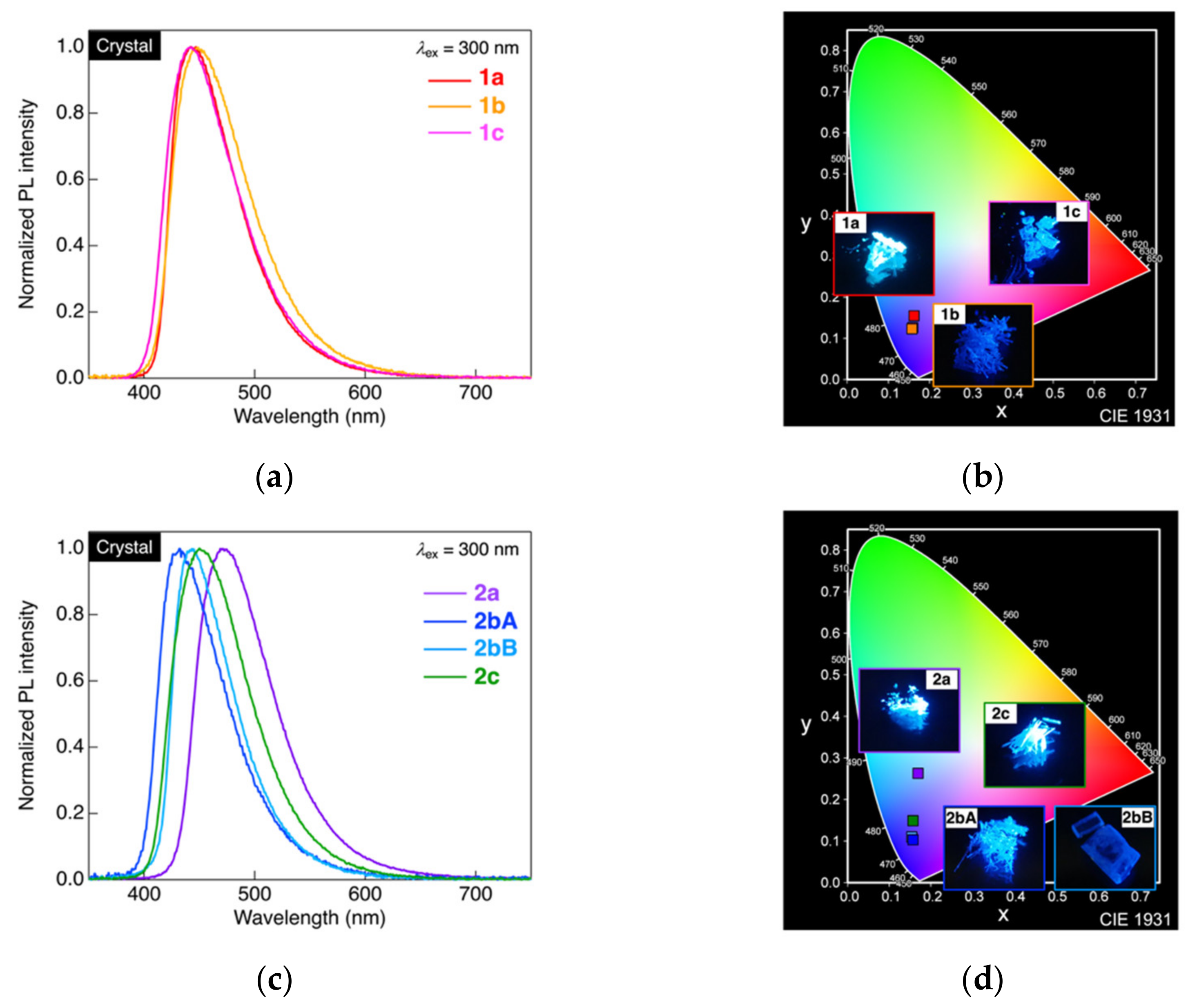
3.4. Crystalline-State Photoluminescence Behavior
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, N.N.M.Y.; Idris, A.; Abidin, Z.H.Z.; Tajuddin, H.A.; Abdullah, Z. White light employing luminescent engineered large (mega) Stokes shift molecules: A review. RSC Adv. 2021, 11, 13409–13445. [Google Scholar] [CrossRef]
- Holden, L.; Burke, C.S.; Cullinane, D.; Keyes, T.E. Strategies to promote permeation and vectorization, and reduce cytotoxity of metal complex luminophores for bioimaging and intracellular sensing. RSC Chem. Biol. 2021, 2, 1021–1049. [Google Scholar] [CrossRef]
- Ansari, A.A.; Thakur, V.K.; Chen, G. Functionalized upconversion nanoparticles: New strategy towards FRET-based luminescence bio-sensing. Coord. Chem. Rev. 2021, 436, 213821. [Google Scholar] [CrossRef]
- Tang, M.C.; Chan, M.Y.; Yam, V.W.W. Molecular design of luminescent gold(III) emitters as thermally evaporable and solution-processable organic light-emitting device (OLED) materials. Chem. Rev. 2021, 121, 7249–7279. [Google Scholar] [CrossRef] [PubMed]
- Bispo-Jr, A.G.; Saraiva, L.F.; Lima, S.A.M.; Pires, A.M.; Davolos, M.R. Recent prospects on phosphor-converted LEDs for lighting, displays, phototherapy, and indoor farming. J. Lumines. 2021, 237, 118167. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Pan, A.; Yang, B.; He, L.; Wu, Y. Rare earth-free luminescent materials for WLEDs: Recent progress and perspectives. Adv. Mater. Technol. 2021, 6, 2000648. [Google Scholar] [CrossRef]
- Kim, H.S.; Park, S.R.; Suh, M.C. Concentration quenching behavior of thermally activated delayed fluorescence in a solid film. J. Phys. Chem. C 2017, 121, 13986–13997. [Google Scholar] [CrossRef]
- Birks, J.B. Photophysics of Aromatic Molecules; Birks, J.B., Ed.; Wiley: London, UK, 1970. [Google Scholar]
- Thomas, S.W.; Joly, G.D.; Swager, T.M. Chemical sensors based on amplifying fluorescent conjugated polymers. Chem. Rev. 2007, 107, 1339–1386. [Google Scholar] [CrossRef] [PubMed]
- Jenekhe, S.A.; Osaheni, J.A. Excimers and exciplexes of conjugated polymers. Science 1994, 265, 765–768. [Google Scholar] [CrossRef]
- Han, T.; Yan, D.; Wu, Q.; Song, N.; Zhang, H.; Wang, D. Aggregation-induced emission: A rising star in chemistry and materials science. Chin. J. Chem. 2021, 39, 677–689. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, H.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission: New vistas at the aggregate level. Angew. Chem. Int. Ed. 2020, 59, 9888–9907. [Google Scholar] [CrossRef]
- Leung, N.L.C.; Xie, N.; Yuan, W.; Liu, Y.; Wu, Q.; Peng, Q.; Miao, Q.; Lam, J.W.Y.; Tang, B.Z. Restriction of intramolecular motions: The general mechanism behind aggregation-induced emission. Chem. Eur. J. 2014, 20, 15349–15353. [Google Scholar] [CrossRef]
- Li, M.; Niu, Y.; Zhu, X.; Peng, Q.; Lu, H.Y.; Xia, A.; Chen, C.F. Tethrahydro[5]helicene-based imide dyes with intense fluorescence in both solution and solid state. Chem. Commun. 2014, 50, 2993–2995. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Liu, Q.; Dong, L.; Cai, Z.; Shi, J.; Zhi, J.; Tong, B.; Dong, Y. The dual-state luminescent mechanism of 2,3,4,5-Tetraphenyl-1H-pyrrole. Chem. Eur. J. 2018, 24, 14269–14274. [Google Scholar] [CrossRef]
- Qiu, Q.; Xu, P.; Zhu, Y.; Yu, J.; Wei, M.; Xi, W.; Feng, H.; Chen, J.; Qian, Z. Rational design of dual-state emission luminogens with solvatochromism by combining a partially shared donor-acceptor pattern and twisted structures. Chem. Eur. J. 2019, 25, 15983–15987. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ren, L.; Dang, D.; Zhi, Y.; Wang, X.; Meng, L. A strategy of “Self-Isolated Enhanced Emission” to achieve highly emissive dual-state emission for organic luminescent materials. Chem. Eur. J. 2018, 24, 10383–10389. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, X.; Peng, Z.; Zhang, Y.; Liu, P.; Cai, Z.; Tong, B.; Shi, J.; Dong, Y. A “Turn-On” fluorescent chemosensor with the aggregation-induced emission characteristics for high-sensitive detection of Ce ion. Sens. Actuators B 2018, 267, 351–356. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, P.C.; Xu, H.B.; Tang, M.J.; Yang, X.P.; Huang, S.; Deng, J.G. Switchable sensitizers stepwise lighting up lanthanide emissions. Sci. Rep. 2015, 5, 9335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Singh, P.; Kumar, P.; Srivastava, R.; Pal, S.K.; Ghosh, S. Exploring an emissive charge transfer process in zero-twist donor-acceptor molecular design as a dual-state emitter. J. Phys. Chem. C 2016, 120, 12723–12733. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, M.; Zhu, X.; Wan, Y. One-pot synthesis of dual-state emission (DSE) luminogens containing the V-shape furo[2,3-b]furan scaffold. Chin. Chem. Lett. 2021, 32, 445–448. [Google Scholar] [CrossRef]
- Yamada, S.; Nishizawa, A.; Morita, M.; Hosokai, T.; Okabayashi, Y.; Agou, T.; Hosoya, T.; Kubota, T.; Konno, T. Synthesis and characterization of bent fluorine-containing donor-p-acceptor molecules as intense luminophores with large Stokes shifts. Org. Biomol. Chem. 2019, 17, 6911–6919. [Google Scholar] [CrossRef]
- CrysAlisPro 1.171.39.43a. Rigaku Oxford Diffraction, Rigaku Corporation: Akishima, Japan, 2015. Available online: https://www.rigakuxrayforum.com/ (accessed on 30 September 2021).
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Okuno, K.; Shigeta, Y.; Kishi, R.; Nakano, M. Non-empirical tuning of CAM-B3LYP functional in time-dependent density functional theory for excitation energies of diarylethene derivatives. Chem. Phys. Lett. 2013, 585, 201–206. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Andzelm, J.; Kölmel, C.; Klamt, A. Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J. Chem. Phys. 1995, 103, 9312–9320. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Grad, J.; Yan, Y.J.; Mukamel, S. Time-dependent self-consistent field approximation for semiclassical dynamics using Gaussian wavepackets in phase space. Chem. Phys. Lett. 1987, 134, 291–295. [Google Scholar] [CrossRef]
- Makri, N. Time-dependent self-consistent field approximation with explicit two-body correlations. Chem. Phys. Lett. 1990, 169, 541–548. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Mataga, N.; Kaifu, Y.; Koizumi, M. The solvent effect on fluorescence spectrum, change of solute-solvent interaction during the lifetime of excited solute molecule. Bull. Chem. Soc. Jpn. 1955, 28, 690–691. [Google Scholar] [CrossRef] [Green Version]
- Mataga, N.; Kaifu, Y.; Koizumi, M. Solvent effects upon fluorescence spectra and the dipole moments of excited molecules. Bull. Chem. Soc. Jpn. 1956, 29, 465–470. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Solvent | λabs [ε (103, L mol−1 cm−1)] 2 | λPL (ΦPL) 3 | CIE Coordinate (x, y) |
|---|---|---|---|---|
| 1a | CH2Cl2 | 257 [25.5], 315sh [39.8], 331 [27.7] | 522 (0.99) | (0.3, 0.47) |
| Hexane | 255 [16.1], 315 [18.2], 332 [21.6] | 422 (0.7) | (0.16, 0.07) | |
| CHCl3 | 260 [28.9], 316sh [21.7], 331 [23.2] | 438 (0.94) | (0.2, 0.34) | |
| EtOAc | 256 [21.1], 314sh [23.3], 330 [27.2] | 534 (0.55) | (0.36, 0.5) | |
| 1b | CH2Cl2 | 259 [30.1], 314sh [24.9], 332 [42.8] | 402sh, 493 (0.90) | (0.22, 0.38) |
| 1c | CH2Cl2 | 261 [38.6], 300 [41.9], 311sh [41.4], 328 [39.9] | 465 (0.57) | (0.17, 0.22) |
| Molecule | λabs [ε (103, L mol−1 cm−1)] 2 | λPL (ΦPL) 3 | CIE Coordinate (x, y) |
|---|---|---|---|
| 2a | 259 [19.2], 335 [25.8] | 409, 571 (0.35) | (0.4, 0.42) |
| 2bA | 259 [21.6], 334 [27.1] | 410, 533 (0.78) | (0.31, 0.43) |
| 2bB | 259 [29.9], 336 [33.2] | 535 (0.26) | (0.35, 0.51) |
| 2c | 261 [23.0], 332 [27.6] | 518 (0.88) | (0.28, 0.44) |
| Molecule | λPL1 | ΦPL2 | CIE Coordinate (x, y) |
|---|---|---|---|
| 1a | 449 | 0.98 | (0.16, 0.16) |
| 1b | 443 | 1.0 | (0.16, 0.12) |
| 1c | 443 | 1.0 | (0.16, 0.13) |
| 2a | 471 | 1.0 | (0.17, 0.26) |
| 2bA | 432 | 0.52 | (0.16, 0.1) |
| 2bB | 443 | 0.99 | (0.15, 0.11) |
| 2c | 450 | 1.0 | (0.16, 0.15) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, S.; Nishizawa, A.; Kobayashi, K.; Yoshida, K.; Morita, M.; Agou, T.; Hosoya, T.; Fukumoto, H.; Konno, T. Unsymmetrical Hexafluorocyclopentene-Linked Twisted ?-Conjugated Molecules as Dual-State Emissive Luminophores. Symmetry 2021, 13, 1885. https://doi.org/10.3390/sym13101885
Yamada S, Nishizawa A, Kobayashi K, Yoshida K, Morita M, Agou T, Hosoya T, Fukumoto H, Konno T. Unsymmetrical Hexafluorocyclopentene-Linked Twisted ?-Conjugated Molecules as Dual-State Emissive Luminophores. Symmetry. 2021; 13(10):1885. https://doi.org/10.3390/sym13101885
Chicago/Turabian StyleYamada, Shigeyuki, Akito Nishizawa, Kazuki Kobayashi, Keigo Yoshida, Masato Morita, Tomohiro Agou, Takaaki Hosoya, Hiroki Fukumoto, and Tsutomu Konno. 2021. "Unsymmetrical Hexafluorocyclopentene-Linked Twisted ?-Conjugated Molecules as Dual-State Emissive Luminophores" Symmetry 13, no. 10: 1885. https://doi.org/10.3390/sym13101885
APA StyleYamada, S., Nishizawa, A., Kobayashi, K., Yoshida, K., Morita, M., Agou, T., Hosoya, T., Fukumoto, H., & Konno, T. (2021). Unsymmetrical Hexafluorocyclopentene-Linked Twisted ?-Conjugated Molecules as Dual-State Emissive Luminophores. Symmetry, 13(10), 1885. https://doi.org/10.3390/sym13101885