
Synthesis of C2-Symmetrical Bis-(β-Enamino-Pyran-2,4-dione) Derivative Linked via 1,6-Hexylene Spacer: X-ray Crystal Structures, Hishfeld Studies and DFT Calculations of Mono- and Bis-(Pyran-2,4-diones) Derivatives


 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
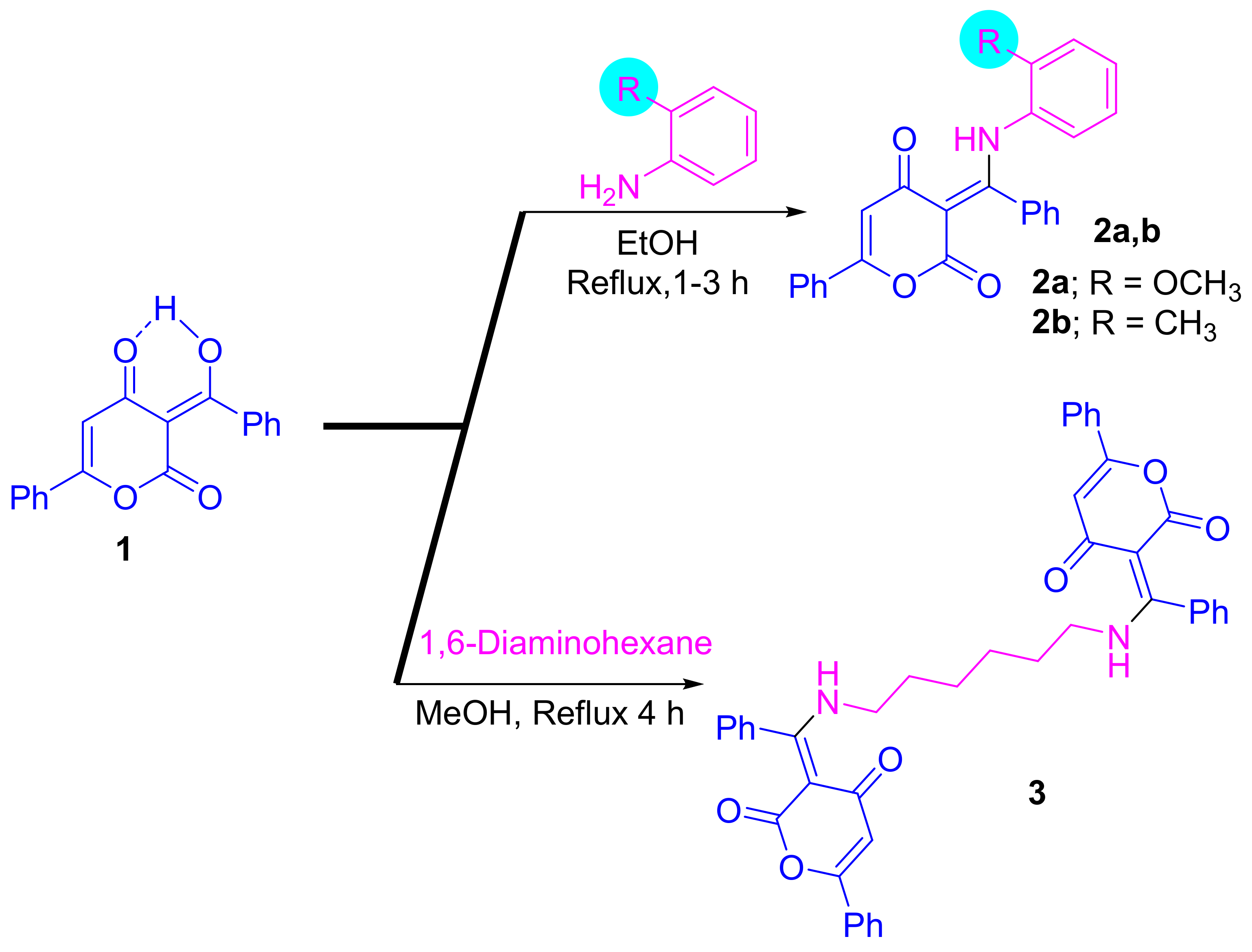
2.1. Synthesis
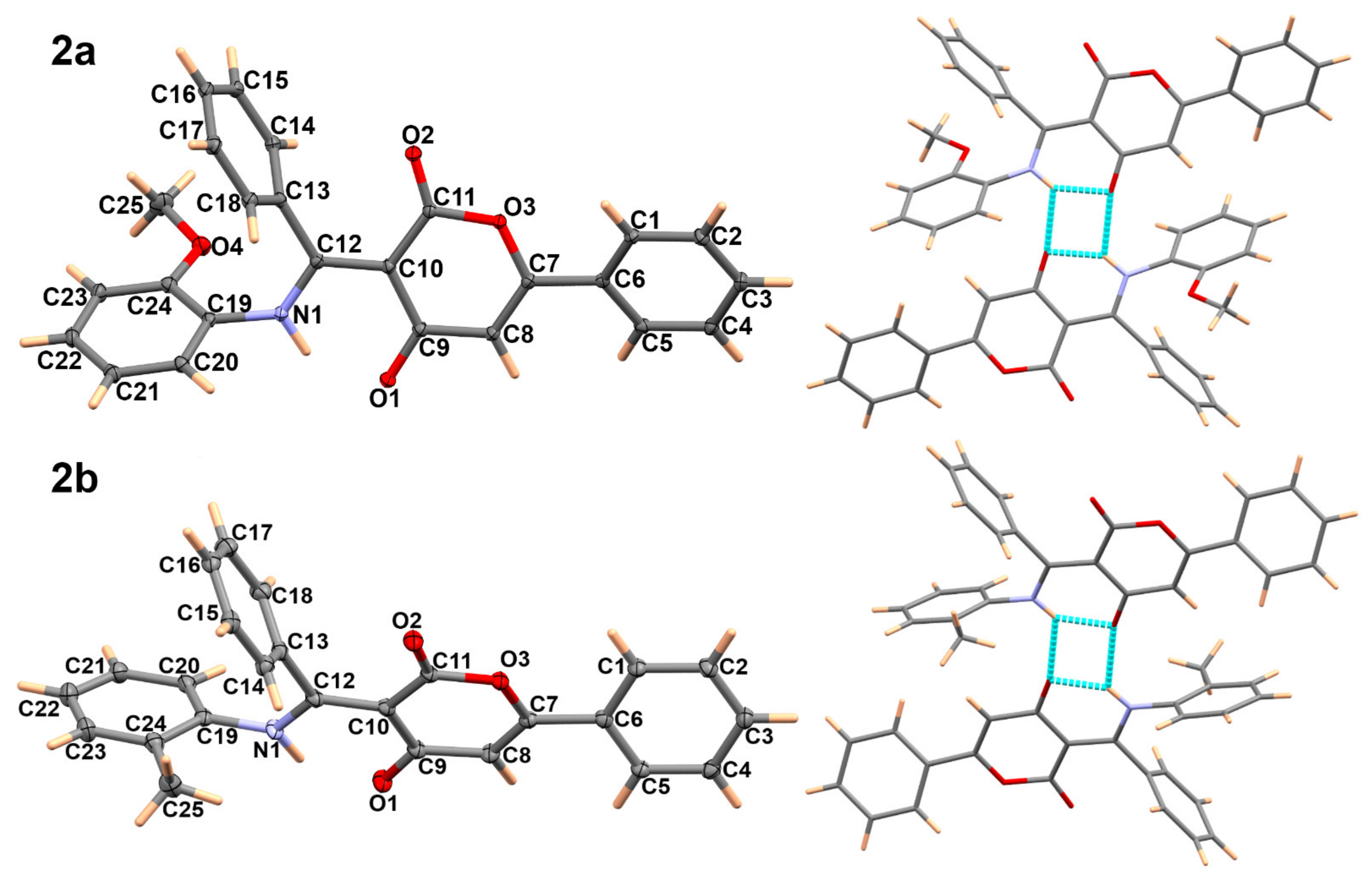
2.2. X-ray Single Crystal Determination of 2a,b and C2-Symmetrical Compound 3
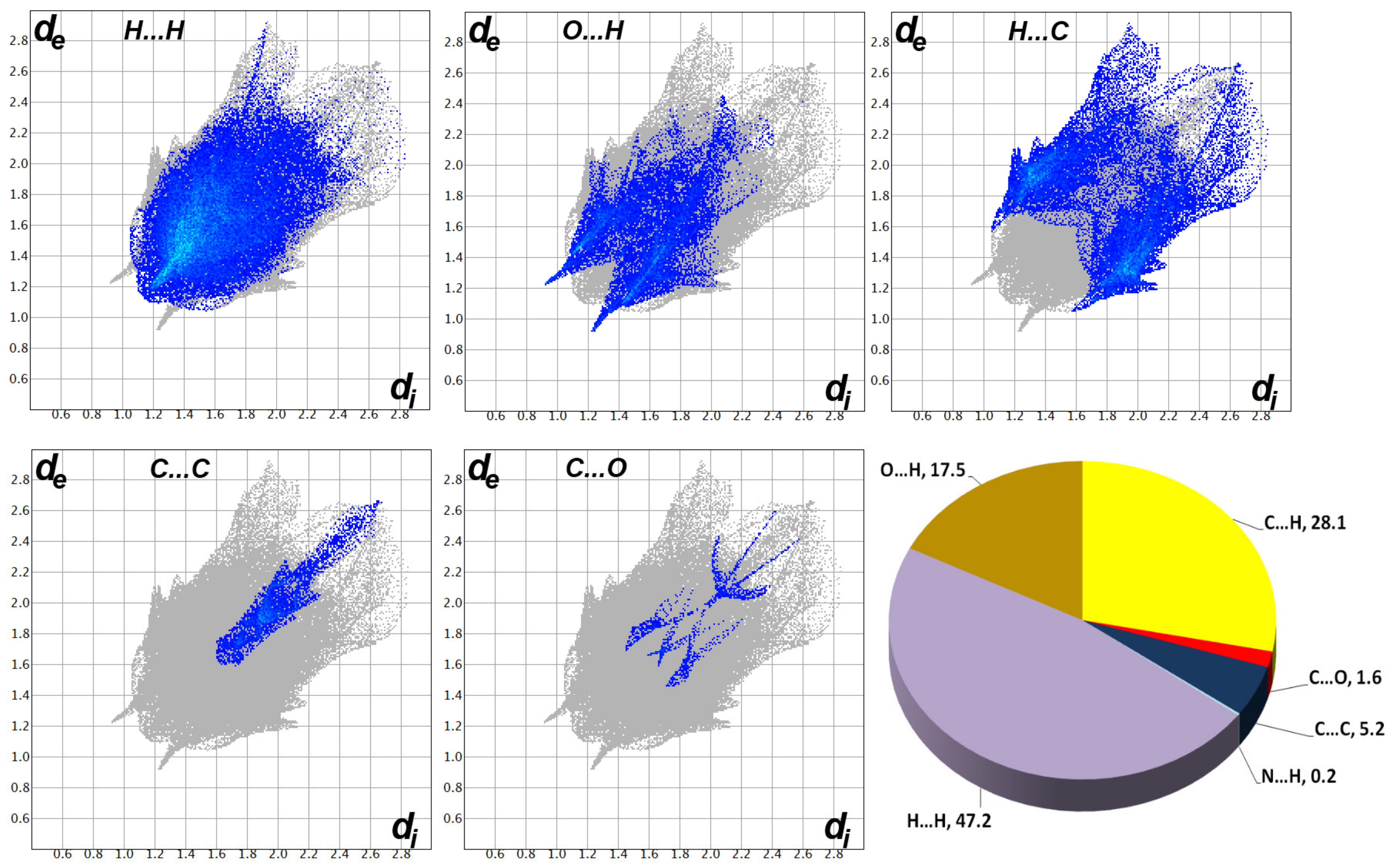
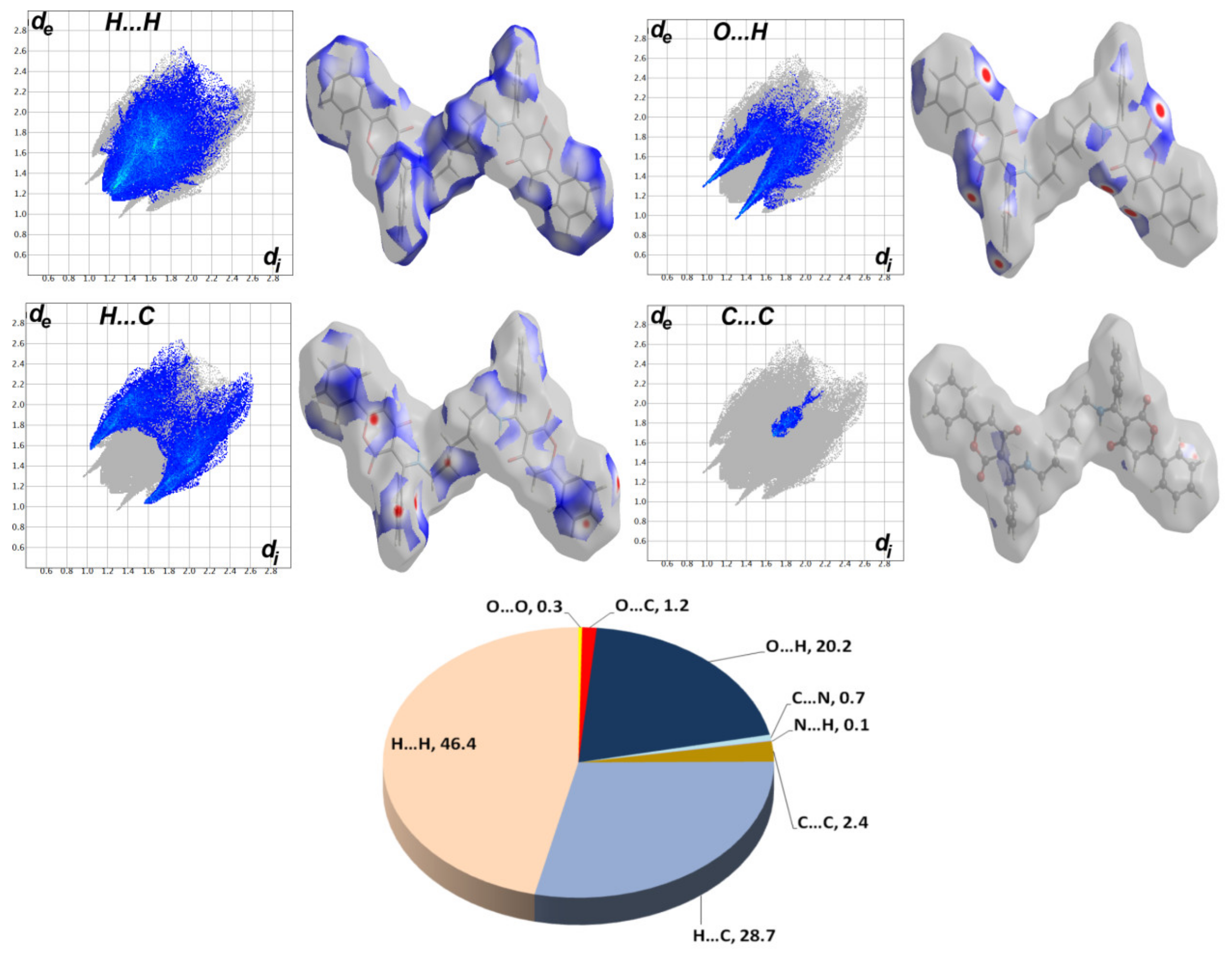
2.3. Hishfeld Analysis
3. Results and Discussion
3.1. Synthesis
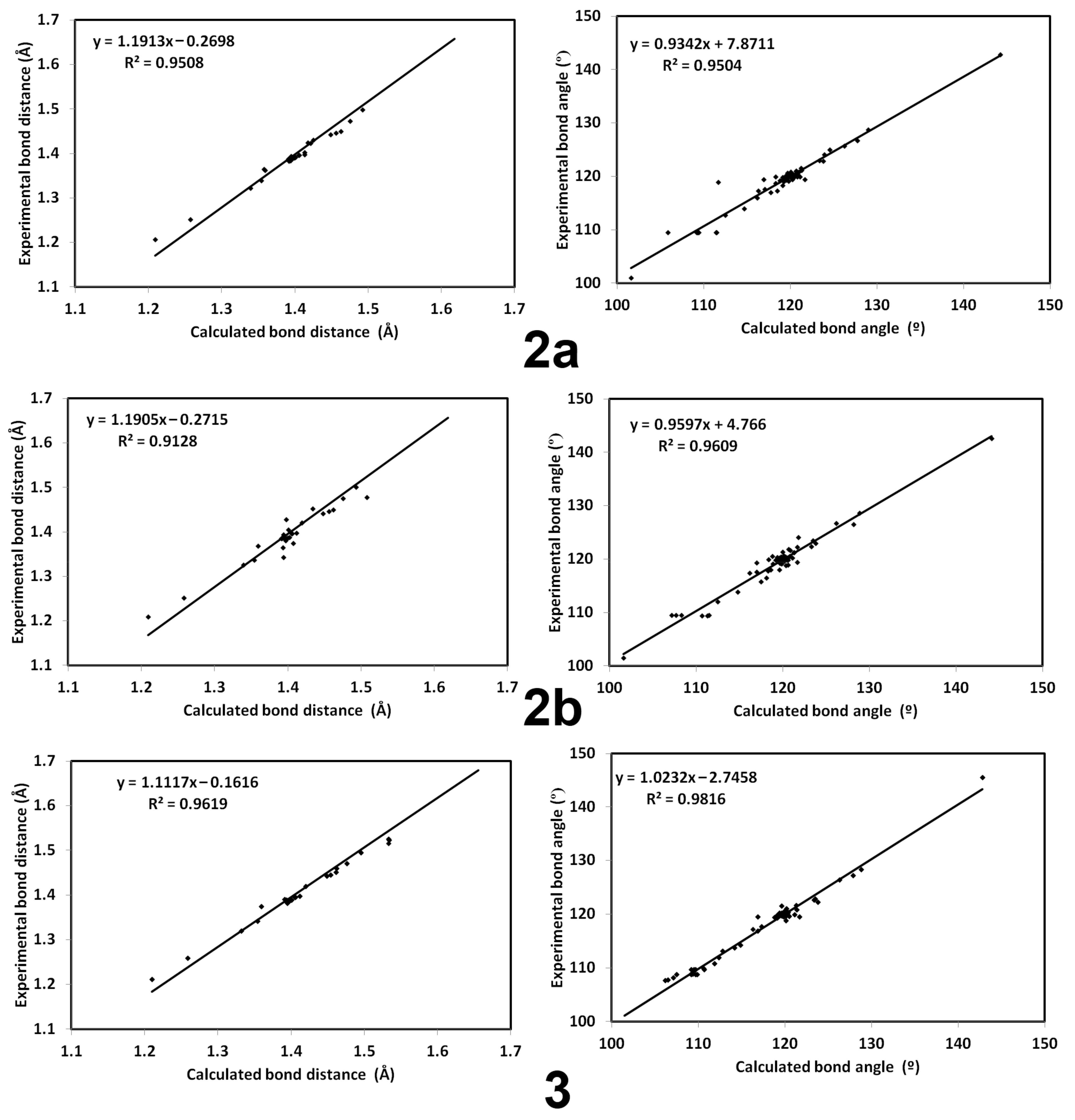
3.2. Crystal Structure Description of 2a,b and 3
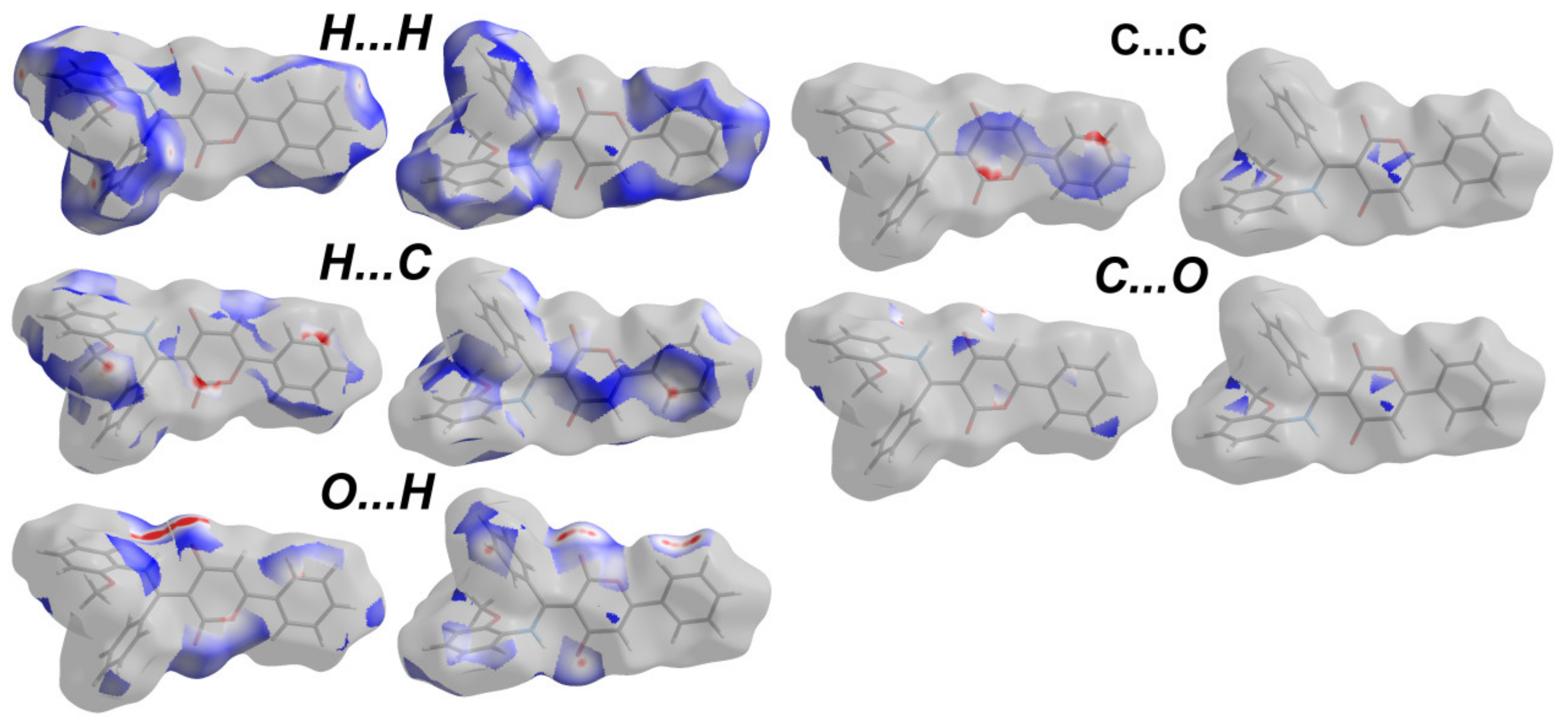
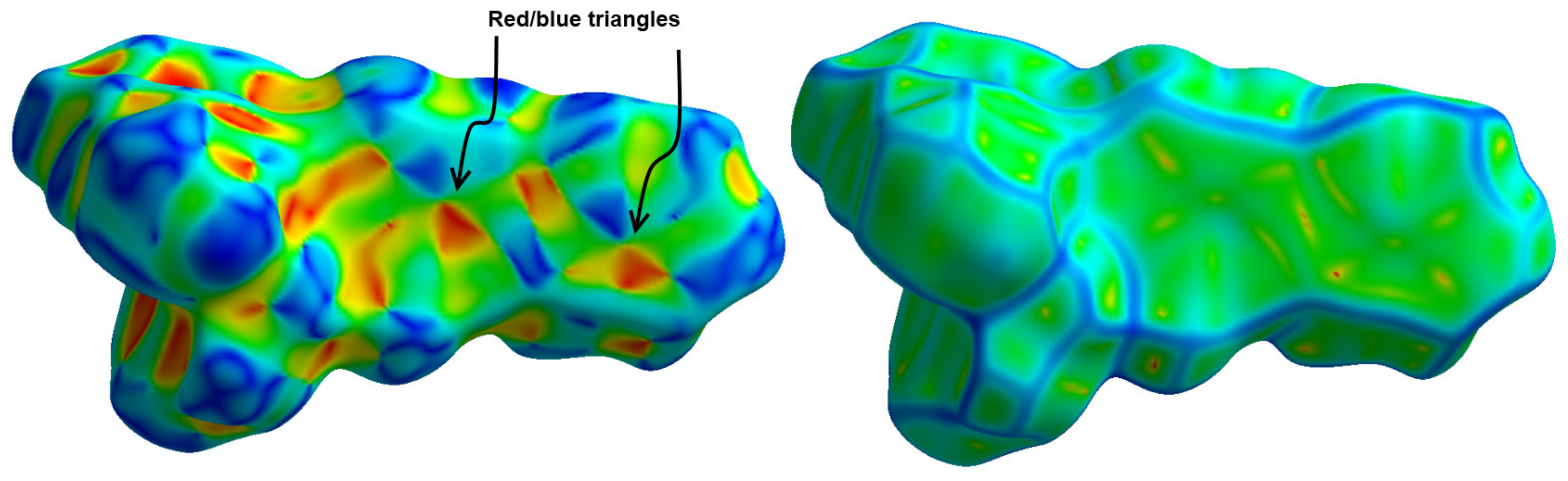
3.3. Analysis of Molecular Packing
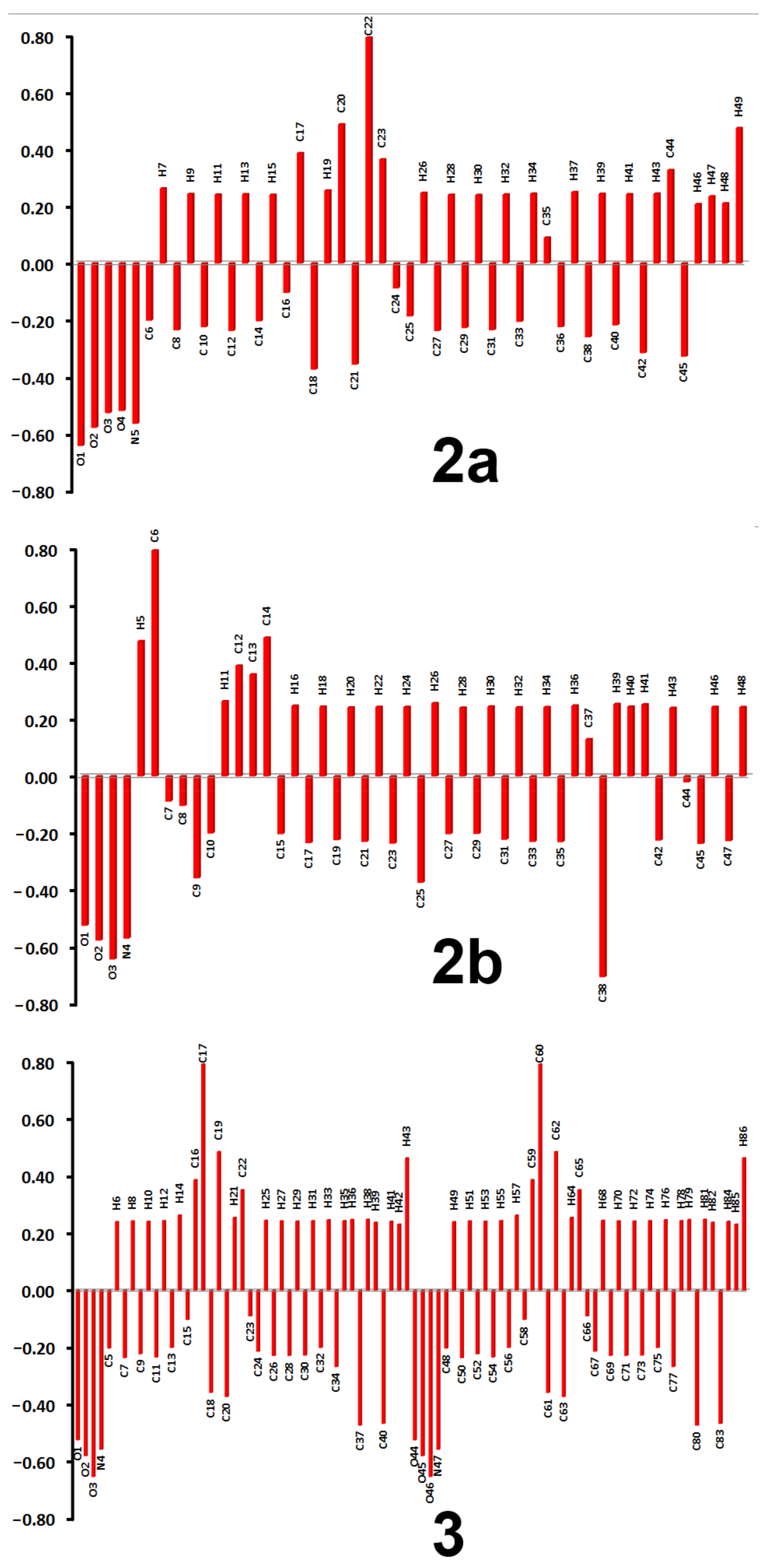
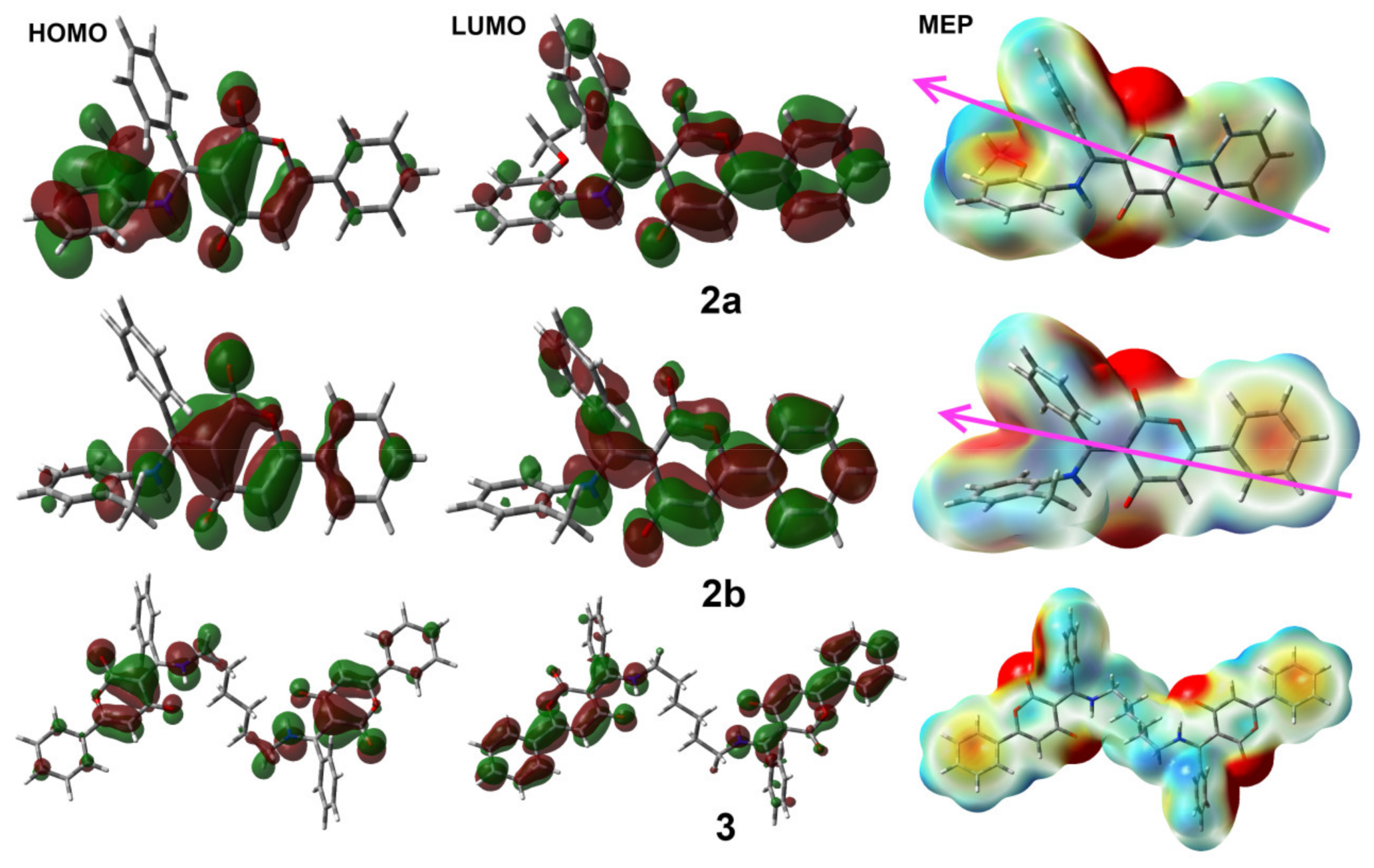
3.4. DFT Studies
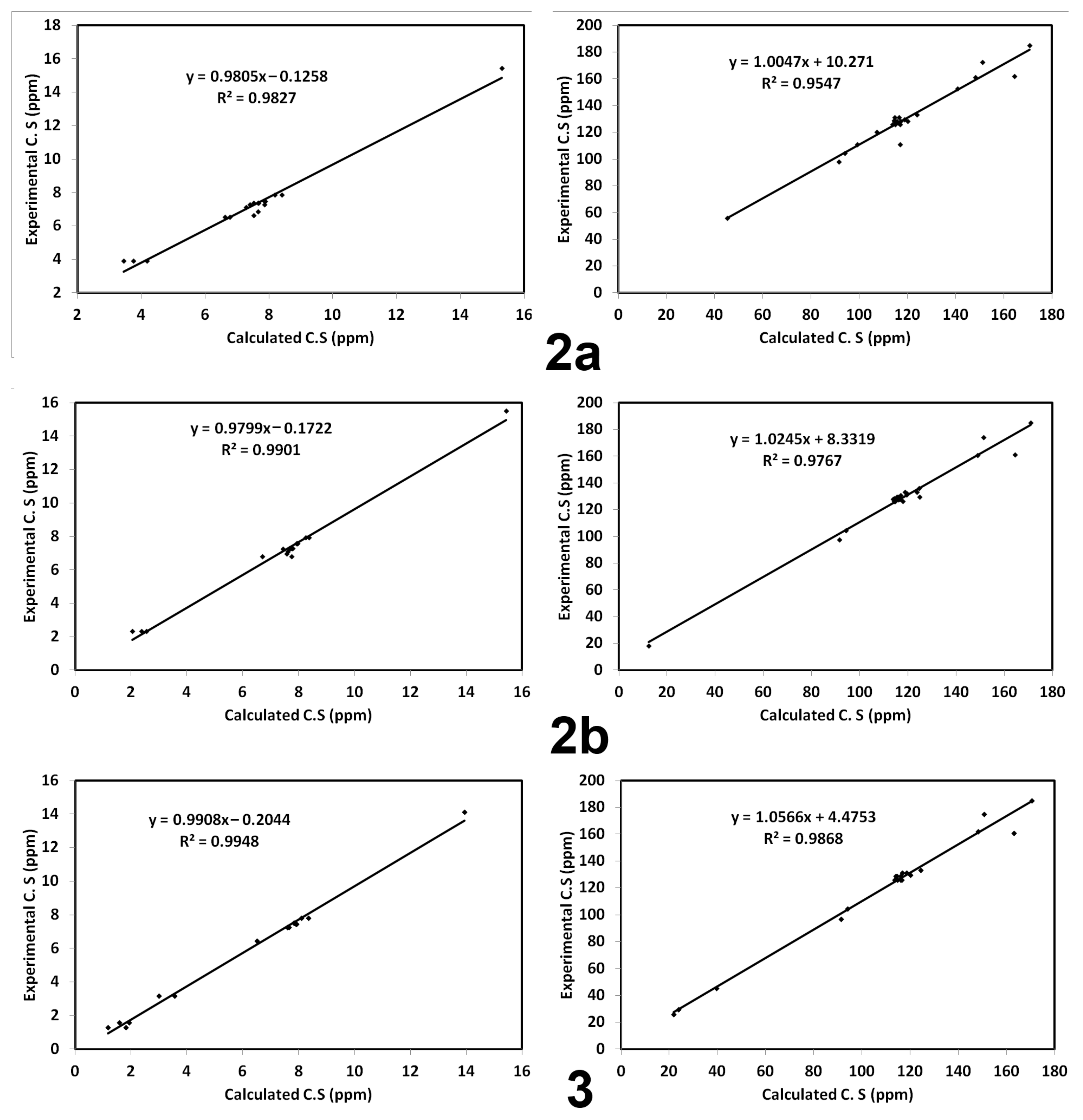
3.5. NMR Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-K.; Yun, B.-S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S. Recent advances in the synthesis of 2-pyrones. Mar. Drugs 2015, 13, 1581–1620. [Google Scholar]
- McGlacken, G.P.; Fairlamb, I.J.S. 2-Pyrone natural products and mimetics: Isolation, characterisation and biological activity. Nat. Prod. Rep. 2005, 22, 369–385. [Google Scholar] [CrossRef]
- Nagai, K.; Kamigiri, K.; Matsumoto, H.; Kawano, Y.; Yamaoka, M.; Shimoi, H.; Watanabe, M.; Suzuki, K. YM-202204, a new antifungal antibiotic produced by marine fungus Phoma sp. J. Antibiot. 2002, 55, 1036–1041. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, C.; Swenson, D.C.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. Novel antiinsectan oxalicine alkaloids from two undescribed fungicolous Penicillium spp. Org. Lett. 2003, 5, 773–776. [Google Scholar] [CrossRef]
- Nair, M.G.; Chandra, A.; Thorogood, D.L. Griseulin, a new nitro-containing bioactive metabolite produced by Streptomyces spp. J. Antibiot. 1993, 46, 1762–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitachitta, N.; Gadepalli, M.; Davidson, B.S. New α-pyrone-containing metabolites from a marine-derived actinomycete. Tetrahedron 1996, 52, 8073–8080. [Google Scholar] [CrossRef]
- Kalaitzis, J.A.; Cheng, Q.; Thomas, P.M.; Kelleher, N.L.; Moore, B.S. In vitro biosynthesis of unnatural enterocin and wailupemycin polyketides. J. Nat. Prod. 2009, 72, 469–472. [Google Scholar] [CrossRef] [Green Version]
- Nair, M.S.R.; Carey, S.T. Metabolites of phyrenomycetes II: Nectriapyrone, an antibiotic monoterpenoid. Tetrahedron Lett. 1975, 16, 1655–1658. [Google Scholar] [CrossRef]
- Fu, P.; Liu, P.; Qu, H.; Wang, Y.; Chen, D.; Wang, H.; Li, J.; Zhu, W. α-Pyrones and diketopiperazine derivatives from the marine-derived actinomycete Nocardiopsis dassonvillei HR10-5. J. Nat. Prod. 2011, 74, 2219–2223. [Google Scholar] [CrossRef]
- Kim, M.C.; Kwon, O.W.; Park, J.S.; Kim, S.Y.; Kwon, H.C. Nocapyrones H-J, 3,6-disubstituted α-pyrones from the marine actinomycete Nocardiopsis sp. KMF-001. Chem. Pharm. Bull. 2013, 61, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Ogura, H.; Akasaka, K.; Oikawa, T.; Matsuura, N.; Imada, C.; Yasuda, H.; Igarashi, Y. Nocapyrones: α- and γ-pyrones from a marine-derived Nocardiopsis sp. Mar. Drugs 2014, 12, 4110–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graziani, E.I.; Ritacco, F.V.; Bernan, V.S.; Telliez, J.-B. Phaeochromycins A–E, anti-inflammatory polyketides isolated from the soil actinomycete Streptomyces phaeochromogenes LL-P018. J. Nat. Prod. 2005, 68, 1262–1265. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Cao, Y.; Liu, J.; Zheng, D.; Chen, X.; Han, L.; Jiang, C.; Huang, X. Violapyrones A–G, α-pyrone derivatives from Streptomyces violascens isolated from Hylobates hoolock feces. J. Nat. Prod. 2013, 76, 2126–2130. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Lee, H.S.; Lee, J.S.; Shin, J.; Lee, M.A.; Lee, H.S.; Lee, Y.J.; Yun, J.; Kang, J.S. Violapyrones H and I, new cytotoxic compounds isolated from Streptomyces sp. associated with the marine starfish Acanthaster planci. Mar. Drugs 2014, 12, 3283–3291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, E.P.; Hasemeyer, A.P.; Lane, A.L.; Davenport, T.M.; Engel, S.; Hay, M.E.; Fairchild, C.R.; Prudhomme, J.; le Roch, K.; Aalbersberg, W.; et al. Antibacterial neurymenolides from the fijian red alga Neurymenia fraxinifolia. Org. Lett. 2008, 11, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Li, X.M.; Meng, L.; Li, C.S.; Gao, S.S.; Shang, Z.; Proksch, P.; Huang, C.G.; Wang, B.G. Nigerapyrones A–H, alpha-pyrone derivatives from the marine mangrove-derived endophytic fungus Aspergillus niger MA-132. J. Nat. Prod. 2011, 74, 1787–1791. [Google Scholar] [CrossRef]
- Sarhan, A.A.; Haukka, M.; Barakat, A.; Boraei, A.T. A novel synthetic approach to pyran-2,4-dione scaffold production: Microwave-assisted dimerization, cyclization, and expeditious regioselective conversion into β-enamino-pyran-2, 4-diones. Tetrahedron Lett. 2020, 61, 152660. [Google Scholar] [CrossRef]
- Xue, F.; Li, X.; Wan, B. A class of benzene backbone-based olefin-sulfoxide ligands for Rh-catalyzed enantioselective addition of arylboronic acids to enones. J. Org. Chem. 2011, 76, 7256–7262. [Google Scholar] [CrossRef]
- Gianni, J.; Pirovano, V.; Abbiati, G. Silver triflate/p-TSA co-catalysed synthesis of 3-substituted isocoumarins from 2-alkynylbenzoates. Org. Biomol. Chem. 2018, 16, 3213–3219. [Google Scholar] [CrossRef] [Green Version]
- Thibonnet, J.; Abarbri, M.; Parrain, J.L.; Duchene, A. One-step synthesis of alpha-pyrones from acyl chlorides by the Stille reaction. J. Org. Chem. 2002, 67, 3941–3944. [Google Scholar] [CrossRef]
- Chaladaj, W.; Corbet, M.; Furstner, A. Total synthesis of neurymenolide a based on a gold-catalyzed synthesis of 4-hydroxy-2-pyrones. Angew. Chem. Int. Ed. 2012, 51, 6929–6933. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Shin, J.; Shin, H.J.; Lee, H.-S.; Lee, Y.-J.; Lee, H.-S.; Won, H. Total synthesis and configurational validation of (+)-violapyrone C. Eur. J. Org. Chem. 2014, 2014, 4472–4476. [Google Scholar] [CrossRef]
- Yeh, P.-P.; Daniels, D.S.B.; Cordes, D.B.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-mediated one-pot synthesis of trifluoromethyl substituted 2-pyrones. Org. Lett. 2014, 16, 964–967. [Google Scholar] [CrossRef] [Green Version]
- Rikagu Oxford Diffraction. CrysAlisPro; Agilent Technologies Inc.: Yarnton, UK, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17 University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 12 June 2017).
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Chang, R. Chemistry, 7th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Kosar, B.; Albayrak, C. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino) methyl] phenol. Spectrochim. Acta 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Koopmans, T.A. Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | 2a | 2b | 3 |
|---|---|---|---|
| empirical formula | C25H19NO4 | C25H19NO3 | C42H36N2O6 |
| fw | 397.41 | 381.41 | 664.73 |
| temp (K) | 120(2) | 120(2) | 120(2) |
| Λ (Å) | 1.54184 | 1.54184 | 1.54184 |
| cryst syst | Triclinic | Triclinic | Triclinic |
| space group | P-1 | P-1 | P-1 |
| a (Å) | 8.4480(2) | 8.5656(2) | 7.3747(4) |
| b (Å) | 11.3775(3) | 11.0766(2) | 10.4849(6) |
| c (Å) | 11.4509(3) | 11.3943(2) | 11.6054(7) |
| α (°) | 62.863(3) | 117.766(2) | 106.422(5) |
| β (°) | 83.527(2) | 93.726(2) | 105.512(5) |
| γ (°) | 82.242(2) | 93.594(2) | 92.416(5) |
| V (Å3) | 968.87(5) | 949.26(4) | 822.64(9) |
| Z | 2 | 2 | 1 |
| ρcalc (Mg/m3) | 1.362 | 1.334 | 1.342 |
| μ (Mo Kα) (mm−1) | 0.753 | 0.705 | 0.726 |
| No. reflns. | 21,259 | 21,678 | 6894 |
| Unique reflns. | 4067 | 3975 | 3388 |
| GOOF (F2) | 1.046 | 1.054 | 1.045 |
| Rint | 0.0238 | 0.0316 | 0.0244 |
| R1a (I ≥ 2σ) | 0.0323 | 0.0382 | 0.0429 |
| wR2b (I ≥ 2σ) | 0.0846 | 0.1024 | 0.1132 |
| CCDC | 2,099,428 | 2,099,429 | 2,099,430 |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| H25B…C5 | 2.702 | H15…H22 | 2.274 |
| H25B…C6 | 2.761 | H4…H14 | 2.142 |
| C1…O1 | 3.099 | O2…H2 | 2.436 |
| C11…C5 | 3.287 | O2…H1A | 2.512 |
| C11…C4 | 3.227 | O1….H17 | 2.537 |
| C10….C4 | 3.563 | O1…H1 | 2.212 |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| H22…C15 | 2.629 | O2…H15 | 2.526 |
| C4…C10 | 3.350 | O1…H17 | 2.523 |
| C4…C11 | 3.232 | O1…H1 | 2.144 |
| H15…H22 | 2.420 | O2…H2 | 2.383 |
| O3…H21 | 2.581 |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| H21B…C2 | 2.784 | O2…H1A | 2.345 |
| H21B…C3 | 2.689 | O2…H16 | 2.407 |
| H4…C17 | 2.589 | O3…H11 | 2.261 |
| H4…C18 | 2.736 | C4…C5 | 3.348 |
| H17…C7 | 2.624 |
| Parameter | 2a | 2b | 3 |
|---|---|---|---|
| EHOMO | −5.8214 | −5.9912 | −5.9357 |
| ELUMO | −1.8452 | −1.9173 | −1.8545 |
| I | 5.8214 | 5.9912 | 5.9357 |
| A | 1.8452 | 1.9173 | 1.8545 |
| η | 3.9762 | 4.0738 | 4.0812 |
| μ | −3.8333 | −3.9543 | −3.8951 |
| ω | 1.8478 | 1.9191 | 1.8587 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boraei, A.T.A.; Haukka, M.; Sarhan, A.A.M.; Soliman, S.M.; Al-Majid, A.M.; Barakat, A. Synthesis of C2-Symmetrical Bis-(β-Enamino-Pyran-2,4-dione) Derivative Linked via 1,6-Hexylene Spacer: X-ray Crystal Structures, Hishfeld Studies and DFT Calculations of Mono- and Bis-(Pyran-2,4-diones) Derivatives. Symmetry 2021, 13, 1646. https://doi.org/10.3390/sym13091646
Boraei ATA, Haukka M, Sarhan AAM, Soliman SM, Al-Majid AM, Barakat A. Synthesis of C2-Symmetrical Bis-(β-Enamino-Pyran-2,4-dione) Derivative Linked via 1,6-Hexylene Spacer: X-ray Crystal Structures, Hishfeld Studies and DFT Calculations of Mono- and Bis-(Pyran-2,4-diones) Derivatives. Symmetry. 2021; 13(9):1646. https://doi.org/10.3390/sym13091646
Chicago/Turabian StyleBoraei, Ahmed T. A., Matti Haukka, Ahmed A. M. Sarhan, Saied M. Soliman, Abdullah Mohammed Al-Majid, and Assem Barakat. 2021. "Synthesis of C2-Symmetrical Bis-(β-Enamino-Pyran-2,4-dione) Derivative Linked via 1,6-Hexylene Spacer: X-ray Crystal Structures, Hishfeld Studies and DFT Calculations of Mono- and Bis-(Pyran-2,4-diones) Derivatives" Symmetry 13, no. 9: 1646. https://doi.org/10.3390/sym13091646