
Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey

Abstract
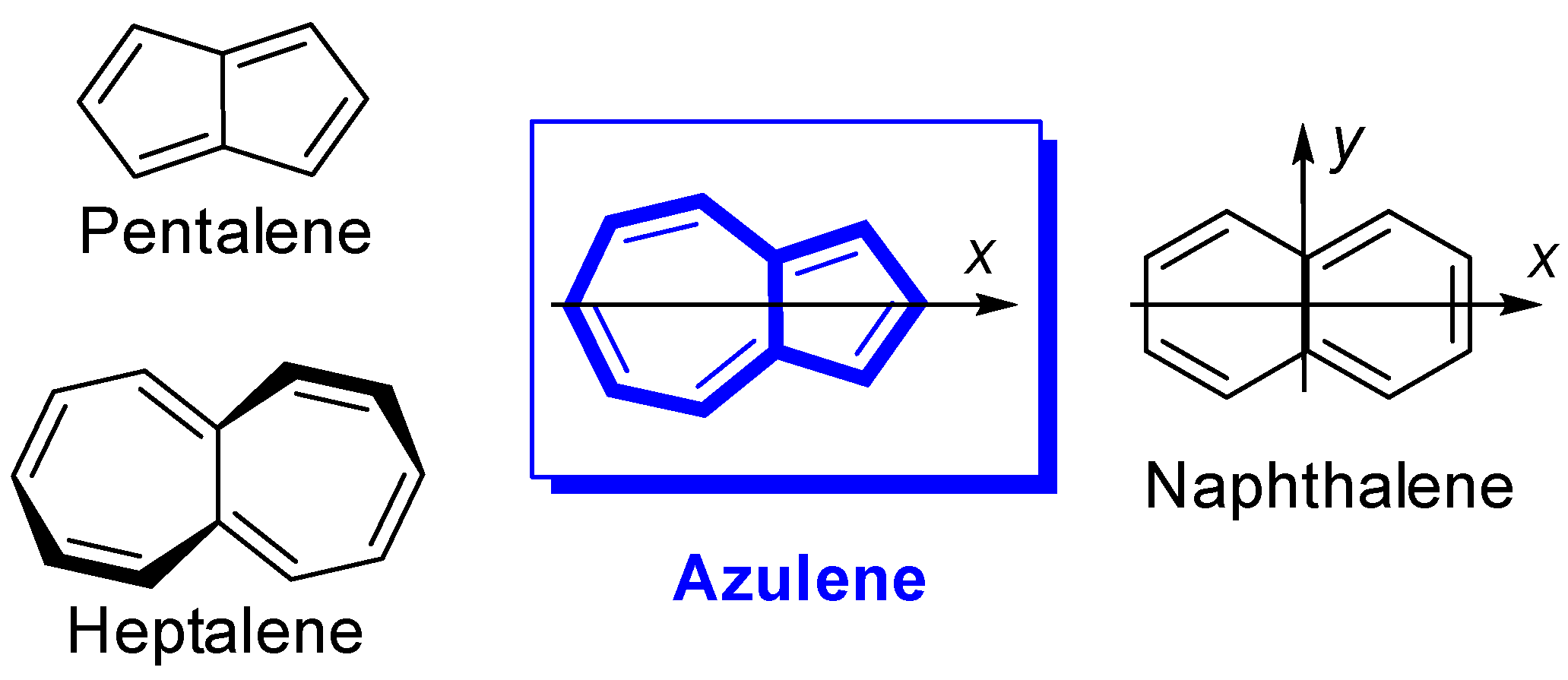
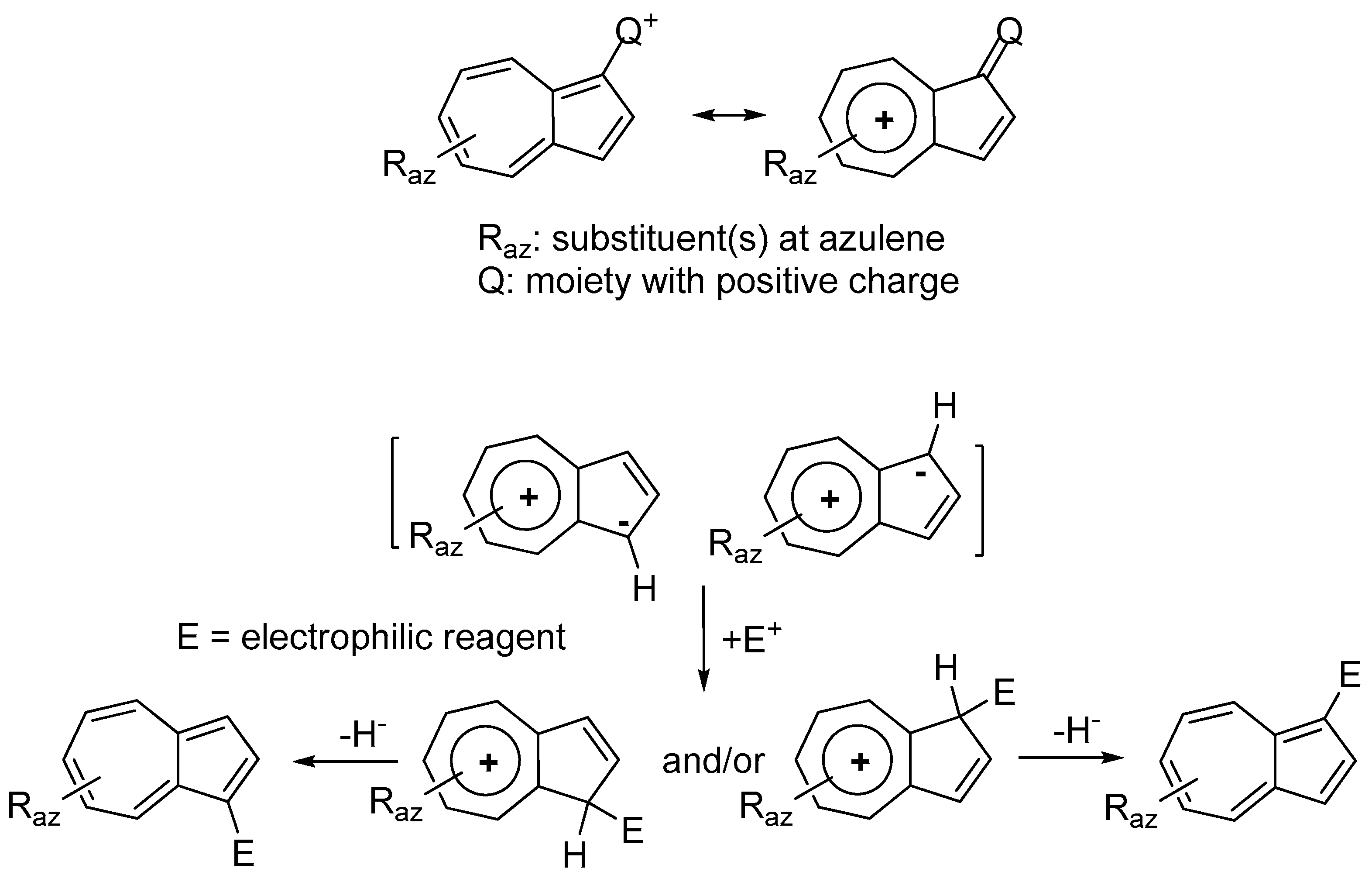
:1. Introduction
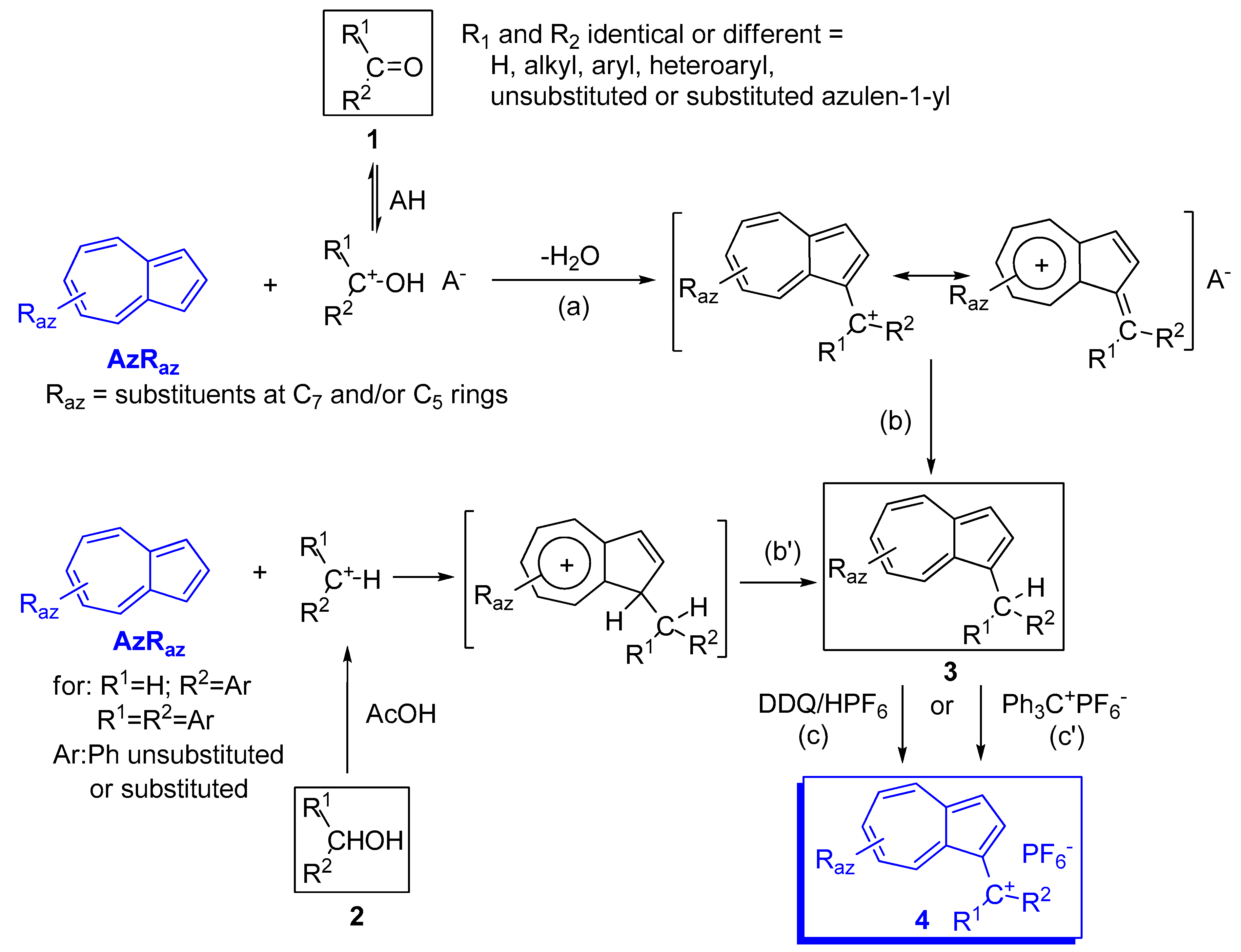
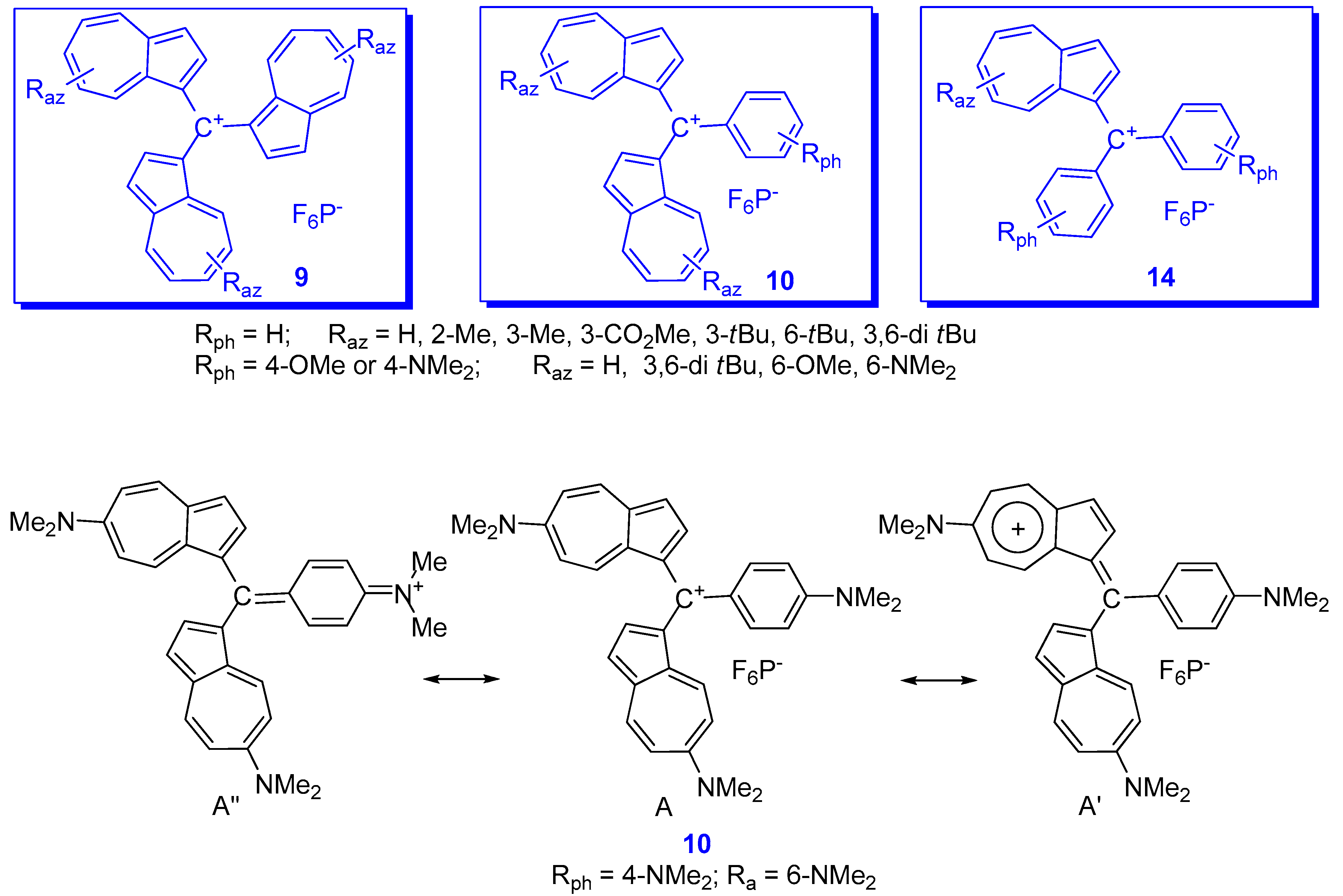
2. Triaryl-methylium Ion with at Least One Azulen-1-yl Moiety
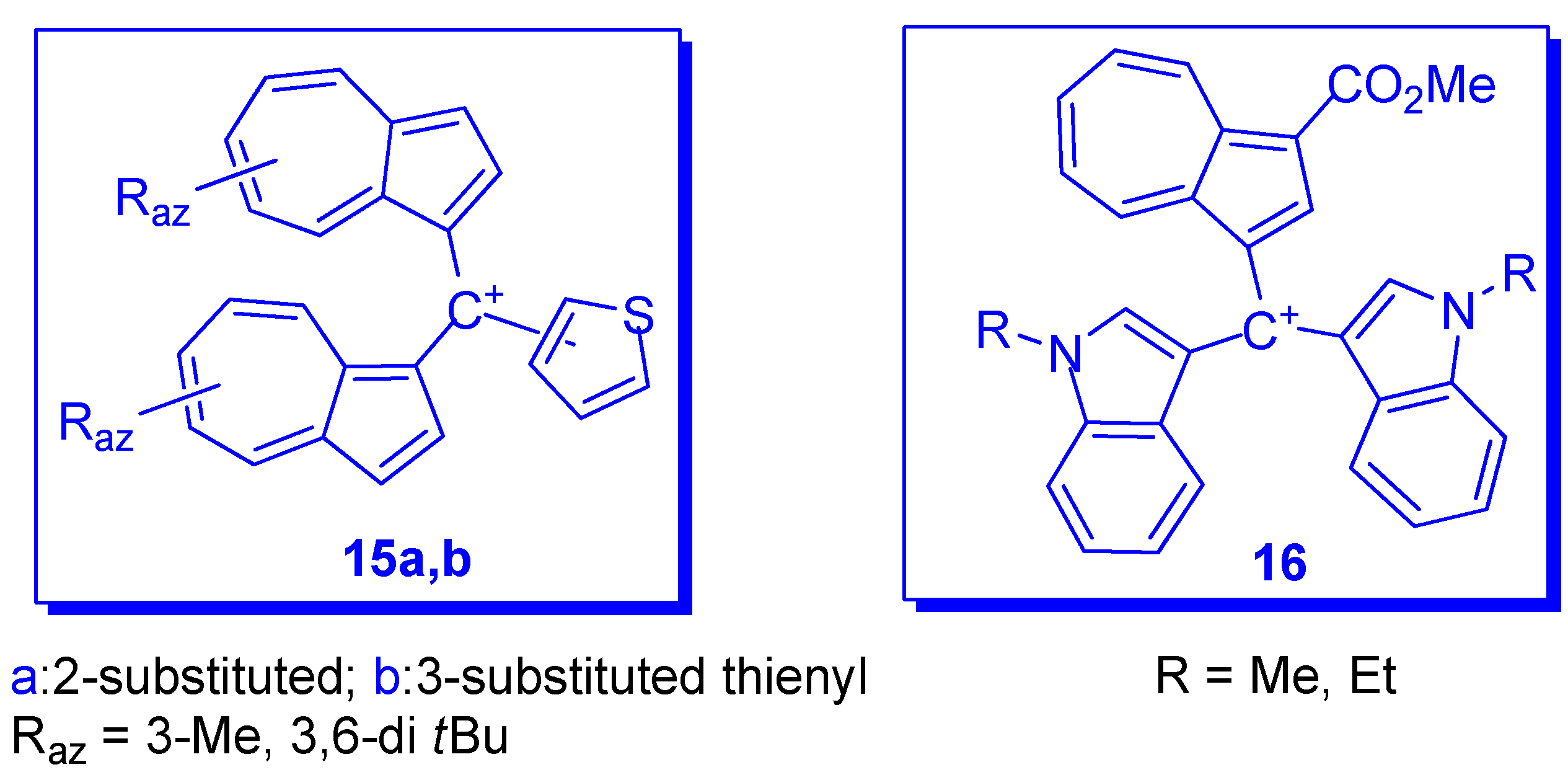
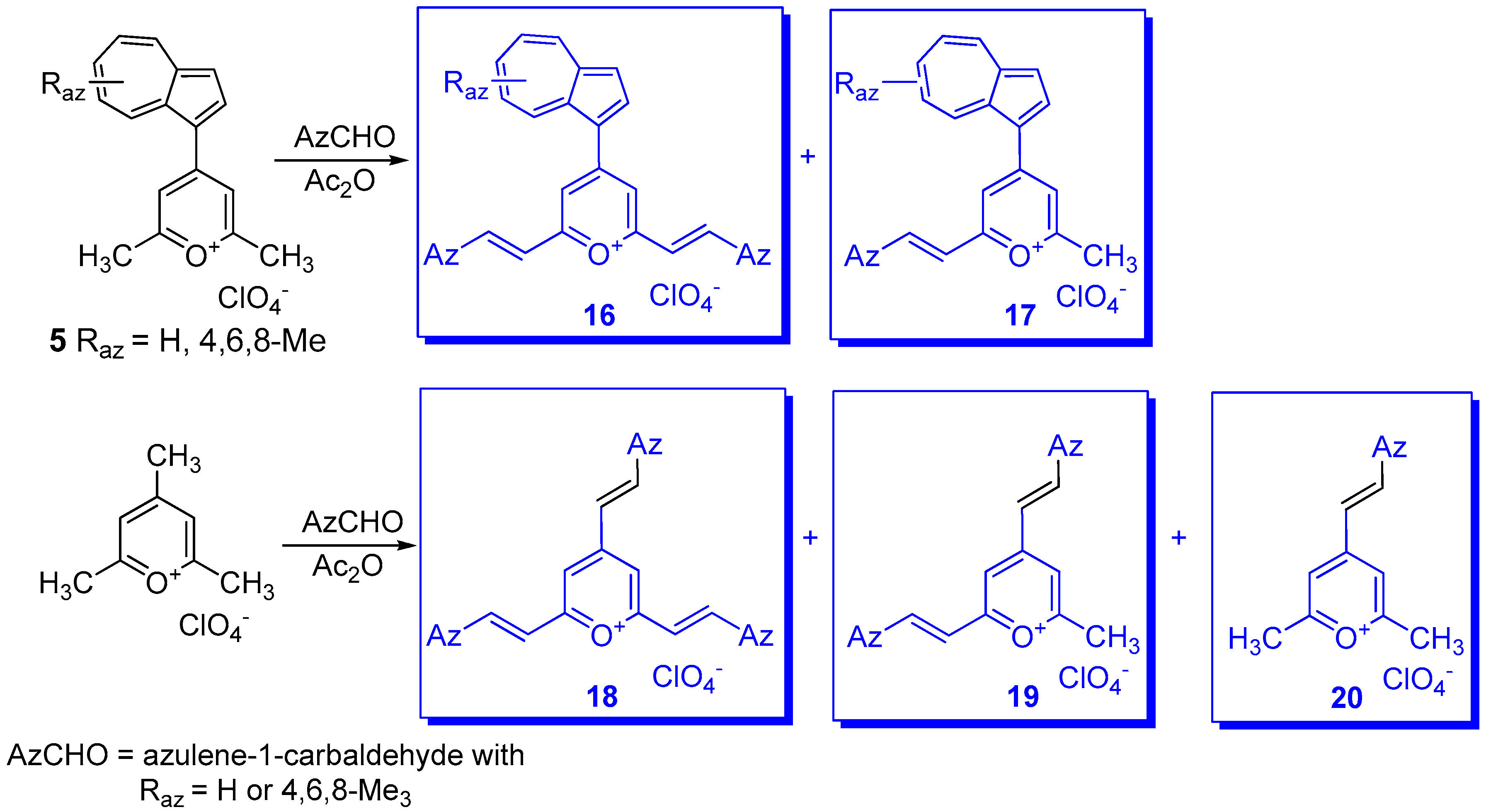
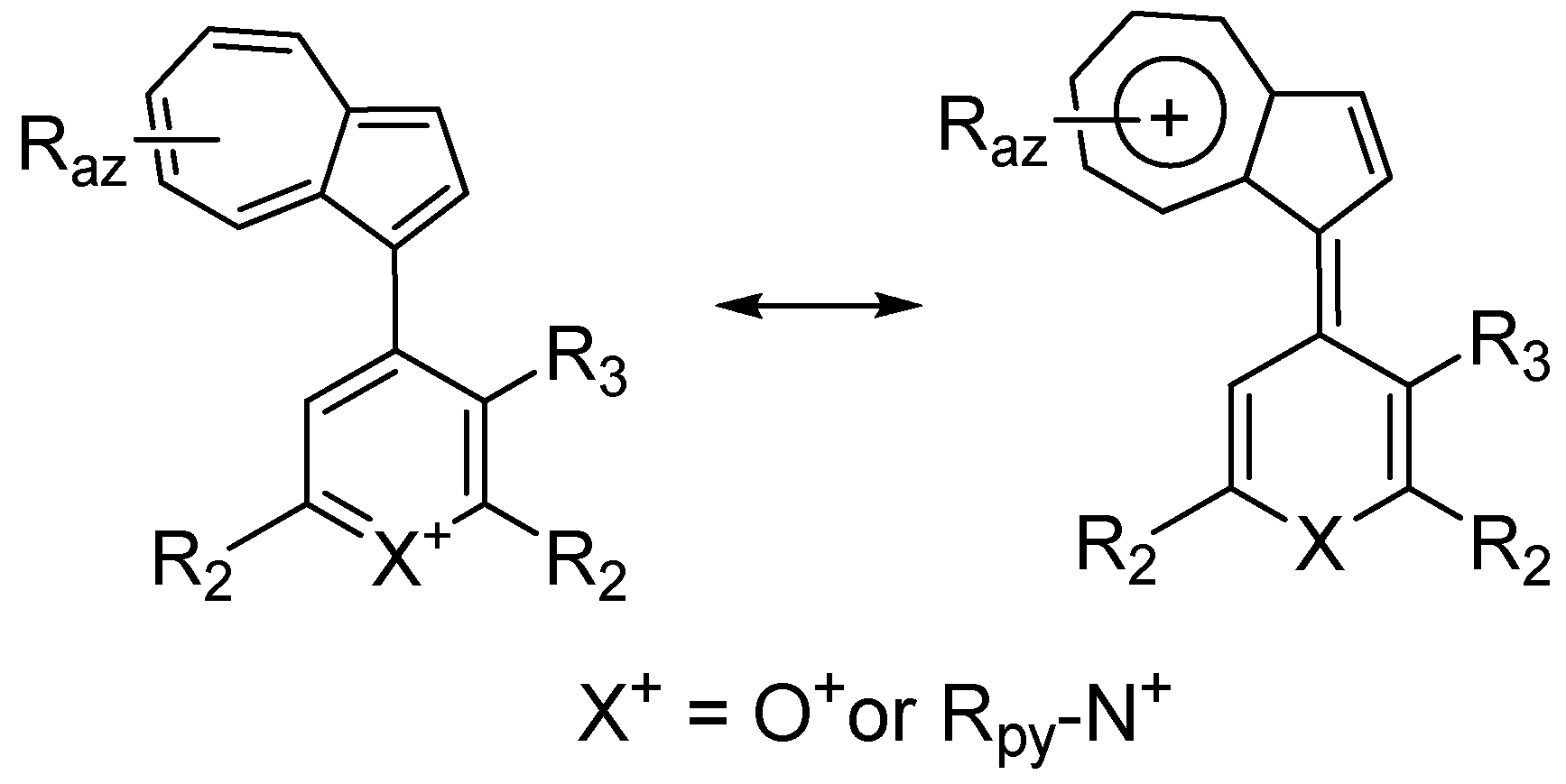
3. Charged Hetero-aryls Stabilized by Azulen-1-yl Moieties
4. Other Systems Stabilized by Azulen-1-yl Moieties
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Solà, M. Forty years of Clar’s aromatic π-sextet rule. Front. Chem. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauben, H.J., Jr.; Bertelli, D.J. Heptalene. J. Am. Chem. Soc. 1961, 83, 4659–4660. [Google Scholar] [CrossRef]
- Bally, T.; Chai, S.; Neuenschwander, M.; Zhu, Z. Pentalene: Formation, Electronic, and Vibrational Structure. J. Am. Chem. Soc. 1997, 119, 1869–1875. [Google Scholar] [CrossRef]
- Mikheev, Y.A.; Guseva, L.N.; Ershov, Y.A. Nature of the aromaticity of azulene and the dimers responsible for its chromaticity. Russ. J. Phys. Chem. 2012, 86, 1875–1880. [Google Scholar] [CrossRef]
- Tobe, Y. Non-Alternant Non-Benzenoid Aromatic Compounds: Past, Present, and Future. Chem. Rec. 2014, 15, 86–96. [Google Scholar] [CrossRef]
- Fischer, L.J.; Dutton, A.S.; Winter, A.H. Anomalous effect of non-alternant hydrocarbons on carbocation and carbanion electronic configurations. Chem. Sci. 2017, 8, 4231–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Morita, N.; Asao, T. Azulene analogues of triphenylmethyl cation; extremely stable hydrocarbon cations. Tetrahedron Lett. 1991, 32, 773–776. [Google Scholar] [CrossRef]
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. Z. Chem. 1962, 2, 275–276. [Google Scholar] [CrossRef]
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. I. J. Prakt. Chem. 1967, 35, 249–261. [Google Scholar] [CrossRef]
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. II. J. Prakt. Chem. 1967, 35, 262–270. [Google Scholar] [CrossRef]
- Zeller, K.-P. Houben Weyl, Methoden der Organischen Chemie; G. Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 1985; Volume V/2c, pp. 258–263. [Google Scholar]
- Kirby, E.C.; Reid, D.H. Conjugated Cyclic Hydrocarbons and Their Heterocyclic Analogues. The Condensation of Azulenes with Homocyclic and Heterocyclic Aromatic Aldehydes in the Presence of Perchloric Acid. Part II. J. Chem. Soc. 1960, 494–501. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated Cyclic Hydrocarbons and Their Heterocyclic Analogues. The Condensation of Azulenes with aliphatic aldehydes in the presence of perchloric acid. Part VI. J. Chem. Soc. 1961, 3579–3593. [Google Scholar] [CrossRef]
- Hafner, K.; Pelster, H.; Schneider, J. Zur kenntnis der azuleneVII. 1- bzw. 3-alkyliden-azulenium salze. Justus Liebigs Ann. Chem. 1961, 650, 62–80. [Google Scholar] [CrossRef]
- Asao, T. Azulenic novel π-electronic compounds. Pure Appl. Chem. 1990, 62, 507–512. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Tris(3,6-di-t-butyl-1-azulenyl)methyl Cation; Hydrocarbon with the highest pKR+. Tetrahedron Lett. 1994, 35, 751–754. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Dynamic stereochemistry of tri(2-methyl-1-azulenyl)methyl cation; steric effect of 2-methyl groups on rotational barriers and mechanism. Tetrahedron Lett. 1994, 35, 3723–3726. [Google Scholar] [CrossRef]
- Ito, S.; Fujita, M.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Bis(3-methyl-1-azulenyl)(1-naphthyl)methyl Hexafluorophosphate. Clear Evidence of the One-Ring Flip Mechanism of Molecular Propeller by Controlling the Flipping Ring. Chem. Lett. 1995, 475–476. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Tris(2-methyl-1-azulenyl)methyl Cation and the Corresponding Methane Derivative. Evidence That the Conjugative Effect Largely Contributes to the Transition State of the Ring Flipping. Bull. Chem. Soc. Jpn. 1995, 68, 2639–2648. [Google Scholar] [CrossRef]
- Ito, S.; Fujita, M.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Di-1-azulenyl(1- and 2-naphthyl)methyl Hexafluorophosphates. Clear Evidence of the One-Ring Flip Mechanism of a Molecular Propeller by Controlling the Flipping Ring. Bull. Chem. Soc. Jpn. 1995, 68, 3611–3620. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Syntheses of Azulene Analogues of Triphenylmethyl Cation: Extremely Stable Hydrocarbon Carbocations and the First Example of a One-Ring Flip as the Threshold Rotation Mechanism for Molecular Propellers. Bull. Chem. Soc. Jpn. 1995, 68, 1409–1436. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. The Most Stable Methylcation. Tris(6-methoxy-1-azulenyl)methyl Hexafluorophosphat. Chem. Lett. 1996, 175–176. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Synthesis and Properties of Extremely Stable Tris(6-methoxy-1-azulenyl)methyl Cation and a Series of Di(1-azulenyl)phenylmethyl and (1-Azulenyl)diphenylmethyl Cations Stabilized by Methoxy Substituents. Bull. Chem. Soc. Jpn. 1999, 72, 839–849. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Syntheses and Properties of Extremely Stable Di(1-azulenyl)phenylmethyl and (1-Azulenyl)diphenylmethyl Cations Having Dimethylamino Substituents on Their Phenyl Groups. Bull. Chem. Soc. Jpn. 1996, 69, 3225–3237. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Tris[6-(dimethylamino)-1-azulenyl]methyl Hexafluorophosphate. Extremely Stable Methyl Cation with the Highest pKR+Value. J. Org. Chem. 1999, 64, 5815–5821. [Google Scholar] [CrossRef]
- Deno, N.C.; Schriesheim, A. Carbonium Ions. II. Linear Free Energy Relationships in Arylcarbonium Ion Equilibria. J. Am. Chem. Soc. 1955, 77, 3051–3054. [Google Scholar] [CrossRef]
- Ito, S.; Kikuchi, S.; Okujima, T. Cations and Dications Composed of Two Di(1-azulenyl)methylium Units Connected with 2,5-Thiophenediyl and 2,5-Thienothiophenediyl Spacers. J. Org. Chem. 2001, 66, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Iki, M.; Ikeda, C.; Imafuku, K. Synthesis and Properties of (1-Azulenyl)di(3-indolyl)methylium Hexafluorophosphates. Heterocycles 2005, 66, 275–283. [Google Scholar] [CrossRef]
- Takekuma, S.; Sasaki, M.; Takekuma, H.; Yamamoto, H. Preparation and Characteristic Properties of 1,4-Bis(3-guaiazulenylmethylium)benzene Bishexafluorophosphate. Chem. Lett. 1999, 28, 999–1000. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behaviors of Di- and Trications Composed of Di(1-azulenyl)methylium Units Connected byp- andm-Phenylene and 1,3,5-Benzenetriyl Spacers. Bull. Chem. Soc. Jpn. 2000, 73, 1865–1874. [Google Scholar] [CrossRef]
- Deuchert, K.; Hünig, S. Multistage Organic Redox Systems-A General Structural Principle. Angew. Chem. Int. Ed. Engl. 1978, 17, 875–886. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Syntheses, Properties, and Redox Behaviors of Di(1-azulenyl)ferrocenylmethyl Cations and 1,3-Bis[(1-azulenyl)ferrocenylmethylium]azulene Dication. J. Org. Chem. 1996, 61, 5077–5082. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S. The Preparation and Properties of Heteroarylazulenes and Hetero-Fused Azulenes. Adv. Heterocycl. Chem. 2018, 1–54. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L. Synthesis of azulenic compounds substituted in the 1-position with heterocycles. Monatsh. Chem. 2018, 150, 139–161. [Google Scholar] [CrossRef]
- Krivun, S.V.; Baranov, S.N.; Buryiak, A.I. Introduction of the pyrylium ring into compounds of the aromatic and heterocyclic series. Chem. Heterocycl. Compd. 1971, 7, 1233–1236. [Google Scholar] [CrossRef]
- Dorofeenko, G.N.; Koblik, A.V.; Polyakova, T.I.; Murad’yan, L.A. Synthesis of 6-hetaryl-substituted azulenes and their reactions with 2,6-diphenylpyrylium perchlorate. Chem. Heterocycl. Compd. 1980, 16, 807–810. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.C.; Enache, C. Azulene-substituted Pyranylium Salts. Syntheses and Products Characterization. J. Heterocycl. Chem. 2006, 43, 963–977. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Zaharia, O.C. Enache. 2-(Azulen-1-yl)-4,6-diphenyl Substituted Six-membered Heteroaromatics. J. Heterocycl. Chem. 2008, 45, 1139–1147. [Google Scholar] [CrossRef]
- Said, S.A.; Fiksdahl, A. Stereoselective transformation of amines via chiral 2,4,6-triphenylpyridinium intermediates. Tetrahedron Asymmetry 2001, 12, 1947–1951. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.; Chiraleu, F.; Enache, C. Azulene-substituted Pyridines and Pyridinium Salts. 1. Azulene-substituted Pyridinium salts. J. Heterocycl. Chem. 2007, 44, 245–251. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.; Chiraleu, F.; Enache, C. Azulene-substituted Pyridines and Pyridinium Salts. 2. J. Heterocycl. Chem. 2007, 44, 251–260. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Cristea, M.; Tecuceanu, V.; Hanganu, A.; Enache, C. 4-(Azulen-1-yl) six-membered heteroaromatics substituted with thiophen-2-yl or furan-2-yl moieties in 2 and 6 positions. J. Heterocycl. Chem. 2011, 48, 1019–1027. [Google Scholar] [CrossRef]
- Razus, A.C.; Cristian, L.; Nica, S.; Zaharia, O.; Hanganu, A. Pyrylium salts with 2-(azulene-1-yl) vinyl substituents in 2-, 4- and/or 6-positions. Arkivoc 2011, 9, 38–50. [Google Scholar] [CrossRef] [Green Version]
- Birzan, L.; Cristea, M.; Draghici, C.; Tecuceanu, V.; Hanganu, A.; Ungureanu, E.-M.; Razus, A.C. 4-(Azulen-1-yl) six-membered heteroaromatics substituted in 2- and 6- positions with 2-(2-furyl)vinyl, 2-(2-thienyl)vinyl or 2-(3-thienyl)vinyl moieties. Tetrahedron 2017, 73, 2488–2500. [Google Scholar] [CrossRef]
- Dykes, G.; Boruski, S.; Heiermann, J.; Korning, J.; Opwis, K.; Henkel, G.; Kocerling, M. First intermolecular palladium catalyzed arylation of an unfunctionalized aromatic hydrocarbon. J. Organomet. Chem. 2000, 606, 108–110. [Google Scholar] [CrossRef]
- Rasala, D.; Bak, T.; Kolehmainen, E.; Styrcz, S.; Gawinecki, R. Transmission of electronic effects in 4-aryl-2, 6-diphenylpyrylium perchlorates and related compounds. J. Phys. Org. Chem. 1996, 9, 631–638. [Google Scholar] [CrossRef]
- Detty, M.R.; McKelvey, J.M.; Luss, H.R. Tellurapyrylium dyes. The electron-donating properties of the chalcogen atoms to the chalcogenapyrylium nuclei and their radical dications, neutral radicals, and anions. Organometallics 1988, 7, 1131–1147. [Google Scholar] [CrossRef]
- Takekuma, S.; Mizutani, K.; Inoue, K.; Nakamura, M.; Sasaki, M.; Minematsu, T.; Takekuma, H. Reactions of azulene and guaiazulene with all-trans-retinal and trans-cinnamaldehyde: Comparative studies on spectroscopic, chemical, and electrochemical properties of monocarbenium-ions stabilized by expanded π-electron systems with an azulenyl or 3-guaiazulenyl group. Tetrahedron 2007, 63, 3882–3893. [Google Scholar] [CrossRef]
- Farrell, T.; Meyer-Friedrichsen, T.; Malessa, M.; Haase, D.; Saak, W.; Asselberghs, I.; Wostyn, K.; Clays, K.; Persoons, A.; Heck, J.; et al. Azulenylium and guaiazulenylium cations as novel accepting moieties in e. xtended sesquifulvalene type D-A NLO chromophores. J. Chem. Soc. Dalton Trans. 2001, 29–36. [Google Scholar] [CrossRef]
- Kerber, R.C.; Hsu, C.-M. Substituent effects on cyclopropenium ions. J. Am. Chem. Soc. 1973, 95, 3239–3245. [Google Scholar] [CrossRef]
- Moss, R.A.; Shen, S.; Krogh-Jespersen, K.; Potenza, J.A.; Schugar, H.J.; Munjal, R.C. Cyclopropyl/phenylcyclopropenyl cations: Studies in stabilization. J. Am. Chem. Soc. 1986, 108, 134–140. [Google Scholar] [CrossRef]
- Agranat, I.; Aharon-Shalom, E. Stabilization of Cyclopropenium Ion and Cyclopropenone by Guaiazulene. J. Org. Chem. 1976, 41, 2379–2383. [Google Scholar] [CrossRef]
- Sprenger, H.-E.; Ziegenbein, W. Kondensationsprodukte aus Quadratsäure und tertiären aromatischen Aminen. Angew. Chem. 1966, 78, 937–938. [Google Scholar] [CrossRef]
- Lynch, D.E.; Hamilton, D.G. The History of Azulenyl Squaraines. Aust. J. Chem. 2017, 70, 857–872. [Google Scholar] [CrossRef]
- Pham, W.; Weissleder, R.; Tung, C.-H. An Azulene Dimer as a Near-Infrared Quencher. Angew. Chem. Int. Ed. 2002, 41, 3659–3662. [Google Scholar] [CrossRef]
- Lash, T.D. Out of the Blue! Azuliporphyrins and Related Carbaporphyrinoid Systems. Acc. Chem. Res. 2016, 49, 471–482, and the references herein. [Google Scholar] [CrossRef]
- Colby, D.A.; Lash, T.D. Calix[4]azulene. J. Org. Chem. 2002, 67, 1031–1033. [Google Scholar] [CrossRef]
- Sprutta, N.; Maćkowiak, S.; Kocik, M.; Szterenberg, L.; Lis, T.; Latos-Grażyński, L. Tetraazuliporphyrin Tetracation. Angew. Chem. Int. Ed. 2009, 48, 3337–3341. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
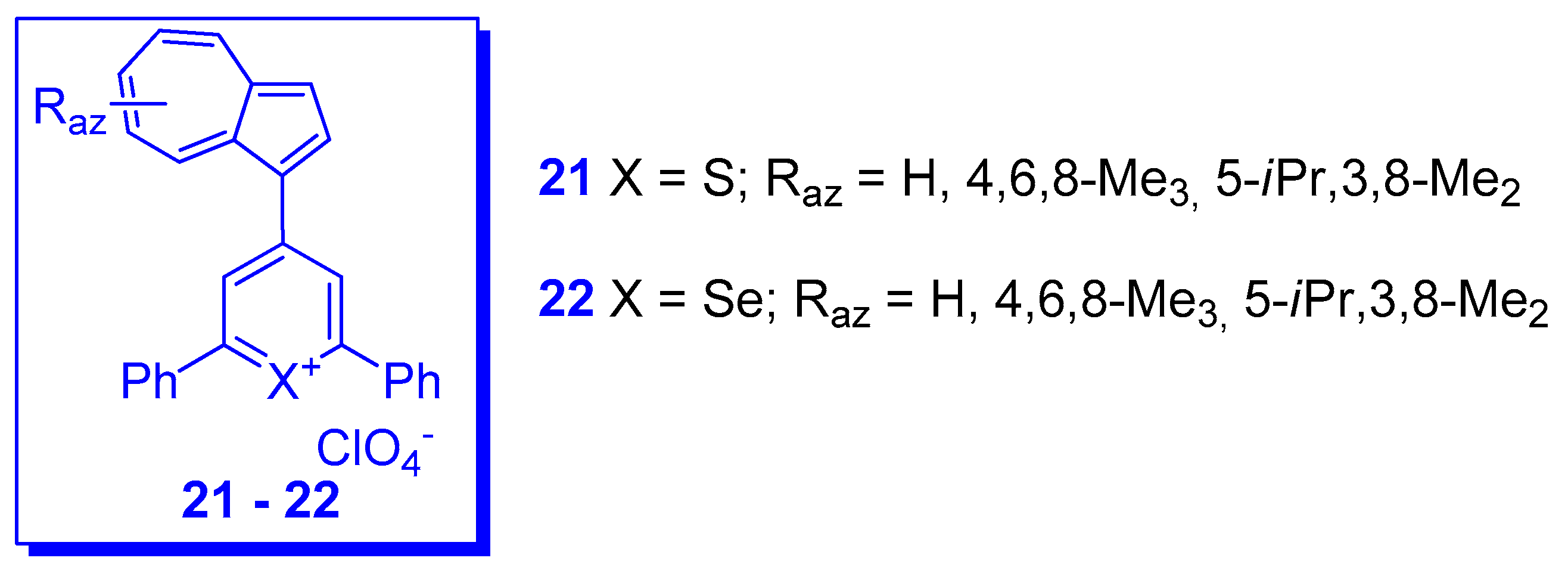{kind=link}
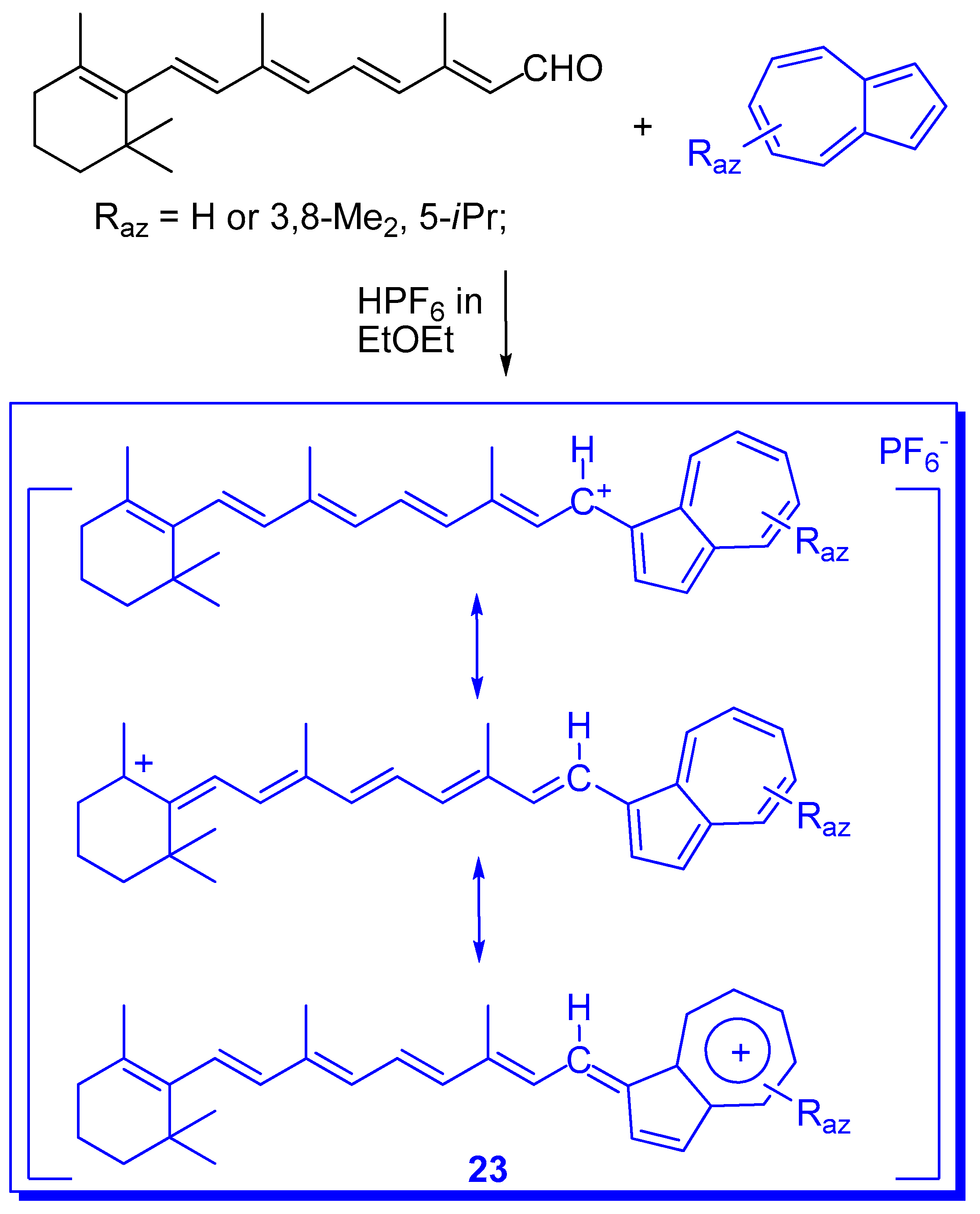{kind=link}
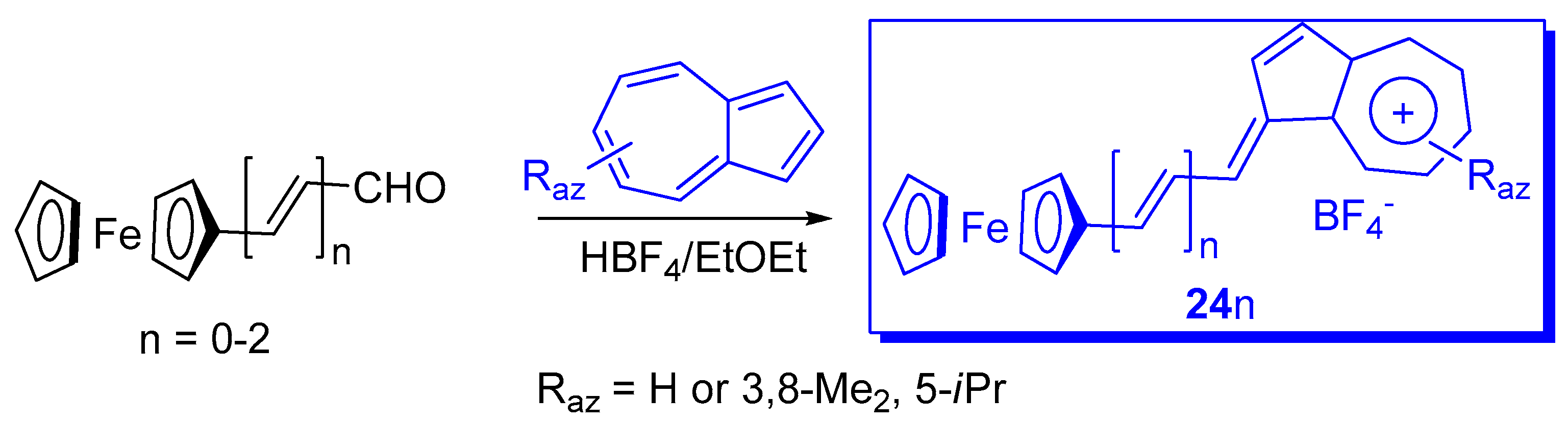{kind=link}
{kind=link}
{kind=link}
{kind=link}
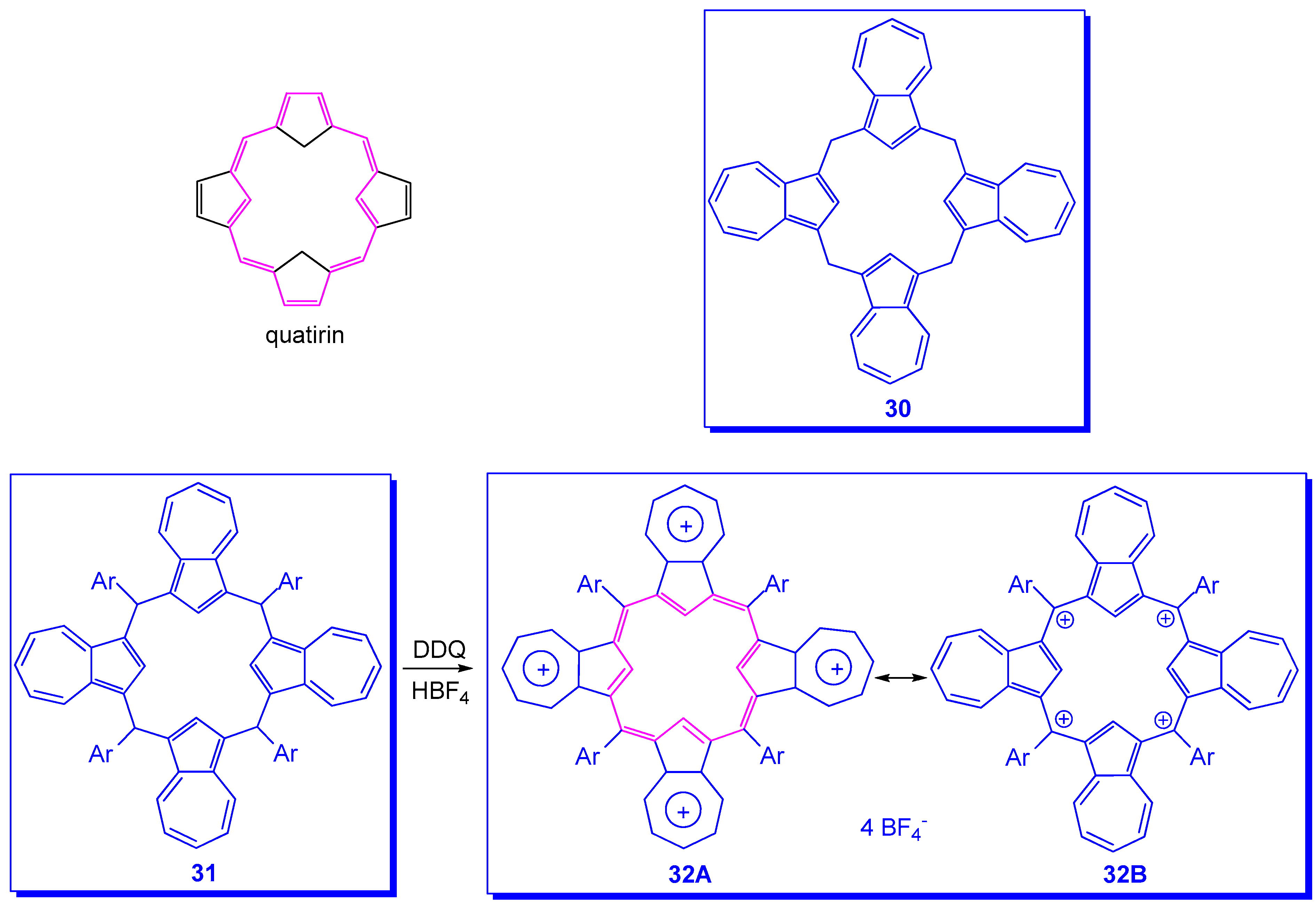{kind=link}
| Raz | Rph for 10 and 14 | pKR+ | ||
|---|---|---|---|---|
| 9 | 10 | 14 | ||
| H | H | 11.3 | 10.5 | 3.0 |
| 3-Me | H | 11.4 | 10.8 | 3.7 |
| 3-tBu | - | 13.2 | - | - |
| 3,6-di tBu | H | 14.3 | 12.4 | 4.6 |
| 6-OMe | - | >14.0 | - | - |
| H | 4-OMe | - | 11.7 | - |
| 6-OMe | 4-OMe | - | >14.0 | - |
| 3,6-di tBu | 4-OMe | - | 13.4 | - |
| H | 4-Me2N | - | 13.2 | - |
| 3,6-di tBu | 4-Me2N | - | 13.8 | - |
| H | 4-Me2N | - | - | 12.6 |
| 3,6-di tBu | 4-Me2N | - | - | 13.3 |
| 6- Me2N | - | 24.3 | - | - |
| 6- Me2N | 4-Me2N | - | 21.5 | - |
| 6- Me2N | 4-Me2N | - | - | 14.0 |
| 3-Me (18a) | 2-thienyl | - | 11.2 | - |
| 3-tBu (18a) | 2-thienyl | - | 11.8 | - |
| 3-Me (18b) | 3-thienyl | - | 11.4 | - |
| 3- tBu (18b) | 3-thienyl | - | 12.4 | - |
| 1H | Azulene | 1-Phenyl- azulene | 4-(Azulen-1-yl)- 2,6-diphenylpyridine | Salt 3 a | Salt 5 b | Salt 10 c |
|---|---|---|---|---|---|---|
| 3′/5′H | - | - | 7.95 | 8.83 | 8.06 | 8.32 |
| 2 | 7.81 | 8.02 | 8.18 | 8.92 | 8.45 | 8.18 |
| 3 | 7.30 | 7.43 | 7.53 | 7.77 | 7.51 | 7.35 |
| 4 | 8.23 | 8.34 | 8.44 | 8.89 | 8.65 | 8.44 |
| 5 | 7.05 | 7.14 | 7.28 | 8.03 | 7.77 | 7.28 |
| 6 | 7.45 | 7.58 | 7.45 | 8.31 | 8.07 | 7.72 |
| 7 | 7.05 | 7.14 | 7.29 | 8.14 | 7.80 | 7.29 |
| 8 | 8.23 | 8.55 | 8.71 | 9.57 | 9.12 | 8.71 |
| Compd. * | Raz | R3 | Dihedral Angle (in °) [48] |
|---|---|---|---|
| 3 (R2 = Ph) | H | H | 24 |
| H | Me | 38 | |
| 2-tBu-6-Me | H | 50 | |
| 5 (R2 = Me) | H | H | 22 |
| 2-tBu-6-Me | H | 45 | |
| 10 (R2 = Me; Rpy = nBu) | H | H | 29 |
| H | Me | 42 | |
| 2-tBu-6-Me | H | 54 | |
| 10 (R2 = Ph; Rpy = Me) | H | H | 39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razus, A.C. Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey. Symmetry 2021, 13, 526. https://doi.org/10.3390/sym13040526
Razus AC. Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey. Symmetry. 2021; 13(4):526. https://doi.org/10.3390/sym13040526
Chicago/Turabian StyleRazus, Alexandru C. 2021. "Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey" Symmetry 13, no. 4: 526. https://doi.org/10.3390/sym13040526
APA StyleRazus, A. C. (2021). Azulene Moiety as Electron Reservoir in Positively Charged Systems; A Short Survey. Symmetry, 13(4), 526. https://doi.org/10.3390/sym13040526