Abstract
Betanodavirus infection induces viral nervous necrosis (VNN) in fish. However, the role of cell death and autophagy in the pathogenesis of VNN remains unknown. This study aimed to investigate the effect of red-spotted grouper nervous necrosis virus (RGNNV) infection on Bcl2 downregulation and overexpression on asymmetric interaction between cell death and autophagy. The mRFP-LC3 reporter system was used to identify autophagosome formation in GF-1 (Grouper fin-1) fish cells. We found that the RGNNV could strongly induce autophagosome formation 36 h post-infection (hpi) after autophagy inhibitor 3-MA had downregulated anti-apoptotic genes such as Bcl2 and Bcl2L1 (Bcl-xL). We proposed that the overexpression of Bcl2 and Bcl2L1 can modulate both cell death and autophagy. Then, we found that it can also reduce either type III cell death or autophagy, which are mildly correlated with reduced viral replication. Our data suggest that RGNNV-induced Bcl2 downregulation correlates with the asymmetrical interaction between cell death induction and the autophagy process, which resembles viral replication.
1. Introduction
Betanodaviruses are the causative agents of viral nervous necrosis (VNN), an infectious neuropathological condition characterized by necrosis of the brain and retina in fish [1]. RGNNV infection causes massive mortality in larval and juvenile populations of several marine teleost species [2,3]. The infection correlates with the modulation of innate or acquired immunity [4,5], but very little is known about the molecular mechanisms underlying the pathogenesis of VNN.
The betanodavirus genome also includes two single-strand RNA molecules with positive polarity: RNA1 (3.1 kb) and RNA2 (1.4 kb), which lacks a 3′ poly (A) extension [4]. RNA1 encodes a 110 kDa protein acting on an RNA-dependent RNA polymerase (protein A) for replication of the viral genome. RNA2 encodes a 42 kDa capsid protein [4,6] that can trigger post-apoptotic necrotic cell death through a cytochrome c release-mediated pathway in GF-1 cells [7]. In the middle–late replication stage, betanodaviruses were also shown to synthesize a sub-genomic RNA3 from the 3′ terminus of RNA1 during genome replication, which encodes two non-structural proteins, B1 (absent in some strains) [8] and B2 (as a death gene) [1,9].
Recently, cell death has acquired multiple meanings. Apoptosis, type I cell death, has taken center stage as the principal mechanism of programmed cell death in mammalian tissues. Autophagy, type II cell death, is characterized by the massive accumulation of autophagic vacuoles in the cytoplasm of cells. The autophagic flow is activated as an adaptive response to a variety of extracellular and intracellular stresses, which include some factor changes such as nutrient deprivation, hormonal or therapeutic treatment, pathogenic infection, aggregated and misfolded proteins, and damaged organelles. Necroptosis, a programmed type III cell death, manifests with a distinctive morphology different from type I and type II [10,11].
A number of studies have investigated the molecular mechanisms by which the red-spotted grouper nervous necrosis virus (RGNNV) induces apoptosis or necrosis in host cells [12,13]. RGNNV infection induces cell death that correlates with the loss of mitochondrial membrane potential (MMP) and the release of cytochrome c at the mid-replication stage. This is regulated by the zebrafish anti-apoptotic protein Bcl-2 [13], a member of the BCL-2 family [14], and blocked by bongkrekic acid, an inhibitor of the mitochondrial permeability transition pore [12,14]. Furthermore, betanodavirus-induced mitochondria-mediated cell death can be blocked by the protein synthesis inhibitor cycloheximide [14], indicating that newly synthesized proteins from the viral genome are necessary for cell necrosis. Recent studies of the viral genome revealed the role of two viral death factors: protein α, which can induce the caspase-3- and caspase-9-dependent signaling pathway [7,15]; and protein B2, a non-structural protein that can induce BAX (BCL-2 associated X, apoptosis regulator)-mediated cell death [9] and cause ATP-depletion-mediated necrosis [16], which is dependent on the expression of the zebrafish anti-apoptotic gene Bcl2L1 (BCL-2 like 1) [9,14]. A recent study showed that B2 can induce DNM1L/Drp1 (dynamin 1 like)-mediated mitochondrial fragmentation [17]. However, little is known as to whether betanodaviruses induce necrosis or necroptosis.
Autophagy is a universal cell defense mechanism against intracellular microbes, which are delivered to lysosomes for degradation [18]. Autophagy contributes to the activation of antiviral innate immunity [19,20] and the adaptive immune response by delivering virus-derived peptides to lymphocytes for presentation to major histocompatibility complex molecules [21,22,23]. Autophagy and autophagy genes likely play both anti-viral and proviral roles in the life cycle and pathogenesis of many different virus families. With respect to anti-viral roles, autophagy proteins function by targeting viral components or virions for lysosomal degradation in a process termed xenophagy. They also play a role in the initiation of innate and adaptive immune system responses to viral infections. Consistent with this anti-viral role of host autophagy, some viruses encode virulence factors that interact with the host autophagy machinery to block its execution. On the other hand, other viruses appear to use components of the autophagic machinery to foster their own intracellular growth or non-lytic cellular egress. As the details of the role(s) of autophagy in viral pathogenesis become clearer, new anti-viral therapies could be developed to inhibit the beneficial and enhance the destructive aspects of autophagy on the viral life cycle [20].
The mechanistic target of rapamycin (MTOR) is a protein kinase that regulates autophagy by stimulating protein synthesis and inhibiting induction of autophagy [24].
The class III phosphatidylinositol 3-kinase (PtdIns3K) plays an important role in many biological processes, including the activation of MTOR. The chemical 3-methyladenine (3-MA) has been reported to inhibit autophagy by blocking autophagosome formation via the inhibition of class III Ptdlns3K [25]. However, 3-MA plays a dual role in autophagy. Prolonged treatment with 3-MA promotes autophagy under nutrient-rich conditions, whereas 3-MA inhibits starvation-induced autophagy [25]. Recently, regarding the molecular pathogenesis of RGNNV infection, the symmetrical or asymmetrical interaction between cell death induction and autophagy during viral replication has become a topic of interest.
Numerous viruses have evolved molecular mechanisms to allow them to escape or inhibit autophagy, thereby increasing their infectivity [20], but little has been discovered about how an RNNV virus induces autophagy. In the present study, we investigated the role of Bcl2 family crosstalk between cell death and autophagy. Our findings in the molecular pathogenesis of RGNNV infection may help to identify potential targets that could be used for the prevention or treatment of RNA viruses.
2. Materials and Methods
2.1. Cell and Virus Lines
Grouper fin-1 (GF-1), cells derived from the fin tissue of a grouper (Epinephelus coioides), were grown at 28 °C in Leibovitz’s L-15 medium (Thermo Fisher Scientic Inc., 11415-114, Waltham, MA, USA) and supplemented with 5% fetal bovine serum and 25 μg/mL gentamycin. Naturally infected red grouper larvae, collected in 2002 from the Tainan Prefecture, were the source of the RGNNV Tainan No. 1 (RGNNV TN1 strain) used to infect GF-1 cells. The virus was purified as described by Mori et al. [26] and stored at −80 °C until use. The viral titer was determined using the TCID50 (50% tissue culture infective dose) assay, as reported by Dobos et al. [27].
2.2. Selection of mRFP-LC3-Producing GF-1 Cells
Vector-producing (pmRFP-C1 as a negative control) (Addgene, 21075; deposited by Tamotsu Yoshimori Lab) and pmRFP-LC3-producing cells were obtained by transfection and selection with G418 (800 μg/mL; Invivogen, ant-gn-1). Transcription of the inserted sequences in these vectors was driven by the immediate-early promoter of human cytomegalovirus. The selection time varied from 2 to 2.5 months according to cell properties [14].
2.3. Autophagosome Formation Assay
The mRFP and mRFP-LC3-producing cells (105 cells/mL) were cultured to monolayer confluence in 60 mm diameter Petri dishes for 20 h, rinsed twice with PBS (pH 7.4; Gibco, 10010023, Waltham, MA, USA), and serum-starved for 48 h as a positive control. Cells were then pre-treated with autophagy inhibitor (2 mM 3-MA; Acros, 379791000, Geel, Belgium) for 2 h, and infected with RGNNV (multiplicity of infection (MOI) = 1) for 0, 24, 36 and 48 h at 28 °C. At the end of each incubation time, the slides were observed under fluorescence microscopy (Olympus IX70). The red fluorescent images were captured at 300× to document the formation of RFP-LC3 puncta or dots indicative of autophagosomes or autolysosomes. The formation of RFP-LC3 puncta or dots was counted to 10 as a positive cell. The number of mRFP dots was determined as previously described [28]. Fluorescent puncta were counted manually in at least 3 independent experiments, and at least 200 cells were examined in each experiment.
2.4. Western Blot Analysis
GF-1 cells were cultured in 60 mm Petri dishes (105/mL) for 20 h to monolayer confluence, rinsed twice with PBS, treated with 3-methyladenine (3-MA, 2 mM) for 2 h, and then infected with RGNNV (MOI = 1) for 0, 24, 36 or 48 h at 28 °C. In other experiments, Flag, Flag-Bcl2, and Flag-Bcl2L1-producing GF-1 cells were cultured in 60 mm Petri dishes (105/mL) for 20 h to monolayer confluence, rinsed twice with PBS, and then infected with RGNNV (MOI = 1) for 0, 24, 36 or 48 h at 28 °C. At the end of each incubation period, the culture medium was aspirated, and the cells were washed with PBS and lysed in 0.3 mL of lysis buffer (10 mM Tris, pH 7.3, 20% glycerol, 10 mM SDS, 2% ß-mercaptoethanol, pH 6.8). Lysates were separated by SDS-PAGE [29] and the proteins were transferred to nitrocellulose. Blots were incubated with polyclonal antibodies to protein α, protein B2, mouse BCL2 (Cell Signaling, 15071, Danvers, MA, USA), BCL2L1 (Cell Signaling, 2764, Danvers, MA, USA), LC3 (GeneTex, GTX127375, Irvine, CA, USA), and ACTB/ß-actin, followed by peroxidase-labeled goat anti-rabbit conjugate (1:7500) (Calbiochem, MAB1501, Darmstadt, Germany). Binding was detected by chemiluminescence; the signals were recorded on Kodak XAR-5 film (Eastman Kodak, Rochester, NY, USA) [30]. Protein expression was quantified using the Personal Densitometer (Molecular Dynamics).
2.5. ANXA5/Annexin-A5–FLUOS-Propidium Iodide (PI) Double Staining
To identify apoptotic cells, phosphatidylserine externalization on the outer layer of the apoptotic cell membranes was analyzed using ANXA5-fluorescein (Invitrogen, V13242, Waltham, MA, USA), and necrotic cells were identified by PI staining [9]. GF-1 cells (105/mL) were cultured to monolayer confluence in 60-mm diameter Petri dishes for 20 h, rinsed twice with PBS, and treated with the autophagy inhibitor 3-MA (2 mM) for 2 h. Cells were then infected with RGNNV (MOI = 1) for 0, 24, 36, and 48 h. In other experiments, Flag-, Flag-Bcl2, and Flag-Bcl2L1-producing GF-1 cells (105/mL) were cultured to monolayer confluence in 60 mm diameter Petri dishes for 20 h, rinsed twice with PBS, and then infected with RGNNV (MOI = 1) for 48 h. At each time point, the cells were harvested, washed with PBS, incubated 10–15 min with 100 µL of a HEPES-based ANAX5–fluorescein solution (Roche, 11858777001, Basel, Switzerland) and 100 μL of a staining solution (PI in HEPES buffer; Merck, H3375, Darmstadt, Germany), and then evaluated by fluorescence microscopy (excitation: 488 nm; emission: 515 nm long-pass filter) [17].
2.6. Regulation of Autophagy by Overexpression of Bcl2 and Bcl2L1 in GF-1 Cells
Flag-Bcl2L1 and Flag-Bcl2-producing cells were obtained by transfection of GF-1 cells were transfected with pFlag (p3XFLAG-Myc-CMV), pFlag-Bcl2, and pFlag-Bcl2L1, respectively as previously described [31] by using Lipofectamine-Plus (Invitrogen, 11668500, Waltham, MA, USA), according to the manufacturer’s instructions. Positive clones were selected with G418 (800 mg/mL). Transcription of the Bcl2 and Bcl2L1 genes in these vectors is driven by the immediate-early promoter of human cytomegalovirus. The selection time was about 2.5–3 months from a single colony, depending on cell properties [32].
2.7. Statistical Analysis
The fraction of cells positive for ANXA5 and PI in each sample was determined from counts of 200 cells. Results are expressed as means ± SEMs. Data were analyzed using the paired or unpaired Student’s t-test or ANOVA with multiple comparisons, as appropriate. p < 0.05 was considered statistically significant.
3. Results
3.1. Betanodavirus Induces Autophagy Process in GF-1 Cells
The mRFP-LC3 reporter system was used to identify autophagosome formation in GF-1 fish cells; cells with serum starvation were used as a positive control. Autophagosome formations were found at 36 h in serum starving cells (Figure 1A) and RGNNV-infected cells (Figure 1A). In RGNNV-infected cells, autophagosome formation was significantly increased in a time-dependent manner, with a ~20% increase at 24 h, ~40% increase at 36 h, and ~60% increase at 48 h (Figure 1B) compared to control cells.
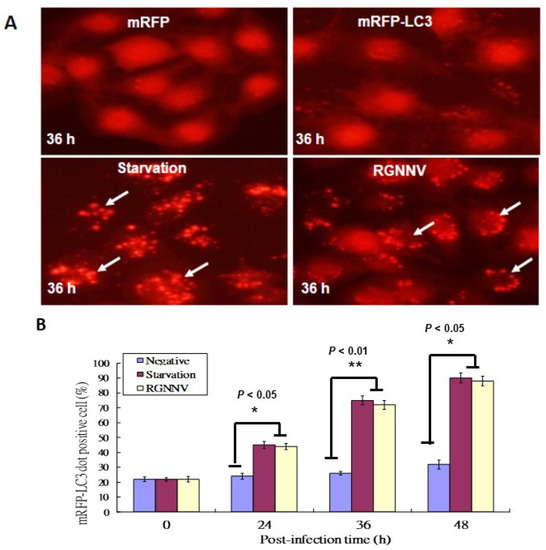
Figure 1.
RGNNV-induced autophagosome formation in GF-1cells. (A) The formation of autophagosomes in GF-1 cells at 36 h detected by the mRFP-LC3 reporter system, with which the formation of RFP-LC3 puncta or dots was counted up to 10 as a positive cell.; mRFP (negative control), mRFP-LC3, serum-starvation treatment, and RGNNV infection. Arrows indicated autosomes. Scale: 10 µM. (B) Quantification of the results in A at 0 h, 24 h, 36 h and 48 h in three separate experiments, based on 200 cells per treatment. Results are shown as means ± SEMs. * p < 0.05, ** p < 0.01.
3.2. 3-Methyladenine (3-MA) Treatment Reduced RGNNV-Induced Autophagy and Blocked Bcl2 Family Downregulation in GF-1 Cells
There was a reduction of autophagosome formation in RGNNV-infected cells treated with 3-MA compared to untreated control by using the mRFP-LC3 reporter system (Figure 2A). The autophagy inhibitor 3-MA underwent titration and received 2 mM as an optimal condition, which did not induce cell damage, and was then further tested. The 3-MA treatment significantly eliminated the autophagosome formation by ~20% at 24 h, ~40% at 36 h, and ~55% at 48 h (p < 0.05) (Figure 2B).
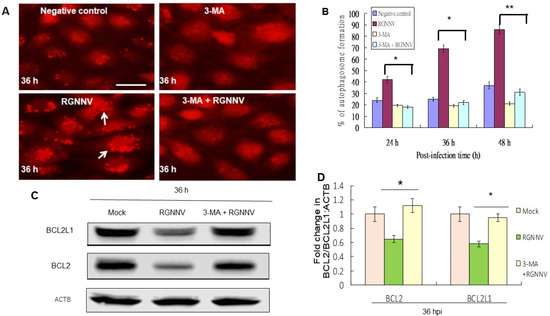
Figure 2.
Elimination of RGNNV-induced autophagy by 3-MA treatment by reducing autophagosome formation and Bcl-2 family downregulation in GF-1 cells. (A) Autophagosome formation in GF-1 cells at 36 h after different treatments. mRFP-LC3 (negative control), 3-MA alone, RGNNV and 3-MA + RGNNV. Arrows indicated autophagosome-positive cells. (B) Percentage of autophagosome-positive cells at 36 h. * p < 0.05; ** p < 0.01. (C) Western blot analysis of BCL2 and BCL2L1 expression at 36 h after different treatments and (D) quantification of these results in three individual experiments. * p < 0.01.
3.3. Overexpression of Zebrafish Bcl2 (Bcl2) and Zebrafish Bcl2 like 1 (Bcl2L1) Reduced Autophagy and Protected against Type III Cell Death
Whether the downregulation of proteins in the BCL2 family correlated with the RGNNV-induced autophagy and cell death was then examined. We designed and produced Bcl2 and Bcl2L1-producing GF-1 cells for these experiments and confirmed that the expression of Bcl2 (Figure 3A) and Bcl2L1 (Figure 3B) were stable in GF-1 cells. Then, the effect of Bcl2 and Bcl2L1 overexpression on the regulation of autophagy-mediated cell death was examined. The results from the LC3-II:LC3-1 ratio at 36 and 48 h showed that the upregulation of Bcl2 and Bcl2L1 had eliminated the RGNNV-induced increase in LC3-II:LC3-1 ratio, indicating that overexpression of Bcl2 and Bcl2L1 may inhibit RGNNV-induced autophagy (Figure 3C,D).
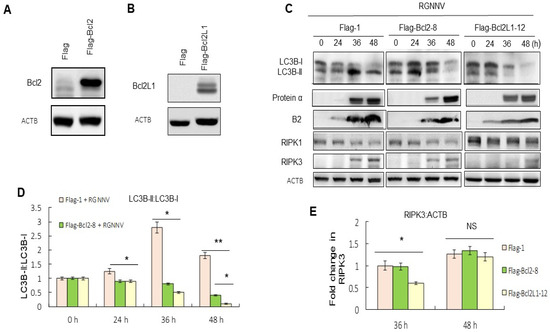
Figure 3.
Overexpression of Bcl2 and Bcl2L1 suppressed autophagy and necroptosis. (A,B) Western blot analysis of Bcl2- and Bcl2L1-producing cells was identified by Bcl2 and Bcl2L1 PolyAbs. (A) Lanes: 1, Flag alone; 2, Bcl2-producing cells. (B) Lanes: 1, Flag alone; 2, Bcl2L1-producing cells. (C) Western blot analysis showed the effect of Bcl2 and Bcl2L1 overexpression on autophagy, necroptosis and viral replication, based on the LC3-II:LC3-I ratio, RIPK3 expression and viral protein expression. Lanes: 1–4, RGNNV-infected Flag-producing GF-1 cells at 0, 24, 36, and 48 h; 5–8, RGNNV-infected Bcl-2-producing GF-1 cells at 0, 24, 36, and 48 h; 9–12, RGNNV-infected Bcl2L1-producing GF-1 cells at 0, 24, 36, and 48 h. (D) Quantitation of the results in C for LC3-II:LC3-I ratio. (E) Quantitation of the results from C showed the effect of Bcl2 and Bcl2L1 overexpression on necroptosis, based on RIPK3 expression at 0, 24, 36, and 48 h in three separate experiments. Results are shown as means ± SEMs. * p < 0.05; ** p < 0.01.
Upregulation of RIPK3 during necroptosis was suppressed more efficiently by Bcl2L1 overexpressed cells than by Bcl2 at 36 h and 48 h, compared with untreated RGNNV-infected cells (Figure 3E).
3.4. Blockage of Cell Death by Bcl2 and Bcl2L1 Overexpression and Reduced Viral Titers
Results from the ANXA5/annexin A5 and PI staining analysis indicated that Bcl2 and Bcl2L1 overexpression also significantly abolished RGNNV-induced cell death (Figure 4). Our data showed that RGNNV-induced autophagy regulated host cell death by increasing signals that promote cell death and decreasing signals that prevent cell death. In addition, Bcl2 and Bcl2L1 overexpression in cells significantly inhibited RGNNV-induced the expression of viral death factors (B2 and protein α) by about 25% (Figure 3C) at 48 h and decreased viral titers (Figure 5) at 24 h (~0.5-fold) and 48 hpi (~0.3-fold).
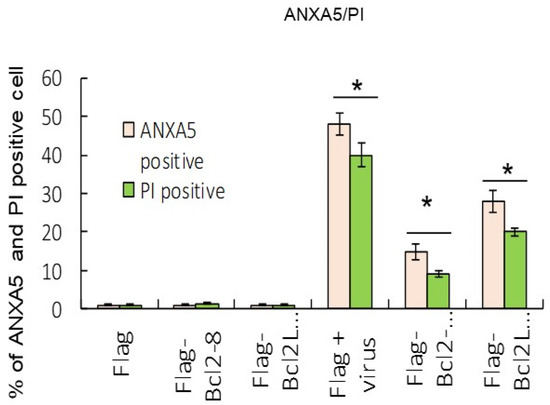
Figure 4.
Overexpression of Bcl2 and Bcl2L1 suppressed cell death markers. Dual labeling with ANXA5 and PI stains indicated that Bcl2 and Bcl2L1 overexpression suppressed RGNNV-mediated autophagy at 48 h. The percentages of ANXA5- and PI-positive cells were determined by the number of green (ANXA5) and red (PI) fluorescent cells in three separate experiments. * p < 0.05.
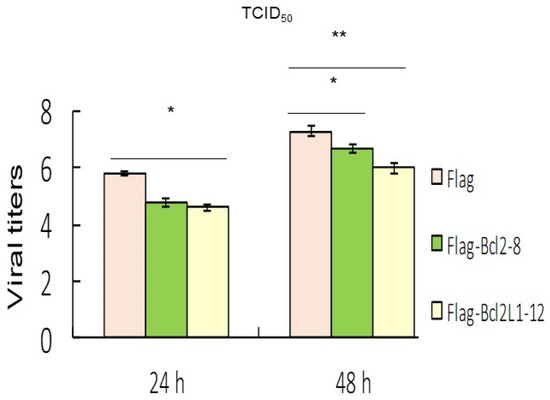
Figure 5.
Overexpression of Bcl2 and Bcl2L1 suppressed viral replication. Viral titers (determined by the TCID50 assay) in the supernatants of different groups at 24 and 48 h, which received best effect on reducing titer between 50 to 100 times by Bcl2 or Bcl2L1 overexpression in three separate experiments. * p < 0.05; ** p < 0.01.
Our study showed that RGNNV induced autophagy of GF-1 cells during the early stage of viral replication and led to a dramatic increase in viral protein expression. Viral infection was correlated with the increased necroptosis in host cells and the downregulation of the anti-apoptotic BCL2 and BCL2L1 proteins. Our finding suggested that overexpression of the Bcl2 family can regulate host cell death and the viral-controlling autophagosome, which correlated to reduced viral expression.
4. Discussion
DNA and RNA viruses have evolved molecular mechanisms to allow them to escape or inhibit autophagy, thereby, increasing their infectivity [20], but RNNV virus-induced autophagy is still rare. As a result, how the autophagy process interacts with necroptosis is relatively unknown. In the present study, we investigated the role of Bcl2 family asymmetric crosstalk between cell death type III and autophagy. Our results in the molecular pathogenesis of RGNNV infection may help to identify potential targets that could be used for the prevention or treatment of RGNNV.
4.1. Betanodavirus-Induced Autophagy
Recently, autophagy has emerged as an important mechanism for controlling intracellular pathogens. A variety of bacteria, including Mycobacteria, Salmonella, Shigella, and Listeria, are recognized by autophagy, but the specific signals that mediate recognition of intracellular pathogens by the autophagy machinery have not yet been well defined. Up to now the ubiquitin associated with intracellular pathogens has promoted targeted autophagosome formation and bacteria pathogen destruction. On the other hand, p62 and NDP52 mediate different pathways of selective autophagy for Shigella and Listeria, which provide novel insight into the mechanisms by which adaptor proteins target bacteria to autophagy [31]. In a viral pathogen infection, such as with DNA virus Herpes simplex type 1 (HSV-1), infection can provoke autophagy under conditions that inhibit viral gene expression. The induction of autophagy occurs very early after infection with HSV-1, and de novo protein synthesis is not required for the response [32,33,34].
Although certain RNA viruses have been previously shown to induce autophagy (e.g., the human immunodeficiency virus (HIV), hepatitis C virus (HCV), and influenza) [35,36,37], there are few reports that examine the effect of betanodaviruses on autophagy. We showed that RGNNV infection induced the formation of autophagosomes (Figure 1) in GF-1 cells by mRFP-LC3 at 36 hpf. In our analysis, 3-MA inhibited phosphatidylinositol 3-kinase (PI3K), which plays an important role in many biological processes, including controlling the activation of mTOR, a key regulator of autophagy. Apparently 3-MA blocked the formation of autophagosomes, which was consistent with previous studies where RGNNV-induced autophagy was inhibited by 3-MA [25]. On the other hand, several lysosomal inhibitors, such as bafilomycin A1 (BafA1), protease inhibitors and chloroquine (CQ), have been used interchangeably to block autophagy in in vitro experiments, assuming that they all primarily block lysosomal degradation. Among them, only CQ and its derivate hydroxychloroquine (HCQ) are FDA-approved drugs and are, thus, currently the principal compounds used in clinical trials aimed to treat tumors through autophagy inhibition. However, the precise mechanism of how CQ blocks autophagy remains to be firmly demonstrated; this will hopefully be uncovered in future testing [38].
4.2. Crosstalk between Autophagy and Necroptosis Cell Death Signals
The MTOR and BECN1 proteins play major roles in the regulation of autophagy [24,39,40]. BECN1 is part of a complex that regulates class III phosphatidylinositol 3-phosphate (Ptdlns3P) and hVps34 (human vacuolar protein sorting 34), whose activity is essential during autophagosome formation. Activators such as UVRAG (UV radiation resistance associated), Bif-1 (Bax-interacting factor) and Ambra-1 (the activating molecule in Beclin-1-regulated autophagy-1), bind with the BECN1 complex to increase phosphatidylinositol 3-monophosphate production. In contrast, proteins in the BCL2 family of anti-apoptotic proteins, such as BCL2 and BCL2L1, bind to BECN1 and act as inhibitors [24,40,41].
The antiapoptotic protein Bcl2 functions either as an antiapoptotic protein or as an anti-autophagy protein via its inhibitory interaction with Beclin 1 [42], which recently functioned in the lysosomal degradation pathway of autophagy and induced autophagic cell death in cancer cells. Furthermore, Bcl2/Bcl2L1 can bind Beclin 1 and inhibit Beclin 1-dependent autophagic cell death in cancer cells. Then, the BH3 domain on Beclin binds to the BH3 binding groove of Bcl2L1 to block Bcl2L1 function.
Viruses, such as γ-herpesvirus-68 (γHV68), modulate this effect [43,44,45]. In our system, RGNNV-induced autophagy correlated with the downregulation of BCL2 and BCL2L1 at the middle stage of viral replication (36–48 h) (Figure 2C), and a subsequent increase in RIPK3-mediated necroptosis of host cells. Interestingly, Bcl2/Bcl2L1 overexpression reduced autophagy, downstream events, the expression of viral death factors and host cell necrosis (Figure 3C), which suggests their potential as therapeutic agents.
In summary (Figure 6), we found that RGNNV infection could induce host cell death and autophagy, which also correlated with anti-Bcl2 member downregulation, such as Bcl2 and Bcl2L1, at the early–middle viral replication stage (12–24 hpi). Then, blockage of autophagy initiation by 3-MA apparently reduced autophagy and Bcl2 downregulation at 36 hpi. On the other hand, overexpression of the Bcl2 family members Bcl2 and Bcl2L1 either dramatically reduced type III cell death or suppressed autophagy initiation. We hypothesized that RGNNV induced cell death and autophagy by asymmetrical interaction, not by direct crosstalk. There seemed to be an upstream or downstream correlation in this system, but this requires further research.
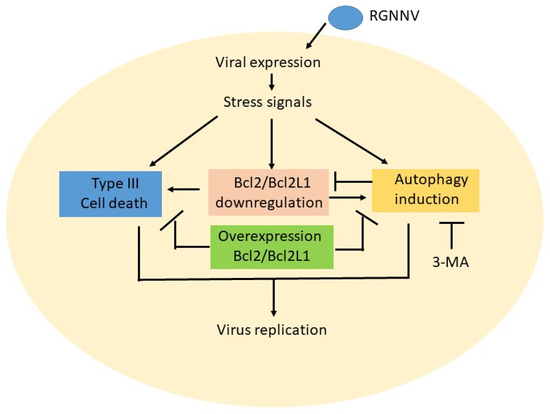
Figure 6.
Hypothesized effect between triggering type III cell death and autophagy induction is through asymmetric crosstalk during RGNNV infection in GF-1 cells. RGNNV infection can also stress signals during viral expression and viral replication at the early replication stage (12–24 hpi). Then, those stress signals can regulate multiple signaling pathways either to induce cell death or trigger an autophagy flow in the middle replication stage that correlates with anti-apoptotic member Bcl2 and Bcl2L1 downregulation. In autophagy inhibitor treatment, 3-MA can suppress autophagy initiation. Furthermore, if overexpression of Bcl2 and Bcl2L1 genes can also either block cell death and reduce the viral titer or suppress the autophagy process. The correlations between cell death and autophagy, appear to be asymmetrical upstream or downstream correlation interactions.
5. Conclusions
RGNNV infection in GF-1 cells can induce autophagosome formation and Bcl-2 downregulation at different post-infection times; however, Bcl-2 downregulation is blocked by the autophagy inhibitor 3-MA. Then, extracellular Bcl-2 and Bcl2L1 overexpression can suppress host cell death type III and autophagy caused by RGNNV infection. Our results suggest that RGNNV-induced cell death and autophagy by asymmetrical interaction was regulated by the anti-apoptotic members Bcl2 and Bcl2L1 and autophagy inhibitor 3-MA via the inhibition of class III Ptdlns3K.
Author Contributions
Conceptualization, J.-R.H.; methodology, A.-J.L. and H.-J.L.; investigation, A.-J.L. and H.-J.L.; formal analysis, J.-R.H.; writing—original draft preparation, J.-R.H.; writing—review and editing, J.-R.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant (NSC 101-3011-P-006-006 and MOST 104-2313-B-006-003) awarded to Jainn-Ruey Hong from the National Science Council, Taiwan, Republic of China.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the manuscript.
Acknowledgments
The authors were grateful to S. C. Chi (Institute of Zoology and Development of Life Science, Taiwan, ROC) for providing the grouper-fin cell line (GF-1). This work was supported by a grant (MOST 104-2313-B-006-003) awarded to Jainn-Ruey Hong from the Ministry of Science and Technology, Taiwan, Republic of China.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ball, L.; Johnson, K. Reverse genetics of nodaviruses. Adv. Virus Res. 1999, 53, 229–244. [Google Scholar] [PubMed]
- Bovo, G.; Nishizawa, T.; Maltese, C.; Borghesan, F.; Mutinelli, F.; Montesi, F.; De Mas, S. Viral encephalopathy and retinopathy of farmed marine fish species in Italy. Virus Res. 1999, 63, 143–146. [Google Scholar] [CrossRef]
- Chi, S.C.; Shieh, J.R. Genetic and antigenic analysis of betanodaviruses isolated from aquatic organisms in Taiwan. Dis. Aquat. Organ. 2003, 55, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Munday, B.; Kwang, J.; Moody, N. Betanodavirus infections of teleost fish: A review. J. Fish Dis. 2002, 25, 127–142. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Ding, S. Induction and Suppression of RNA Silencing by an Animal Virus. Science 2002, 296, 1319–1321. [Google Scholar] [CrossRef]
- Delsert, C.; Morin, N.; Comps, M. A fish encephalitis virus that differs from other nodaviruses by its capsid protein processing. Arch. Virol. 1997, 142, 2359–2371. [Google Scholar] [CrossRef]
- Wu, H.C.; Chiu, C.S.; Wu, J.L.; Gong, H.Y.; Chen, M.C.; Lu, M.W.; Hong, J.R. Zebrafish anti-apoptotic protein zfBcl-xL can block betanodavirus protein alpha-induced mitochondria-mediated secondary necrosis cell death. Fish Shellfish Immunol. 2008, 24, 436–449. [Google Scholar] [CrossRef]
- Su, Y.C.; Reshi, L.; Chen, L.J.; Li, W.H.; Chiu, H.W.; Hong, J.R. Nuclear targeting of the betanodavirus B1 protein via two arginine-rich domains induces G1/S cell cycle arrest mediated by upregulation of p53/p21. Sci. Rep. 2018, 8, 3079. [Google Scholar] [CrossRef]
- Chiu, H.W.; Su, Y.C.; Hong, J.R. Betanodavirus B2 triggers apoptosis and necroptosis in lung cancer cells that suppresses autophagy. Oncotarget 2017, 8, 94129–94141. [Google Scholar] [CrossRef][Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2020, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.P.; Yang, H.L.; Her, G.M.; Lin, H.Y.; Jeng, M.F.; Wu, J.L.; Hong, J.R. Betanodavirus induces phosphatidylserine exposure and loss of mitochondrial membrane potential in secondary necrotic cells, both of which are blocked by bongkrekic acid. Virology 2006, 347, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R. Betanodavirus: Mitochondrial disruption and necrotic cell death. World J. Virol. 2013, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-P.; Wu, J.-L.; Su, Y.-C.; Hong, J.-R. Anti-Bcl-2 family members, zfBcl-x(L) and zfMcl-1a, prevent cytochrome c release from cells undergoing betanodavirus-induced secondary necrotic cell death. Apoptosis 2007, 12, 1043–1060. [Google Scholar] [CrossRef]
- Reshi, L.; Wu, J.L.; Wang, H.V.; Hong, J.R. Aquatic viruses induce host cell death pathways and its application. Virus Res. 2016, 4, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Hong, J.R. Betanodavirus B2 causes ATP depletion-induced cell death via mitochondrial targeting and complex II inhibition in vitro and in vivo. J. Biol. Chem. 2010, 285, 39801–39810. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.; Chiu, H.W.; Hung, J.C.; Hong, J.R. Beta-nodavirus B2 protein induces hydrogen peroxide production, leading to Drp1-recruited mitochondrial fragmentation and cell death via mitochondrial targeting. Apoptosis 2014, 19, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- McClung, C.R. Regulation of catalase in Arabidopsis. Free Radic. Biol. Med. 1997, 23, 489–496. [Google Scholar] [CrossRef]
- Choi, T.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 2010, 22, 157–168. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.-M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Mori, K.I.; Nakai, T.; Muroga, K.; Arimoto, M.; Mushiake, K.; Furusawa, I. Properties of a new virus belonging to nodaviridae found in larval striped jack (Pseudocaranx dentex) with nervous necrosis. Virology 1992, 187, 368–371. [Google Scholar] [CrossRef]
- Dobos, P.; Hill, B.J.; Hallett, R.; Kells, D.T.; Becht, H.; Teninges, D. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 1979, 32, 593–605. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef]
- McNulty, M.S.; Connor, T.J.; McNeilly, F.; McLoughlin, M.F.; Kirkpatrick, K.S. Preliminary characterization of isolates of chicken anemia agent from the United Kingdom. Avian Pathol. 1990, 19, 67–73. [Google Scholar] [CrossRef]
- Kain, S.; Mai, K.; Sinai, P. Human multiple tissue western blots: A new immunological tool for the analysis of tissue-specific protein expression. BioTechniques 1994, 17, 982–987. [Google Scholar]
- Chen, M.-C.; Gong, H.-Y.; Cheng, C.-Y.; Wang, J.-P.; Hong, J.-R.; Wu, J.-L. Cloning and characterization of zfBLP1, zfBcl-xL homolog from the zebrafish, Danio rerio. Biochim. Et Biophys. Acta Gene Struct. Expr. 2001, 1519, 127–133. [Google Scholar] [CrossRef]
- Reshi, L.; Wang, H.V.; Hui, C.F.; Su, Y.C.; Hong, J.R. Anti-apoptotic genes Bcl-2 and Bcl-xL overexpression can block iridovirus serine/threonine kinase-induced Bax/mitochondria-mediated cell death in GF-1 cells. Fish Shellfish Immunol. 2017, 61, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, U.; Huang, S.; Liu, Z.G.; Han, J. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J. Biol. Chem. 2006, 281, 8788–8795. [Google Scholar] [CrossRef] [PubMed]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef]
- Borel, S.; Esper, L.; Biard-Piechaczyk, M. Macroautophagy regulation during HIV-1 infection of CD4þ T cells and macrophages. Front. Immunol. 2012, 3, 97. [Google Scholar] [CrossRef]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER-stress response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef]
- Chen, M.; Hong, M.J.; Sun, H.; Wang, L.; Shi, X.; Gilbert, B.E.; Corry, D.B.; Kheradmand, F.; Wang, J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 2014, 20, 503–510. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Cregan, S.P.; Fortin, A.; MacLaurin, J.G.; Callaghan, S.M.; Cecconi, F.; Yu, S.W.; Dawson, T.M.; Dawson, V.L.; Park, D.S.; Kroemer, G.; et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002, 158, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Hong, H.S.; E, X.; Kim, K.S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Paul, W.H. Richardson, D.C.D. Viral interactions with macroautophagy: A double-edged sword. Virology 2010, 402, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.J.; Levine, B. Selective autophagy and viruses. Autophagy 2011, 7, 260–265. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).