Abstract
One of the current problems in the environmental catalysis is the design of an effective and less costly catalytic system for the oxidation of CO. The nano-sized α-Mn2O3 oxide has been prepared and modified with 0.5 wt.% Pt. The catalysts have been characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), temperature-programmed reduction (TPR) and diffuse-reflectance infrared spectroscopy (DRIFTS). Finely divided PtO and Pt(OH)2 are being formed on the Mn2O3 surface as a result of the strong interaction between platinum and the nano-oxide. Based on DRIFTS investigations and the model calculations, a Langmuir–Hinshelwood type of mechanism is supposed for CO oxidation on Pt/Mn2O3. The CO and oxygen are adsorbed on different types of sites. The Mars–van Krevelen mechanism is the most probable one over pure Mn2O3, thus suggesting that CO2 is adsorbed on the oxidized sites. The CO adsorption in the mixture CO + N2 or in the presence of oxygen (CO + N2 + O2) leads to a partial reduction in the Pt+ surface species and the formation of linear Pt1+−CO and Pt0−CO carbonyls. Both of them take part in the CO oxidation reaction.
1. Introduction
The main global environmental problems include air, water and soil contamination. Water pollution is caused by oil spills, acid rain and urban waste [1,2,3].
Air pollution is caused by various gases and toxins that are released by the industry and by burning fuels. The main air pollutants are PM (particulate matter), SO2, NH3, NOx, CO, volatile organic compounds (VOCs), and CH4. The abatement of volatile organic compounds (VOCs) and carbon monoxide (CO) in the waste gases is an important task in the areas of environmental protection and odor control.
One of the most dangerous air pollutants, CO, is due to its high toxicity. Long-term exposure should not exceed 25 ppm in 8 h or 50 ppm in 4 h [4].
Catalysis plays a key role in solving environmental problems by creating highly active and efficient catalytic systems for environmental protection and reducing greenhouse gas emissions from the industry. Designing an efficient and cheaper catalytic system for CO oxidation is an important problem of actual environmental catalysis. Low-temperature CO oxidation has attracted considerable attention due to its wide application in exhaust gas treatment, automotive emission control, and the preferential oxidation of CO (PROX) for proton exchange membrane fuel cell application [5,6].
According to the literature data, a single α-Mn2O3 phase has been demonstrated to display remarkable catalytic activity in different reactions, such as ethylene oxidation [7], methane oxidation [8,9] and carbon monoxide oxidation [10,11,12]. It has been established that the order of reactivities for CO oxidation over various bulk MnOx is as follows: α-Mn2O3 > Mn3O4 > MnO2 [13]. The α-Mn2O3 shows excellent thermal stability among all manganese oxides within the temperature interval 100–900 °C [13]. Below approximately 300 K, α-Mn2O3 transforms from a cubic to an orthorhombic structure with symmetrical Pbca [14]. The complex crystalline structure of α- Mn2O3 is responsible for its superior catalytic behaviour [15]. The α-Mn2O3 with orthorhombic symmetry crystallizes in a distorted bixbyite structure. The unique characteristic feature of α-Mn2O3 is that MnO6 octahedra can share 12 edges and six corners with neighbouring coordination units. Furthermore, α-Mn2O3 occupies two different types of coordination octahedra. Type I octahedra shows three pairs of Mn−O distances, varying from 1.955 to 2.067 Å, and in type II octahedra, all the distances are different, four short Mn−O bonds are located in a common plane with values ranging from 1.875 to 2.011 Å and two long bonds are found on each side of this plane forming the top of an elongated octahedron with distances varying from 2.192 to 2.306 Å. The α- Mn2O3 phase exhibits a very short O−O distance of 2.55 Å, which could facilitate the O2 generation from these sites [13].
It is well known that supported Au catalysts exhibit higher activity in CO oxidation at a low temperature, while supported Pt and Pd catalysts are quite active at higher temperatures [16]. The low reaction rate of CO oxidation on the supported Pt and Pd catalysts at a low temperature appears to be the result of the strong CO adsorption, which hinders the dissociative oxygen adsorption on the same sites [17]. Catalysts based on Pt have been of considerable interest due to their high efficiency as catalysts in some essential for the environment processes, such as the oxidation of CO and VOC [6,18], PROX [19,20], DeNox [21] and others.
Since Mn2O3 is the most stable oxide within the temperature interval of 100–900 °C and the crystalline Mn2O3 exhibits highest activity in CO oxidation among Mn3O4 and MnO2 [13], as it was shown above, we directed our research efforts to shedding some more light on the behavior of the Pt phase supported on nanosized Mn2O3 during the CO oxidation.
The next purpose of our research work was to obtain more knowledge on the reaction mechanism of CO oxidation on crystalline Mn2O3 and platinum modified Mn2O3. The most probable reaction mechanism of CO oxidation in the presence of non-modified manganese-based catalysts is that of Mars van Krevelen type (MVK) [12,22,23]. There are suggestions in the current literature that other mechanisms are also possible. For example, Wang et al [13] proposed Eley–Rideal (E-R), and Langmuir–Hinshelwood (LH) at temperatures up to 523 K, for CO oxidation over Mn2O3 and MnO2 catalysts and Brooks [24] suggested an Eley–Rideal mechanism on a MnO2 surface.
To elucidate the nature of the interaction between Pt and α- Mn2O3 and its possible influence on the CO oxidation reaction mechanism, nanosized α- Mn2O3 was modified with platinum by impregnation.
2. Materials and Methods
The Mn2O3 was prepared after the calcination of Mn3O4 for 2h at 450 °C in air. The nanosized Mn3O4 was prepared according to the procedure described in [24]. Aqueous solution of Mn(CH3CO2)2·4H2O was mixed with ethanol at a 1:3 ratio in a beaker. The solution was stirred for 3 h and then hydrothermally treated for 12 h at 100 °C. The obtained brownish red precipitate was washed with water and ethanol twice and finally dried at 70 °C [25].
The as-prepared Mn2O3 was modified with Pt (~0.5 wt.% Pt) by impregnation using the aqueous solution of Pt(NH3)4Cl2. The solvent was evaporated quickly after the impregnation [26] in order to coat the oxide particles by a thin layer of Pt. In addition, the sample was calcined for 2 h at 450 °C in air.
The specific surface area of each sample was calculated by the Brunauer–Emmett–Teller (BET) method using a low temperature adsorption of nitrogen and Nova 1200 (Quantachrome) apparatus. The samples were degassed for 5 h at temperatures within the interval of 70–150 °C before the measurement
Powder X-ray diffraction (XRD) patterns were collected at room temperature in a step-scan regime (step of 0.02°, 2θ) on a Bruker D8 Advance diffractometer using Cu Kα radiation and a LynxEye detector. The XRD data processing was performed using the X’Pert HighScore program.
Electron spectrometer ESCALAB MkII (VG Scientific, now Thermofisher Scientific) was used for X-ray photoelectron measurements. The base pressure in the analysis chamber was 5 × 10−10 mbar. The spectrometer was equipped with a twin anode MgKα/AlKα X-ray source with excitation energies of 1253.6 and 1486.6 eV, respectively. The spectra were recorded at the total instrumental resolution of approximately 1 eV for both MgKα and AlKα excitation sources. The C1s line of adsorbed adventitious hydrocarbons with a binding energy of 285.0 eV has been used for energy calibration for the surfaces with electrostatic charging. Casa XPS software has been used for the processing of the measured spectra. Peak positions and areas were evaluated by a symmetrical Gaussian-Lorentzian curve fitting after a subtraction of X-ray satellites and Shirley-type background [27]. The relative concentrations of the elements, present on the surface, were determined based on the normalization of the peak areas to their photoionization cross-sections, calculated by Scofield [28].
Temperature-programmed reduction experiment was conducted by the method described in [29].
High resolution transmission electron microscopy (HRTEM) studies were carried out using the FEI Technai G2 20 (200 kV) instrument. The samples were prepared by dispersion in ethanol and loading on a holey copper grid.
The in situ diffuse–reflectance infrared Fourier transform spectroscopy was used to study CO adsorption on Pt/Me2O3-supported catalysts. The spectra were recorded on the Nicolet 6700 spectrometer (Thermo Electron Corporation, Madison, WI, USA), equipped with a high temperature/vacuum chamber installed in the Collector II accessory (Thermo Spectra-Tech, Thermo Electron Corporation, Madison, WI, USA). CO was adsorbed from the CO + N2 or CO + O2 + N2 mixture. The experiments were carried out on oxidized (“as-prepared”) catalysts, pretreated in N2 for 1 h at 350 °C.
The following experimental testing conditions were applied: Catalyst bed volume of 0.3 cm3, irregular shaped particles having an average size of 0.2–0.4 mm, reactor diameter of 5.5 mm, quartz-glass, the catalyst bed volume was increased to 0.5 cm3 with the same size of inert particles of crushed quartz glass. In order to minimize the external mass transfer limitations, the gaseous hourly space velocity (GHSVSTP) was fixed at 100,000 h–1. The adiabatic effect of the reaction was compensated by keeping the catalyst bed temperature almost constant (the deviations did not exceed +/–±1 °C). The pressure drop of the catalytic bed was established to be less than 2 kPa. The axial dispersion effect was not taken into account since the catalyst bed corresponds to a chain of more than 10 ideal-mixing cells. Thus, the geometric characteristics and the flow conditions of the catalytic reactor are similar to the case of the isothermal plug flow reactor (PFR), except for the effect of the radial velocity profile inside the catalyst bed that has been subtracted by mathematic modelling.
To obtain the experimental data, suitable for the fitting of the reaction parameters, the inlet concentrations of reactants were varied as follows: CO feed concentrations were set on the levels of 5 × 10−5, 1.6 × 10−4 and 1.5 × 10−3 vol.%, oxygen had levels of 0.7, 7.0 and 20.0 vol.%. All gas mixtures were balanced to 100 % with nitrogen (4.0). The reproducibility of the measured conversion degrees was checked by preliminary tests under conditions similar but not identical to those represented in the study. The value of the standard deviation (+/−±1.5%) was calculated using the data from six consecutive measurements at each experimental point. The represented results are based on the average values for the measured conversion degree within two parallel measurements. The gas analysis was performed using an on-line gas-analyzer of CO/CO2/O2 (FTIR, Horiba, HORIBA, Ltd., Tokyo, Japan).
3. Results and Discussion
Figure 1 illustrates the temperature dependencies of the conversion of CO to CO2. Obviously, the activity of the Mn-Pt-sample is higher than that of the pure Mn-catalyst.
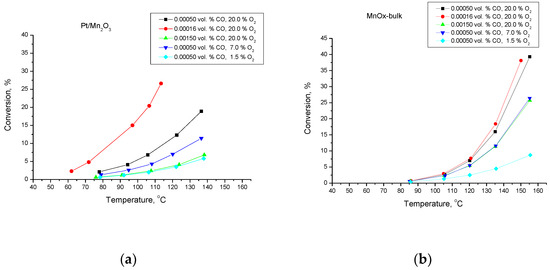
Figure 1.
Temperature dependencies of the conversion degree of CO oxidation for Pt/Mn2O3 (a) and pure Mn2O3 (b).
The XRD, IR, TEM data and surface area of as-prepared manganese oxide and after its calcination are discussed in [29]. According to these analyses, the formation of the Mn3O4 phase with tetragonal symmetry was suggested after synthesis following the procedure described above. The particles size is within the range ~15–20 nm and SBET = 76 m2/g. Calcination for 2h at 450 °C leads to the transformation of Mn3O4 into α-Mn2O3 (bixbyite, PDF 01-071-0636). The transformation of Mn3O4 with tetragonal symmetry into α-Mn2O3 is accompanied by an increase in the particle size (30–40 nm) and a decrease in the surface area (20 m2/g) [29]. The form of α-Mn2O3 particles remains spherical (Figure 2A). The platinum loading and next calcination does not further change the manganese oxide phase, its symmetry and structure, surface area and manganese oxide particle size (Figure 2B).

Figure 2.
TEM image of: (A) Mn2O3 after Mn3O4 calcination; (B) after Pt deposition figure.
Platinum or platinum oxide was not detected in Pt/Mn2O3 catalysts revealing a finely divided phase of Pt (XRD data are not represented here). Additional information concerning manganese oxide phases were obtained from the infrared spectroscopy. The amorphous components and those with short-range order could be established by means of IR analysis [30]. Figure 3 exhibits the IR spectrum of pure Mn2O3 and Pt/Mn2O3.
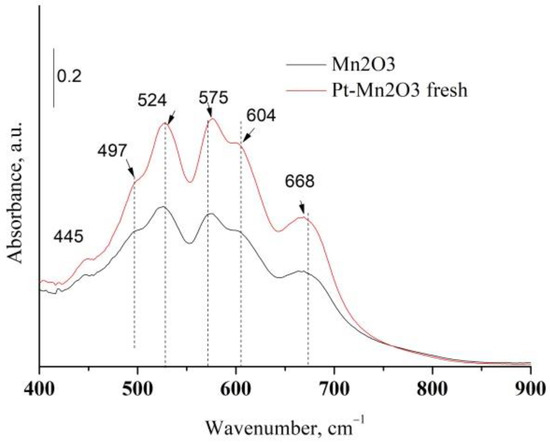
Figure 3.
IR spectra of pure Mn2O3 and Pt/Mn2O3.
The characteristic IR bands of manganese oxides are appearing in the next three regions: 200–450, 450–600, and 600–750 cm−1. They are related to spectral regions where wagging, bending and stretching vibrations are active [31]. Table 1 compares the IR bands of pure manganese oxide after calcination with those of various types of manganese oxides. As is visible in Figure 3 and Table 1, the IR spectrum of our mono-component manganese sample exhibits the bands characteristic of α-Mn2O3 and γ-Mn2O3, thus suggesting that, after calcination, a part of Mn2O3 is in the form of amorphous γ-Mn2O3.

Table 1.
IR frequency vibrations of manganese oxides.
The TPR patterns of pure Mn2O3 and after Pt deposition are represented in Figure 4. The reduction profile of Mn2O3 consists of two overlapping peaks with a maxima at 420 and 473 °C (Figure 4) [29]. They are related to a subsequent reduction of Mn2O3 to Mn3O4 and MnO. The reduction of Mn2O3 in hydrogen is influenced by the preparation procedure and the Mn2O3 of higher crystallinity is reduced at a higher temperature [32]. The reduction of Pt/Mn2O3 takes place at a significantly lower temperature. The hydrogen consumption is observed at the following temperatures in the TPR profile of Pt/Mn2O3: 144, 206 and 261 °C. The peak at 144 °C most likely result from the reduction in platinum oxide [33], and the other two peaks result from the promoted reduction of manganese oxide due to the presence of platinum. It is well known in the current literature that noble metals promote the reduction of oxides by supplying hydrogen via spillover from the pre-reduced noble metal particles to the oxides [33,34].
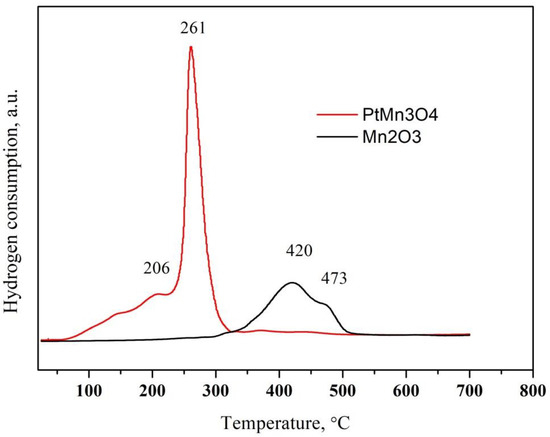
Figure 4.
TPR profile of pure oxides and after Pt modification.
X-ray photoelectron spectroscopy was applied to study the chemical state of Pt and Mn and the atomic concentrations of the different elements on the catalytic surfaces (Figure 5). The binding energy of the Mn2p peak and the Pt/Mn2O3 samples is within the range of 641.3–641.9 eV, which is characteristic of Mn2O3 [35,36].
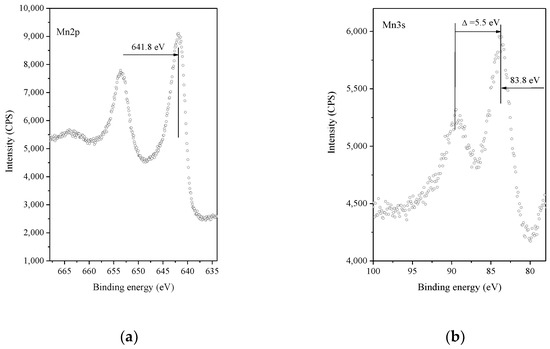
Figure 5.
XPS spectra of Pt/Mn2O3 after calcination for 2 h at 450 °C: (a) Mn 2p region; (b) Mn3s region.
The Mn3s exchange splitting is accepted as a more precise means for determining the oxidation state of manganese ions [36,37]. According to the data in the literature, the exchange splitting of the 3s level is 6.5 eV for Mn2+, 5.5 eV for Mn3+ and 4.5 eV for Mn4+ [38]. The Mn3s spectrum of Pt/Mn2O3 shows a broad 3s line at 83.8 eV and a satellite line at 89.3 eV, which is in accordance with the existence of Mn3+ (5.5 eV) species.
The XPS spectra of the calcined Pt/Mn2O3 sample in the region of Pt4f (Figure 6a) show a peak at 74.6 eV attributed to Pt2+ and another peak at 72.8 eV assigned to Pt(OH)2 [39,40,41]. Obviously, during the calcination, both Pt(OH)2 and PtO are being formed. According to the data for thermal decomposition of [Pt(NH3)4]Cl2 above 320 °C [42], the main decomposition product is Pt0. The formation of platinum in an oxidized state only on the surface of Mn2O3 could be associated with the interaction between the Pt(II)tetraamine complex and the Mn2O3 surface. A similar phenomenon was observed by the deposition of platinum from [Pt(NH3)4]Cl2 on hexaniobate nanoscrolls [43] and Pt on SnO2 [44].
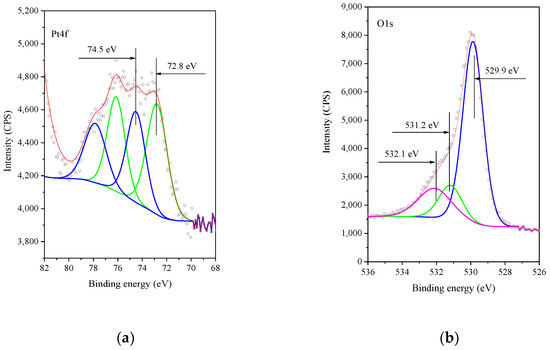
Figure 6.
XP Spectra of Pt4f core level (a) and of the O1s core level (b) of the 0.5%Pt/Mn2O3 sample as prepared. Legend: (a) dots-represent the experiment; red color is used for resultant; grenn line–Pt2+; blue line–Pt4+; (b) dots–experiment; red line–resultant; blue and green lines–lattice oxygen; magenta line–OH− groups.
The O1s spectrum is shown in Figure 6. It contains a main peak at ca. 529.4 eV, which is characteristic of the lattice oxygen (O2−) [27] along with a distinct shoulder at ca. 531.5 eV, which is assigned to the OH¯-groups, and 532.1 eV is assignable to a mixture of C-H-O groups, residues from the preparation procedure, and adsorbed water on the surface of the catalysts [44,45,46].
In situ DRIFTS was used to acquire some additional information about the type, stability and reactivity of the active sites, chemical state of the surface under static and dynamic conditions. Since the ν(C−O) vibration is sensitive to the chemical state of the metal atom(s), to which the CO molecule is coordinated, it is the most used probe molecule for studying supported catalysts. IR spectra can provide information on the relative strength of the adsorption of the reactants, whose strength is closely related to the catalytic activity of the respective sites, and it allows information to be gained regarding the mechanism of the CO oxidation reaction [47,48]. The DRIFTS experiments were carried out to investigate the CO adsorption on the surface of Pt supported on manganese oxide and the behavior of adsorbed CO surface species in feed flow mixtures of CO, N2 and O2. The IR spectra of adsorbed CO from CO + N2 flow on the calcined sample are represented in Figure 7.
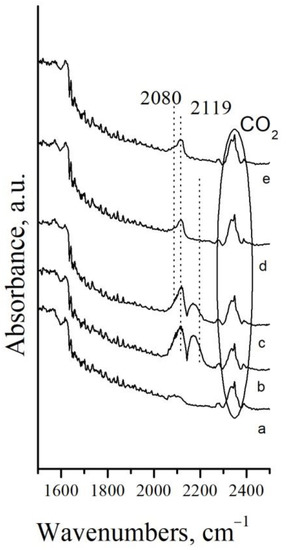
Figure 7.
DRIFTS spectra of Pt/Mn2O3 after: a–in N2 flow; b–10 min in a flow mixture 10% CO in N2, at Troom; c–stay 21 h in the mixture 10% CO in N2 at Troom; d–5 min desorption of CO in N2 flow; e–10 min desorption in N2 flow.
The CO gas is demonstrated by a doublet with a minimum centered at 2142 cm−1 [49]. It is visible that the wing of the CO gas phase at higher wavenumbers overlaps with the band at 2119 cm−1, attributed to the linear CO species adsorbed on Pt1+ [50]. The occurrence of such species was confirmed after CO gas removal by a short purge with nitrogen (Figure 7d,e). Low intensive shoulder at 2080 cm−1 is visible after 10 min in a nitrogen flow as well. It is ascribed to the Pt0−CO species [50].
Considering the XPS results (see Figure 6a), the Pt/Mn2O3 sample initially contains Pt2+ surface species only. The formation of Pt1+−CO and Pt0−CO linear carbonyls proves the reduction of the platinum surface as a result of CO adsorption.
The IR spectra of Pt/Mn2O3 were also recorded after exposure to CO/O2 mixture at different temperatures (Figure 8). The formation of Pt1+−CO and Pt0−CO linear carbonyls at room temperature is seen as well. As a result of a further increase in the temperature, the band at 2078 cm−1 related to linearly adsorbed CO on metallic Pt increases, thus showing a gradual reduction in the platinum surface.
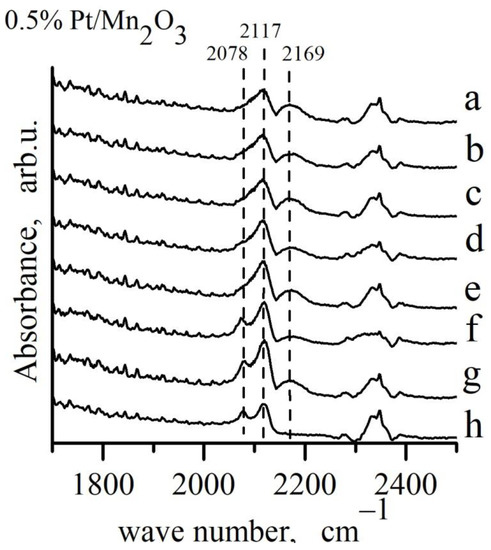
Figure 8.
Adsorption of CO on 0.5% Pt/Mn2O3 from a mixture of 4% CO + 10% O2 + N2 at different temperatures: a–Troom; b–100 °C; c–cooling after reaction at 100 °C; d–150 °C; e–cooling after reaction at 150 °C; f–200 °C; g–cooling after reaction at 200 °C; h–67 h in mixture.
The reactivity of the different carbonyl species was studied after adsorption of CO at a longer contact time. The spectra in oxygen flow at elevated temperatures are shown in Figure 9.
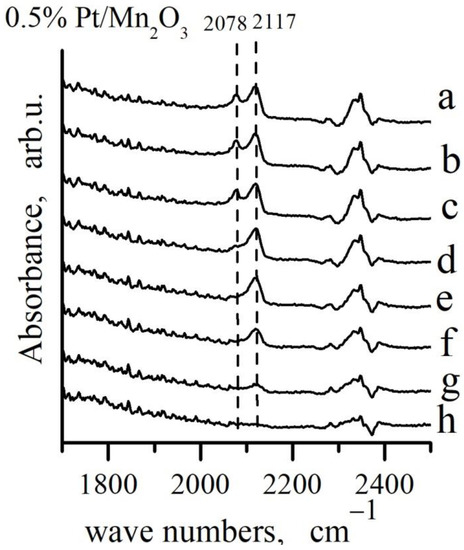
Figure 9.
FTIR spectra during oxidation of CO adsorbed on 0.5% Pt/Mn2O3: a–CO adsorption from a flow mixture (4% CO + 10% O2)/N2 at Troom, waiting 67 h at Troom and desorption in: b–10 min N2; c–30 min at Troom in N2 + O2; d–30 min at 50 °C in N2 + O2; e–30 min at 100 °C in N2 + O2 flow; f–30 min at 150 °C in N2 + O2 flow; g–30 min at 200 °C in N2 + O2 flow; h–30 min at 250 °C in N2 + O2 flow.
The highest initial rate of depletion was observed in case of the Pt0−CO species and the band intensity decreases to zero after 30 min in N2 + O2 flow at 100 °C. The high frequency band at 2117 cm−1, assigned to Pt1+−CO species decreased in intensity more slightly and it disappeared completely after 30 min in a He + O2 flow at 250 °C.
The DRIFTS of CO adsorption from the mixtures 10 vol.% CO in N2 and 1 vol.% CO + 10 vol.% O2 in N2 indicates that: (i) CO linear carbonyls, formed on Pt/Mn2O3, are stable at ambient temperature in N2 and O2; (ii) under oxidation conditions part of the platinum is in the reduced (Pt0) state.
The Langmuir–Hinshelwood mechanism regarding the reaction between the adsorbed CO and oxygen species was well established as the main reaction pathway over the platinum catalysts [51]. As was mentioned above, the linear Pt1+−CO and Pt0−CO carbonyls were observed in the IR spectra during the CO oxidation. The CO adsorbed on Pt0 is stable up to 100 °C and Pt1+−CO up to 250 °C. Based on this, it is possible to assume that the reaction mechanism below 100 °C involves both types of linear carbonyls (Pt1+−CO and Pt0−CO), while at temperatures higher than 100 °C, the reaction pathway most likely involves the CO linear carbonyl adsorbed on the oxidized sites (Pt1+−CO). Due to the fact that the CO species are not stable at higher temperatures, the great part of the Pt surface will be accessible for O2 adsorption and dissociation. This allows the pre-adsorbed oxygen atoms to react with gaseous CO via the Eley-Rideal (denoted “E-R”) mechanism. A so-called bifunctional reaction pathway involving the interaction between CO adsorbed on the noble metal and oxygen from the oxide is also possible, due to the high oxygen storage capacity of the manganese oxides [29].
In searching for the most probable mechanism, kinetic experiments were performed. The corresponding kinetic parameters were obtained by applying the method described by Duprat [52]. More details on the calculation procedure were reported in earlier publications [53,54,55]. The method consists of a direct integration of the reaction rate when operating at conversions higher than the limits of 5–8%, which is typical of the case for a differential type of reactor. The kinetic parameters fitting was performed by applying an integrated computer program for the simultaneous solving of the material balance in an isothermal reactor and numerical nonlinear optimization procedure. Residuals squared sums (RSS) between the experimental data and the model predictions are minimized and the square of correlation coefficient (R2) was calculated. The optimization procedure was carried out by different sets of starting conditions in order to find the global minimum.
Power-law kinetic model (PWL) was applied as a first approach for further selection among the chosen mechanistic models.
The value for the reaction order towards CO is relatively close to unity in the case of MnOx catalysts, which supposed the inclusion of the consideration of Eley–Rideal mechanism. The negative reaction order towards the CO2 shows an inhibition effect and it is the highest in the Mn-bulk sample. It should be pointed out that the impact of the CO2 formed during the reaction is only taken into account. The consumption of oxygen is also accounted for along the reactor length.
Based on the values of the observed reaction orders given by the PWL model, one could suggest to evaluate several mechanistic models for their consistence with the obtained experimental results, i.e., the following specification could be given:
- Langmuir-Hinshelwood (LH-D-DS-1), adsorption of CO and oxygen on different types of sites (DS), dissociative (D) adsorption of oxygen, CO2 molecules compete with oxygen molecules for the same type of adsorption sites [56].
- Langmuir-Hinshelwood (LH-D-DS-2), adsorption of CO and oxygen on different types of sites (DS), CO2 molecules compete with CO molecules for the same type of adsorption sites [56].;
- Mars-van Krevelen [57] (MVK-1), CO2 molecules compete with the CO molecules for the oxidized adsorption sites;
- Mars-van Krevelen (MVK-2), CO2 molecules compete with oxygen molecules for the reduced adsorption sites
- Eley-Rideal mechanism, CO2 molecules compete with the oxygen molecules for the same type of adsorption sites, O2 reacts directly from gas phase.
The calculated kinetics parameters (pre-exponential factor, activation energy, heat of adsorption and reaction order) are represented in Table 2, Table 3, Table 4 and Table 5.
Table 2.
Reaction rate expressions and kinetics parameters for the applied power–law kinetic model.
Table 3.
Reaction rate expressions and kinetics parameters for the applied Mars-van Krevelen models.
Table 4.
Reaction rate expressions and kinetics parameters for the applied Langmuir–Hinshelwood models.
Table 5.
Reaction rate expressions and kinetics parameters for the applied Elley-Rideal model.
Regarding the Mn-bulk sample, the best fit was achieved when using the MVK-1 model, suggesting that CO2 is adsorbed on the oxidized sites. In the case of the Pt/MnOx sample, the most probable mechanism is LH-DS-D-2, where the adsorption of CO and oxygen proceed on different types of sites (DS), the CO2 molecules being in competition with CO molecules for the same type of adsorption sites. Regarding the Eley–Rideal mechanism, the probability is relatively lower, and the Pt/Mn2O3 sample still cannot be excluded.
4. Conclusions
The deposition of platinum on the surface of nanosized Mn2O3 having cubic symmetry leads to the formation of finely divided PtO and Pt(OH)2. The CO adsorption in an inert atmosphere or in the presence of oxygen leads to a partial reduction in the Pt2+ surface species and the formation of linear Pt1+−CO and Pt0−CO carbonyls.
Based on the combination of the experimental results and model calculations, we can conclude that the Mars–van Krevelen mechanism is the most probable over pure Mn2O3, thus suggesting that CO2 is adsorbed on the oxidized sites. The addition of platinum to Mn2O3 changes the reaction mechanism to the Langmuir–Hinshelwood type, where the adsorption of CO and oxygen proceeds on different types of sites, CO2 molecules being in competition with CO molecules for the same type of adsorption sites. The results, obtained by the DRIFTS of CO adsorption from the mixtures CO in N2 and CO+O2+N2, revealed that both linear carbonyls, formed on Pt1+ and Pt0 centers took part in the reaction on the surface of Pt/Mn2O3.
Author Contributions
S.T., A.N.-results analysis, writing—original draft preparation, conceptualization and discussion; A.N.-catalytic test, experiments and analysis; H.K. performed and discussed XPS analysis; M.S. performed and discussed FTIR analysis; I.Y.-sample synthesis, TEM and BET measurements and analysis; K.T.-H2-TPR measurements; S.T., A.N.-supervision and project administration. All authors contributed to discussion of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported financially by National Science Fund of Bulgaria; Grant number KP-06-H49/4 and by the European Regional Development Fund within the framework of OP “Science and Education for Smart Growth 2014–2020”, Project CoE “National center of mechatronics and clean technologies”, No.BG05M2OP001-1.001-0008-C01.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- El-Bindary, M.A.; El-Desouky, M.G.; El-Bindary, A.A. Adsorption of industrial dye from aqueous solutions onto thermally treated green adsorbent: A complete batch system evaluation. J. Mol. Liq. 2021, 346, 117082. [Google Scholar] [CrossRef]
- Kiwaan, H.A.; Mohamed, F.S.; El-Ghamaz, N.A.; Beshry, N.M.; El-Bindary, A.A. Experimental and electrical studies of Na-X zeolite for the adsorption of different dyes. J. Mol. Liq. 2021, 332, 115877. [Google Scholar] [CrossRef]
- Altalhi, T.; Ibrahim, M.M.; Mersal, G.A.M.; Mahmoud, M.H.H.; Mohamed, T.K.; Ashraf, G.E.-D.; Mohamed, A.E.-B.; El-Bindary, A. Adsorption of doxorubicin hydrochloride onto thermally trea ted green adsorbent: Equilibrium, kinetic and thermodynamic studies. J. Mol. Struct. 2022, 1263, 133160. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D.L. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent Advances in Preferential Oxidation of CO Reaction over Platinum Group Metal Catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Spivey, J.J. Complete catalytic oxidation of volatile organics. Ind. Eng. Chem. Res. 1987, 26, 2165–2180. [Google Scholar] [CrossRef]
- Dmuchovsky, B.; Freerks, M.C.; Zienty, F.B. Metal oxide activities in the oxidation of ethylene. J. Catal. 1965, 4, 577–580. [Google Scholar] [CrossRef]
- Han, Y.-F.; Ramesh, K.; Chen, L.W.; Widjaja, E.; Chilukoti, S.; Chen, F. Observation of the reversible phase-transformation of α-Mn2O3 nanocrystals during the catalytic combustion of methane by in situ Raman spectroscopy. J. Phys. Chem. C 2007, 111, 2830–2833. [Google Scholar] [CrossRef]
- Han, Y.-F.; Chen, L.W.; Ramesh, K.; Widjaja, E.; Chilukoti, S.; Surjami, I.K.; Chen, J. Kinetic and spectroscopic study of methane combustion over α-Mn2O3 nanocrystal catalysts. J. Catal. 2008, 253, 261–268. [Google Scholar] [CrossRef]
- Frey, K.; Iablokov, V.; Sáfrán, G.; Osán, J.; Sajό, I.; Szukiewicz, R.; Chenakin, S.; Kruse, N. Nanostructured MnOx as highly active catalyst for CO oxidation. J. Catal. 2012, 287, 30–36. [Google Scholar] [CrossRef]
- Cracium, R.; Nentwick, B.; Hadjiivanov, K.; Knözinger, H. Structure and redox properties of MnOx/Yttrium-stabilized zirconia (YSZ) catalyst and its used in CO and CH4 oxidation. Appl. Catal. A Gen. 2003, 243, 67–79. [Google Scholar] [CrossRef]
- Wenge, L.; Deyong, G.; Xin, X. Research progress of palladium catalysts for methane combustion. China Petrol. Proc. Petrochem. Technol. Rev. 2012, 14, 1–9. [Google Scholar]
- Wang, L.-C.; Liu, Q.; Huang, X.-S.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Re-investigating the CO oxidation mechanism over unsupported MnO, Mn2O3 and MnO2 catalysts. Appl. Catal. B 2009, 88, 204–212. [Google Scholar] [CrossRef]
- Cockayne, E.; Levin, I.; Hui, W.H.; Llobet, A. Magnetic structure of bixbyite α-Mn2O3: A combined DFT+U and neutron diffraction study. Phys. Rev. B 2013, 87, 184413. [Google Scholar] [CrossRef]
- Ramírez, A.; Hillebrand, P.; Stellmach, D.; Matthias, M.M.; Peter Bogdanoff, P.; Sebastian Fiechter, S. Evaluation of MnOx, Mn2O3, and Mn3O4 Electrodeposited Films or the Oxygen Evolution Reaction of Water. J. Phys. Chem. C 2014, 118, 14073–14081. [Google Scholar] [CrossRef]
- Haruta, M. Gold as a novel catalyst in the 21st century: Preparation, working mechanism and applications. Gold Bull. 2004, 37, 27–36. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Iglesias-Juez, A.; Anderson, J.A.; Conesa, J.C. Light-off behaviour of PdO/γ-Al2O3 catalysts for stoichiometric CO–O2 and CO–O2–NO reactions: A combined catalytic activity–in situ DRIFTS study. J. Catal. 2004, 221, 85–92. [Google Scholar] [CrossRef]
- Tsou, J.; Magnoux, P.; Guisnet, M.; Órfão, J.J.M.; Figueiredo, J.L. Catalytic oxidation of volatile organic compounds: Oxidation of methyl-isobutyl-ketone over Pt/zeolite catalysts. Appl. Catal. B 2005, 57, 117–123. [Google Scholar] [CrossRef]
- Teschner, D.; Wootsch, A.; Pozdnyakova-Tellinger, O.; Kröhnert, J.; Vass, E.M.; Hävecker, M.; Zafeiratos, S.; Schnörch, P.; Jentoft, P.C.; Knop-Gericke, A.; et al. Partial pressure dependent in situ spectroscopic study on the preferential CO oxidation in hydrogen (PROX) over Pt/ceria catalysts. J. Catal. 2007, 249, 318–327. [Google Scholar] [CrossRef]
- Wu, J.C.-S.; Lin, Z.; Pan, J.; Rei, M. A novel boron nitride supported Pt catalyst for VOC incineration. Appl. Catal. A 2000, 219, 117–124. [Google Scholar] [CrossRef]
- Olympiou, G.G.; Efstathiou, A.M. Industrial NOx control via H2-SCR on a novel supported-Pt nanocatalyst. Chem. Eng. J. 2011, 170, 424–432. [Google Scholar] [CrossRef]
- Mooi, P.W. Selwood, Structure and Catalytic Activity of Supported Manganese, Copper and Iron Oxides. J. Am. Chem. Soc. 1952, 74, 2461. [Google Scholar] [CrossRef]
- Cracium, R. Structure/activity correlation for unpromoted and CeO2-promoted MnO2/SiO2 catalysts. Catal. Lett. 1998, 55, 25–31. [Google Scholar]
- Brooks, C.S. The kinetics of hydrogen and carbon monoxide oxidation over a manganese oxide. J Catal. 1967, 8, 272–282. [Google Scholar] [CrossRef]
- Song, R.; Feng, S.; Wang, H.; Hou, C. Effect of organic solvents on particle size of Mn3O4 nanoparticles synthesized by a solvothermal method. J. Solid State Chem. 2013, 202, 57–60. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis a Practical Approach; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Shirley, D. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron. Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Todorova, S.; Naydenov, A.; Kolev, H.; Ivanov, G.; Ganguly, A.; Mondal, S.; Saha, S.; Ganguli, A.K. Reaction kinetics and mechanism of complete methane oxidation on Pd/Mn2O3 catalyst. Reac. Kinet. Mechan. Catal. 2018, 123, 585–605. [Google Scholar] [CrossRef]
- Kang, L.; Zhang, M.; Liu, Z.H.; Ooi, K. IR spectra of manganese oxides with either layered or tunnel structures. Spectrochim. Acta Part A 2007, 67, 864–869. [Google Scholar] [CrossRef]
- Julien, C.M.; Massot, M.; Poinsignon, C. Lattice vibrations of manganese oxides: Part I. Periodic structures. Spectrochim. Acta Part A 2004, 60, 689–700. [Google Scholar] [CrossRef]
- Stobbe, E.R.; De Boer, B.A.; Geus, J.W. The reduction and oxidation behaviour of manganese oxides. Catal. Today 1999, 47, 161–167. [Google Scholar] [CrossRef]
- K. Reddy, G.; C. Peck, T.; A. Roberts, C. “PdO vs. PtO”—The Influence of PGM Oxide Promotion of Co3O4 Spinel on Direct NO Decomposition Activity. Catalysts 2019, 9, 62. [Google Scholar] [CrossRef]
- Bianchi, C. TPR and XPS investigations of Co/Al2O3 catalysts promoted with Ru, Ir and Pt. Catal. Lett. 2001, 76, 155–159. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Banerjee, D. Interpretation of XPS Mn (2p) spectra of Mn oxyhydroxides and constraints on the mechanism of MnO2 precipitation. Am. Mineral. 1998, 83, 305–315. [Google Scholar] [CrossRef]
- Oku, M.; Hirokawa, K.; Ikeda, S. X-ray photoelectron spectroscopy of manganese-oxygen systems. J. Electron. Spectrosc. Relat. Phenom. 1975, 7, 465–472. [Google Scholar] [CrossRef]
- Barrio, I.; Legorburu, I.; Montes, M.; Dominguez, M.I.; Centeno, M.A.; Odriozola, J.A. New redox deposition-precipitation method for preparation of supported manganese oxide catalysts. Catal. Lett. 2005, 101, 151–157. [Google Scholar] [CrossRef]
- Brabers, V.A.M.; Van Setten, F.M.; Knapen, P.S.A. X-ray photoelectron spectroscopy study of the cation valences in nickel manganite. J. Solid State. Chem. 1983, 49, 93–98. [Google Scholar] [CrossRef]
- Raddi de Araujo, L.R.; Shamal, M. The calcination effects on Pt/HZSM-5 catalysts in the aromatization of propane. Appl. Catal. A 2000, 203, 275–284. [Google Scholar] [CrossRef]
- Kiss, G.; Josepovits, V.K.; Kovacs, K.; Ostrick, B.; Fleischer, M.; Meixner, H.; Reti, F. CO sensitivity of the PtO/SnO2 and PdO/SnO2 layer structures: Kelvin probe and XPS analysis. Thin Solid Films 2003, 436, 115–118. [Google Scholar] [CrossRef]
- Drawdy, J.E.; Hoflund, G.B.; Gardner, S.D.; Yngvadottir, E.; Schryer, D.R. Effect of pretreatment on a platinized tin oxide catalyst used for low-temperature CO oxidation. Surf. Interface Anal. 1990, 16, 369. [Google Scholar] [CrossRef]
- Kinoshita, K.; Routsis, K.; Bett, J.A.S. The thermal decomposition of platinum(II) and (IV) complexes. Thermochim. Acta 1974, 10, 109–117. [Google Scholar] [CrossRef]
- Nunes, B.N.; Haisch, C.; Emeline, A.V.; Bahnemann, D.W.; Patrocinio, A.O.T. Photocatalytic properties of layer-by-layer thin films of hexaniobate nanoscrolls. Catal. Today 2019, 326, 60–67. [Google Scholar] [CrossRef]
- Gardner, S.D.; Hoflund, G.B.; Davidson, M.R.; Laitinen, H.A.; Schryer, D.R.; Upchurch, B.T. Catalytic behavior of noble metal/reducible oxide materials for low-temperature carbon monoxide oxidation. 2. Surface characterization of gold/manganese oxide. Langmuir 1991, 7, 2140–2145. [Google Scholar] [CrossRef]
- Lee, S.J.; Gavriilidis, A.; Pankhurst, Q.A.; Kyek, A.; Wagner, F.E.; Wong, P.C.L.; Yeung, K.L. Effect of drying conditions of Au–Mn co-precipitates for low-temperature CO oxidation. J. Catal. 2001, 200, 298–300. [Google Scholar] [CrossRef]
- Iablokov, V. Manganese and Cobalt Oxides as Highly Active Catalysts for CO Oxidation. Ph.D. Thesis, Faculte des Sciences, Chimie Physique des Matériaux (Catalyse-Tribologie), Universite Libre de Bruxelles, Bruxelles, Belgium, 2011. [Google Scholar]
- Zeinalipour-Yazdi, C.D.; Cooksy, A.L.; Efstathiou, A.M. CO adsorption on transition metal clusters: Trends from density functional theory. Surf. Sci. 2008, 602, 1858. [Google Scholar] [CrossRef]
- Monteiro, S.R.; Dieguez, L.C.; Schmal, M. The role of Pd precursors in the oxidation of carbon monoxide over Pd/Al2O3 and Pd/CeO2/Al2O3 catalysts. Catal. Today 2001, 65, 77. [Google Scholar] [CrossRef]
- Little, L.H. Infrared Spectra of Adsorbed Species; Academic Press Inc.: New York, NY, USA, 1966. [Google Scholar]
- Hadjiivanov, K.; Vayssilov, G. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Engel, T.; Ertl, G. Oxidation of carbon monoxide. In The Chemical Physics of Solid Surfaces and Heterogeneous Catalysis; King, D.A., Woodruff, D.P., Eds.; Elsevier: Amsterdam, The Netherlands, 1982; Volume 4, p. 73. [Google Scholar]
- Duprat, F. Light-off curve of catalytic reaction and kinetics. Chem. Eng. Sci. 2002, 57, 901–911. [Google Scholar] [CrossRef]
- Todorova, S.; Naydenov, A.; Kolev, H.; Holgado, J.P.; Ivanov, G.; Kadinov, G.; Caballero, A. Mechanism of complete n-hexane oxidation on silica supported cobalt and manganese catalysts. Appl. Catal. A 2012, 413–414, 43–51. [Google Scholar] [CrossRef]
- Markova-Velichkova, M.; Lazarova, T.; Tumbalev, V.; Ivanov, G.; Kovacheva, D.; Stefanov, P.; Naydenov, A. Complete oxidation of hydrocarbons on YFeO3 and LaFeO3 catalysts. Chem. Eng. J. 2013, 231, 236–244. [Google Scholar] [CrossRef]
- Stefanov, P.; Todorova, S.; Naydenov, A.; Tzaneva, B.; Kolev, H.; Atanasova, G.; Stoyanova, D.; Karakirova, Y.; Alexieva, K. On the development of active and stable Pd-Co/γ-Al2O3 catalyst for complete oxidation of methane. Chem. Eng. 2015, 266, 329–338. [Google Scholar] [CrossRef]
- Harriott, P. Chemical Reactor Design; Marcel Dekker, Inc.: New York, NY, USA; Taylor & Francis Group LLC: Oxford, UK, 2003; pp. 52–55. [Google Scholar]
- Mars, P.; Van Krevelen, D.W. Oxidations carried out by the means of vanadium oxide catalysts. Chem. Eng. Sci. Spec. Suppl. 1954, 3, 41–59. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).