
Synthesis and Evaluation of C2-Symmetric SPIROL-Based bis-Oxazoline Ligands


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Introduction
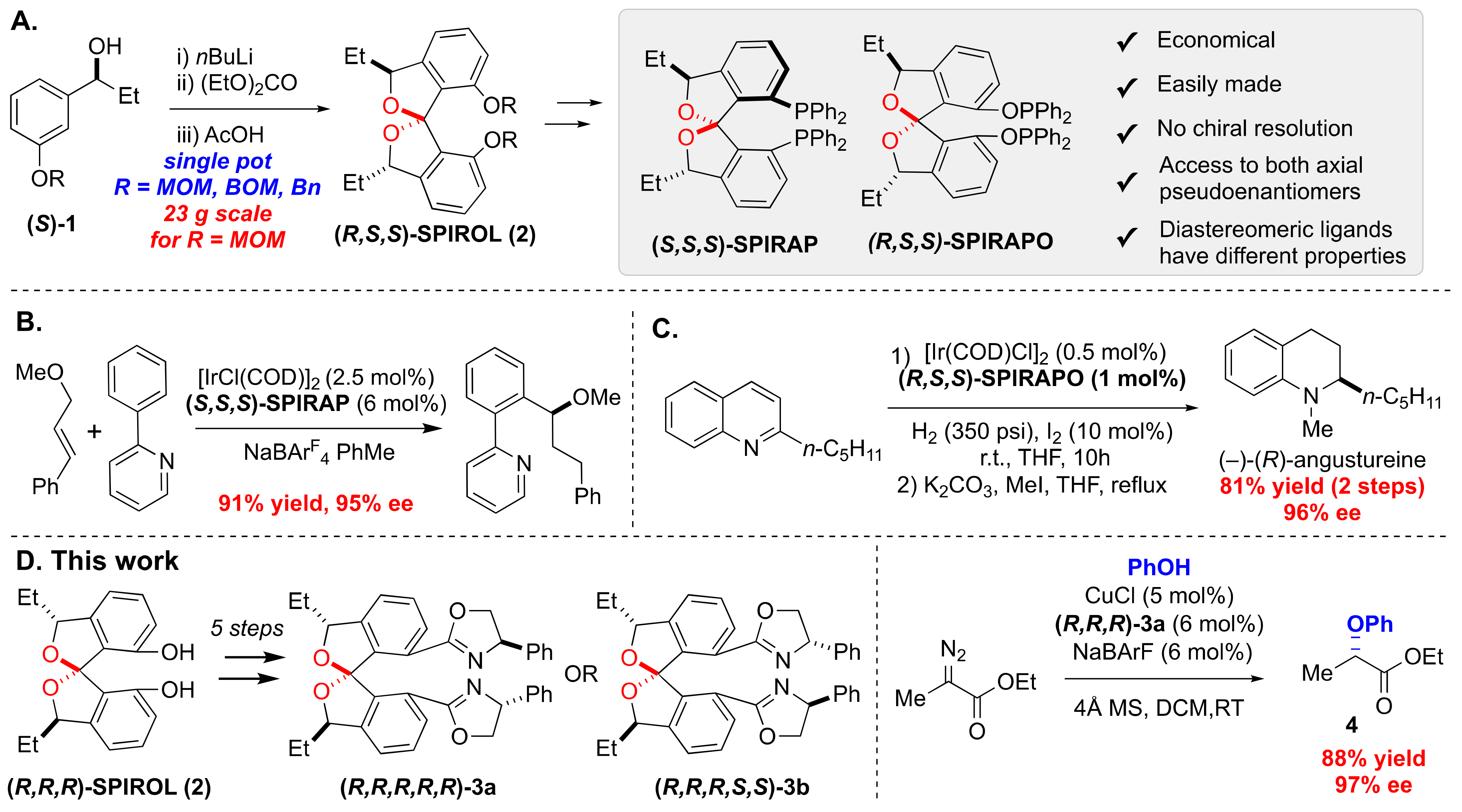
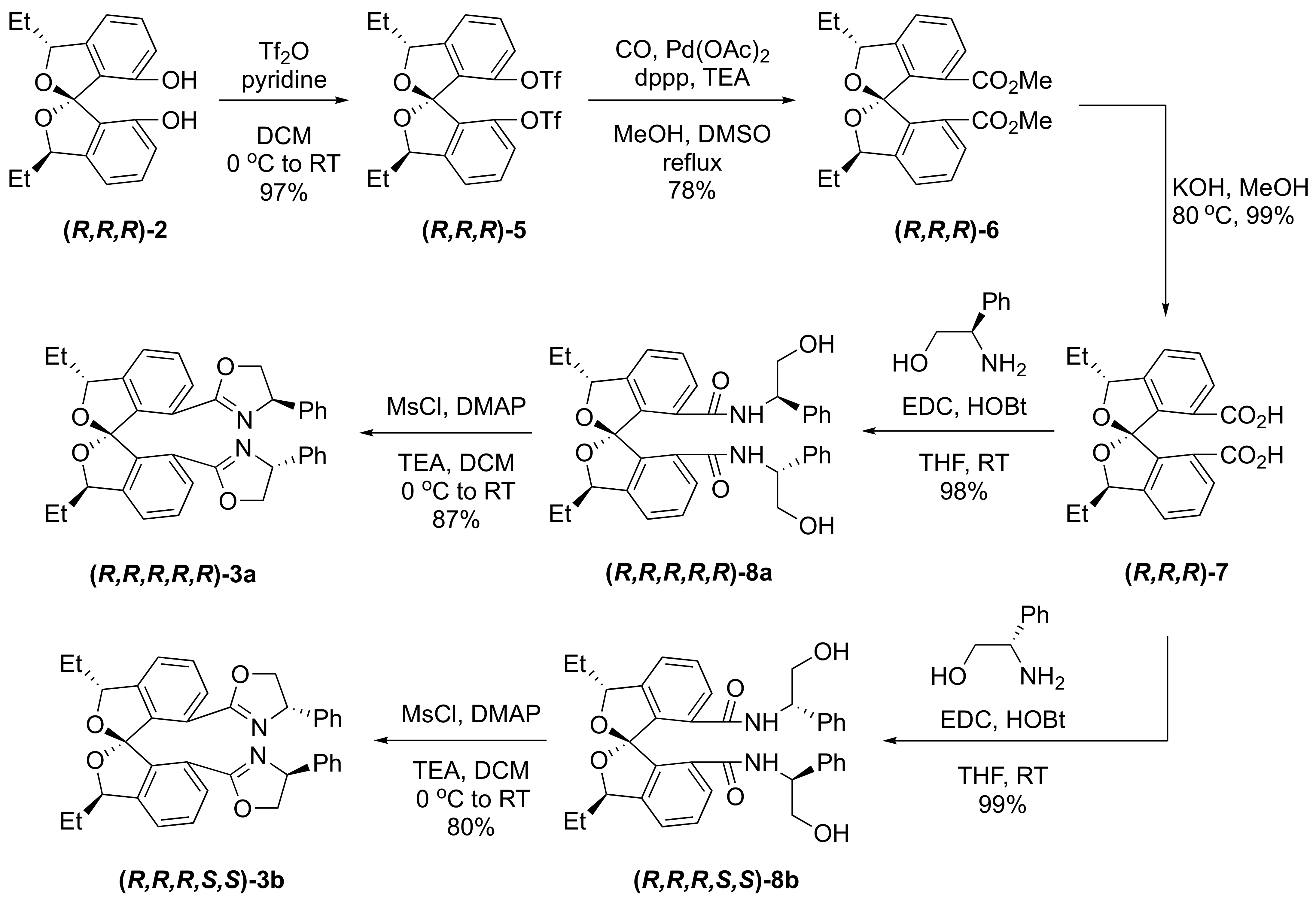
3.2. Synthesis of C2-Symmetric SPIROL-Based Bisoxazoline Ligands (SPIROX)
3.3. Application of SPIROL-Based Bisoxazoline Ligands in Insertion of Carbenoids into O–H Bonds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, A.W.; Lackner, S.; Stoltz, B.M. Modularity: Adding new dimensions to total synthesis. Trends Chem. 2019, 1, 630–643. [Google Scholar] [CrossRef] [Green Version]
- Khatri, H.R.; Carney, N.; Rutkoski, R.; Nagorny, P. Recent progress in total synthesis triggered by the emergence of new catalytic methods. Eur. J. Org. Chem. 2020, 7, 755–776. [Google Scholar] [CrossRef] [Green Version]
- Krautwald, S.; Sarlah, D.; Schafroth, M.A.; Carreira, E.M. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science 2013, 340, 1065–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shugrue, C.R.; Miller, S.J. Applications of nonenzymatic catalysis to the alteration of natural products. Chem. Rev. 2017, 117, 11894–11951. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Han, Z.; Wang, Z. Spiro skeletons: A class of privileged structures for chiral ligand design. Chem. Asian J. 2009, 4, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-H.; Zhou, Q.-L. Chiral diphosphine and monodentate phosphorus ligands on a spiro scaffold for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res. 2008, 41, 581–593. [Google Scholar] [CrossRef]
- Zhu, S.-F.; Zhou, Q.-L. Spiro Ligands for Asymmetric Catalysis. In Ligand Design in Metal Chemistry: Reactivity and Catalysis, 1st ed.; Stradiotto, M., Lundgren, R.J., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2016; Chapter 4; pp. 66–103. [Google Scholar]
- Zhou, Q.-L.; Xie, J.-H. Chiral Spiro Catalysts. In Topics in Organometallic Chemistry; Ma, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 36, pp. 1–28. [Google Scholar]
- Sun, Z.; Winschel, G.A.; Borovika, A.; Nagorny, P.J. Chiral phosphoric acid-catalyzed enantioselective and diastereoselective spiroketalizations. Am. Chem. Soc. 2012, 134, 8074–8077. [Google Scholar] [CrossRef]
- Borovika, A.; Nagorny, P. Chiral Bronsted acid-catalyzed enantioselective ionic [2+4] cycloadditions. Tetrahedron 2013, 69, 5719–5725. [Google Scholar] [CrossRef]
- Sun, Z.; Winschel, G.A.; Zimmerman, P.; Nagorny, P. Enantioselective synthesis of piperidines through the formation of chiral mixed phosphoric acid acetals: Experimental and theoretical studies. Angew. Chem. Int. Ed. 2014, 53, 11194–11198. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Borovika, A.; Khomutnyk, Y.; Nagorny, P. Chiral phosphoric acid-catalyzed desymmetrizative glycosylation of 2-deoxystreptamine and its application to aminoglycoside synthesis. Chem. Commun. 2017, 53, 8976–8979. [Google Scholar] [CrossRef]
- Wang, S.; Arguelles, A.J.; Tay, J.-H.; Hotta, M.; Zimmerman, P.M.; Nagorny, P. Experimental and computational studies on regiodivergent chiral phosphoric acid catalyzed cycloisomerization of mupirocin methyl ester. Chem. Eur. J. 2020, 26, 4583–4591. [Google Scholar] [CrossRef]
- Arguelles, A.J.; Sun, S.; Budaitis, B.; Nagorny, P. Design, synthesis and application of chiral C2-symmetric spiroketal-containing ligands in transition metal catalysis. Angew. Chem. Int. Ed. 2018, 57, 5325–5329. [Google Scholar] [CrossRef]
- Sun, S.; Nagorny, P. Exploration of chiral diastereomeric spiroketal (SPIROL)-based phosphinite ligands in asymmetric hydrogenation of heterocycles. Chem. Commun. 2020, 56, 8432–8435. [Google Scholar] [CrossRef] [PubMed]
- Le Maux, P.; Carrie, D.; Jehan, P.; Simonneaux, G. Asymmetric O–H insertion reaction of carbenoids catalyzed by chiral bicyclo bisoxazoline copper(I) and (II) complexes. Tetrahedron 2016, 72, 4671–4675. [Google Scholar] [CrossRef]
- Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley-Interscience: New York, NY, USA, 1998. [Google Scholar]
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern organic synthesis with α-diazocarbonyl compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, G.; Loiodice, F.; Tortorella, V.; Conte-Camerino, D.; Mambrini, M.; Ferrannini, E.; Bryant, S.H.J. Stereospecificity of the chloride ion channel: The action of chiral clofibric acid analogues. Med. Chem. 1987, 30, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Chimichi, S.; Boccalini, M.; Cravotto, G.; Rosati, O. A new convenient route to enantiopure 2-coumarinyloxypropanals: Application to the synthesis of optically active geiparvarin analogues. Tetrahedron Lett. 2006, 47, 2405–2408. [Google Scholar] [CrossRef]
- Maier, T.C.; Fu, G.C. Catalytic enantioselective O–H insertion reactions. J. Am. Chem. Soc. 2006, 128, 4594–4595. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, S.-F.; Liu, B.; Wang, L.-X.; Zhou, Q.-L. Highly enantioselective insertion of carbenoids into O–H bonds of phenols: An efficient approach to chiral α-aryloxycarboxylic esters. J. Am. Chem. Soc. 2007, 129, 12616–12617. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-F.; Chen, C.; Cai, Y.; Zhou, Q.-L. Catalytic asymmetric reaction with water: Enantioselective synthesis of alpha-hydroxyesters by a copper-carbenoid O–H insertion reactions. Angew. Chem. Int. Ed. 2008, 47, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-F.; Song, X.-G.; Li, Y.; Cai, Y.; Zhou, Q.-L. Enantioselective copper-catalyzed intramolecular O–H insertion: An efficient approach to chiral 2-carboxy cyclic ethers. J. Am. Chem. Soc. 2010, 132, 16374–16376. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-F.; Cai, Y.; Mao, H.-X.; Xie, J.-H.; Zhou, Q.-L. Enantioselective iron-catalysed O–H bond insertions. Nat. Chem. 2010, 2, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Osako, T.; Panichakul, D.; Uozumi, Y. Enantioselective carbenoid insertion into phenolic O–H bonds with a chiral copper(I) imidaoindolephosphine complex. Org. Lett. 2012, 14, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-G.; Cheng, X.; Ma, J.-A.; Zhou, Q.-L.J. Synthesis of chiral 2-alkyl-8-quinolinyl-oxazoline ligands: Reversal of enantioselectivity in the asymmetric palladium-catalyzed allylic alkylation. Organomet. Chem. 2001, 640, 65–71. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
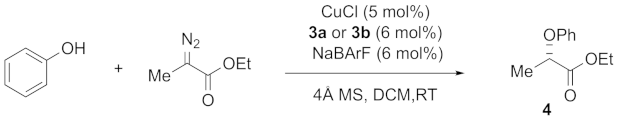
| Entry | Ligand | [Cu] | Conversion (%) b | ee (%) c |
|---|---|---|---|---|
| 1 | (R,R,R,R,R)-3a | CuCl | 99(88% yield) | 97 |
| 2 | (R,R,R,R,R)-3a | Cu(OTf)2 | 99 | 95 |
| 3 | (R,R,R,S,S)-3b | CuCl | 99 | −22 |
| 4 | (R,R,R,S,S)-3b | Cu(OTf)2 | 99 | −62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, S.; Diaz, N.A.; Nagorny, P. Synthesis and Evaluation of C2-Symmetric SPIROL-Based bis-Oxazoline Ligands. Symmetry 2021, 13, 1667. https://doi.org/10.3390/sym13091667
Sun S, Diaz NA, Nagorny P. Synthesis and Evaluation of C2-Symmetric SPIROL-Based bis-Oxazoline Ligands. Symmetry. 2021; 13(9):1667. https://doi.org/10.3390/sym13091667
Chicago/Turabian StyleSun, Siyuan, Nicolas A. Diaz, and Pavel Nagorny. 2021. "Synthesis and Evaluation of C2-Symmetric SPIROL-Based bis-Oxazoline Ligands" Symmetry 13, no. 9: 1667. https://doi.org/10.3390/sym13091667
APA StyleSun, S., Diaz, N. A., & Nagorny, P. (2021). Synthesis and Evaluation of C2-Symmetric SPIROL-Based bis-Oxazoline Ligands. Symmetry, 13(9), 1667. https://doi.org/10.3390/sym13091667