3.1. Structural Properties
The diameter size trend with respect to the chirality in each nanotube is shown in
Figure 1. It is evident that for both armchair- and zigzag-type nanotubes, as the chiral index increases, the diameter increases almost linearly, whose coefficients of determination (termed as R2) are 0.9984 and 0.9996 for the (
n,
n) BPNT and (
n,0) BPNT, respectively. These R2 values indicate a very good linear fit since values are close to unity.
Table 1 shows in detail the diameter size corresponding to each nanotube.
The axial length of each nanotube shows no considerable variation with respect to the chirality, with an average value of 19.94 Ȧ and 17.98 Ȧ for armchair- and zigzag-type nanotubes, respectively.
Table 2 and
Table 3 summarize the relaxed bond lengths and angles. In both cases, the B-P bond length decreases as the chirality index increases. The average length of the B-P bond for both chiralities is ~1.88 Ȧ. In addition, the boron–hydrogen bond length is shorter than that of the phosphorus–hydrogen bond (
), because of the electronegativity difference in the B-H, which is greater than in the P-H, inducing a more strengthened bond. The average value of the angle formed by the P-B-P atoms is 120.08° and 120.45° for armchair- and zigzag-type BPNTs, respectively. The angles of the H-P-B and H-B-P bonds at the ends of armchair and zigzag nanotubes increase slightly with increasing chirality. This small increase occurs because of chirality increase, as there is a higher density of hydrogen atoms at the end of the tube, causing a repulsion effect between them, which in turn generates an increase in the formed angle. The cohesion energy values are reported in
Table 4 (armchair type) and
Table 5 (zigzag type). Negative values were obtained for each nanotube, with an average value of
for armchair-type and
for zigzag-type BPNTs, indicating that they are all stable structures. These values suggest that pristine BPNTs are less stable than CNTs and BNNTs, since the latter have cohesion energies of −8.72 and −7.27 eV, respectively [
33]. For both types of chirality, the cohesion energy decreases as the chiral index (
) increases, indicating that the greater the diameter is, the better the stability of the system is. However, the decrease in
is very small, since the energy difference between the maximum and the minimum value is only
, both in the armchair- and zigzag-type. Vibrational frequency calculations showed that all nanotubes have non-imaginary frequencies.
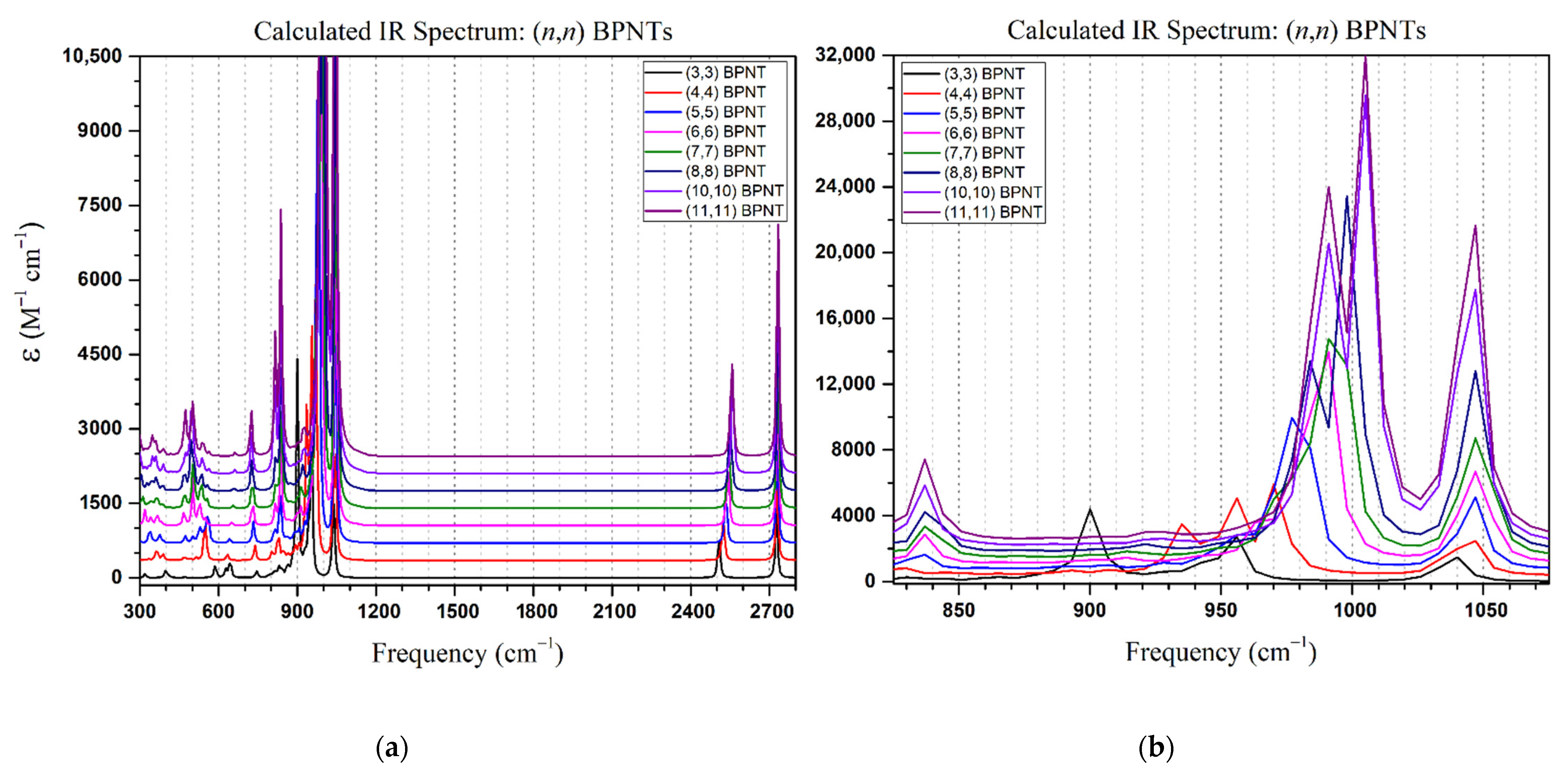
The calculated IR spectra of all systems are shown in
Figure 2 and
Figure 3 for the armchair- and zigzag-type boron phosphide nanotubes, respectively. In both cases, a pair of sets of characteristic peaks are noted in the frequency region of >2400 cm
−1, corresponding to the P-H stretch vibration (2503–2521 cm
−1) and the B-H stretch vibration (~2725 cm
−1). The highest
ε intensity peaks occur in the frequency range between 850–1100 cm
−1, again for both cases, attributed to a P-B-P stretch vibration (900–955 cm
−1) and to the B-H bending vibration (1031–1040 cm
−1). It is noted that the P-B-P stretch vibration mode is affected by a blue shift when the chiral index increases (+10 cm
−1/
n,
Figure 2b and
Figure 3b). The armchair- and zigzag-type BPNTs display a very similar IR spectra; however, the armchair-type nanotubes exhibit a characteristic small peak in the frequency range of 820–840 cm
−1 corresponding to the P-H bond bending mode. This characteristic signal allows differentiating between both chiralities. In general, the peak intensity increases with the chiral index, as induced by the number of bonds present in the structure.
3.2. Electronic Properties
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are discussed in this section.
Figure 4 shows the |HOMO-LUMO| energy gaps as functions of the chirality as obtained in this work and compared with those obtained in the previously mentioned reports (see Introduction Section). The energy gap of the (
n,
n) BPNTs decreases as the index (diameter) increases, reaching stabilization when
n ≥ 10, (10,10) BPNT. This characteristic has also been reported for BNNT and SiCNT, where starting at a certain diameter the band gap energy is chirality independent [
16,
24]. On the other hand, zigzag nanotubes transform from a metallic character (5,0) BPNT with 0.08 eV to semiconductor, with (10,0) BPNT being the one with a larger energy gap value (0.98 eV). Thus, low chirality zigzag BPNTs can be used in electronic and energy transport devices. It can be noted that, in the case of zigzag BPNTs, our values have a similar trend and are close to those reported by Srivastava et al. [
24], particularly with those calculated by the LDA/PZ/DZP method. However, our results differ largely from those described by Azizi et al. [
25] for both armchair- and zigzag-type nanotubes. Other authors have also reported the value of the energy gap for some chiralities in studies about the functionalization of boron phosphide nanotubes.
Table 6 summarizes these values reported by different authors [
9,
10,
11,
12,
14,
15,
17,
18,
20,
21,
22,
34,
35,
36,
37] for the nanotubes (5,0), (6,0), (7,0), (8,0) and (4,4), comparing them with the values obtained in this work. Considerable different values of molecular gap are noted for the same chirality, however, all the values reported by other authors are calculated with the B3LYP or BLYP functional. This suggests that there is an underestimation or overestimation when calculating the HOMO-LUMO gap when the functionals B3LYP/BLYP and the M06-2X are used, respectively. Another factor that could be responsible for this difference in the molecular gap is the number of atoms that make up the nanotube, that is, its molecular formula. For all the cases shown in
Table 6, our models of BPNTs have a larger number of atoms and, therefore, a greater length, because chirality determines the diameter of the nanotube but not its length. This would be a great challenge to synthesize BPNTs in a controlled way, since their electronic properties would present a strong dependence on the length of the nanotube.
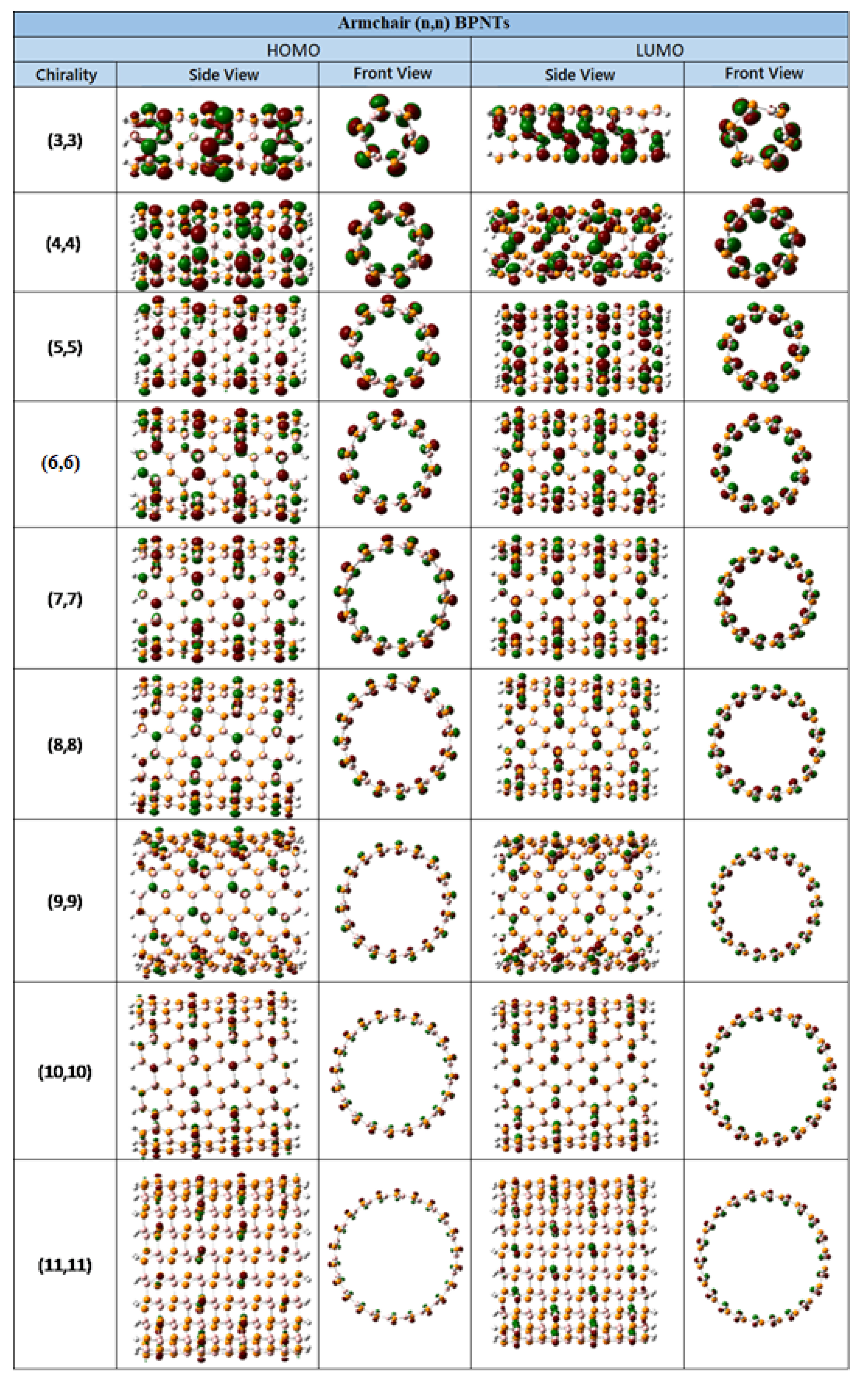
The distribution of the HOMO and LUMO orbitals through the nanotubes are graphically depicted in
Figure 5 and
Figure 6 for the armchair and zigzag BPNTs, respectively. It is evident that the HOMO orbital is uniformly distributed throughout the entire nanotube in the armchair type, however, different features display the zigzag type, since this is concentrated at one end of the nanotube. The LUMO orbital, in the zigzag-type BPNTs, is uniformly distributed until chirality (8,0). At larger index, this is concentrated at one of the extremes. In the armchair-type BPNTs, analogous to HOMO, the LUMO is distributed throughout the entire nanotube.
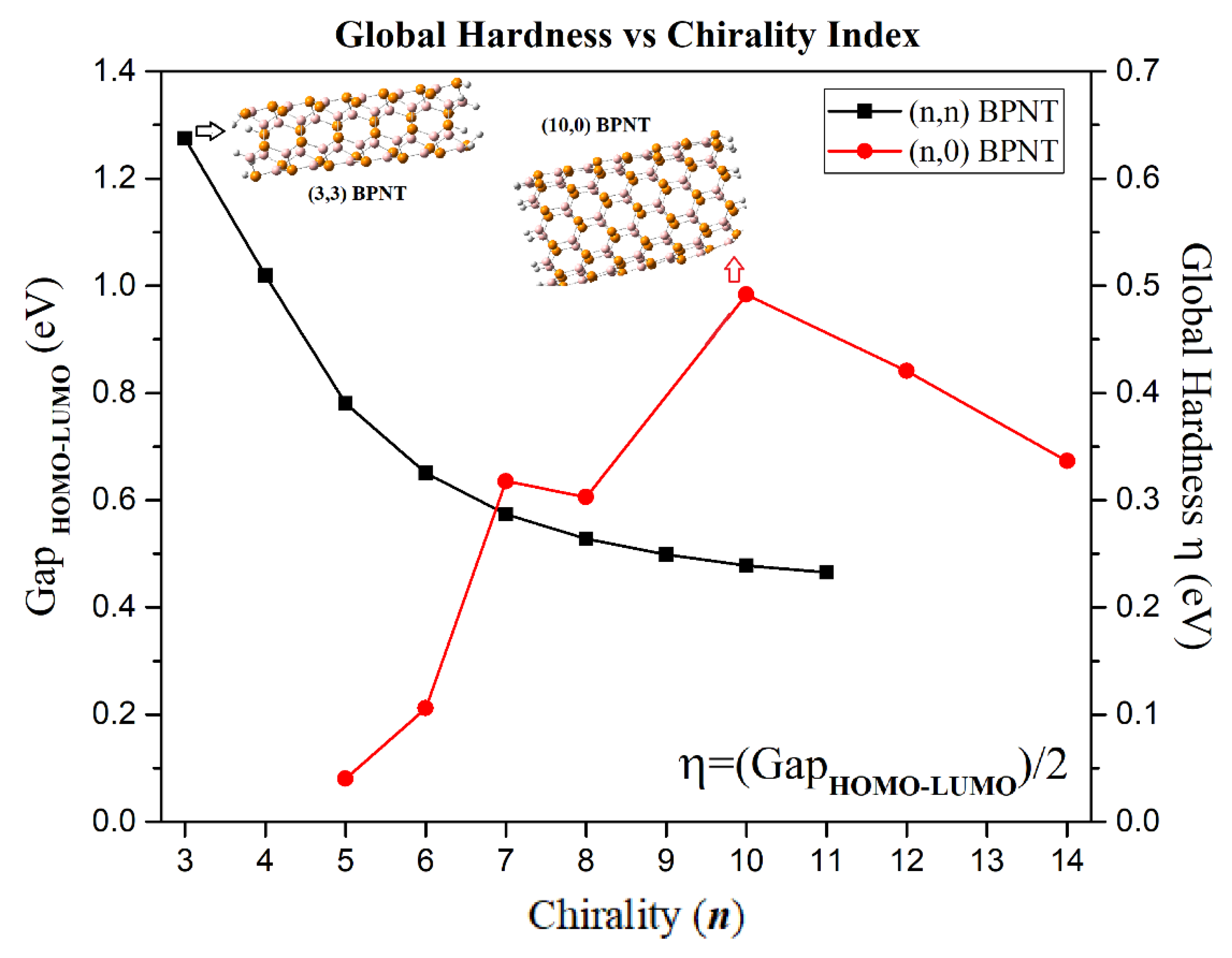
The reactivity and chemical stability of the nanotubes was measured through global quantum molecular descriptors such as chemical potential (
μ), global hardness (
η) and electrophilicity index (
ω), energy gap (E
g), ionization potential (
I) and electronic affinity (A), as summarized in
Table 4 and
Table 5. These parameters have been widely used for many years in computational chemistry studies [
38,
39,
40,
41] and, recently, to study the functionalization and stability of some III-V nanotubes [
42,
43,
44,
45,
46]. The global hardness represents the system resistance to a charge transfer, that is, high values of
indicate greater electronic stability. Thus, the electronic stability of the armchair-type BPNTs decreases as chirality increases, with (3,3) BPNT being the most electronically stable system (see
Figure 7). The zigzag-type BPNTs exhibit an arbitrary variation in the global hardness with respect to the chirality, showing a maximum energy value of ~0.492 eV in the (10,0) BPNT. In addition, the zigzag nanotubes display the highest (most negative)
values and these decrease with increasing chirality, so that the zigzag BPNTs with the highest chirality are the least chemically reactive, since the chemical potential is a property that characterizes the tendency of electrons to escape from a system in equilibrium. No significant variation in the value of
of the armchair-type BPNTs, −6.34 eV <
<−6.26 eV, is found, indicating chirality independence (see
Figure 8). However, the chemical potential of both chiralities tend to converge towards the same value, i.e.,
for
. Comparing the global hardness and chemical potential values of boron phosphide nanotubes with their most studied structural analogues, CNTs and BNNTs, it is noted that BPNTs show
values in a range very close to that of CNTs, however, this later structure is much smaller than those reported for the BNNTs. Moreover, in the case of the chemical potential,
, the BPNT values are similar to those of the BNNTs, being much smaller than those reported for CNTs [
46,
47,
48]. Therefore, we can establish that
and
.
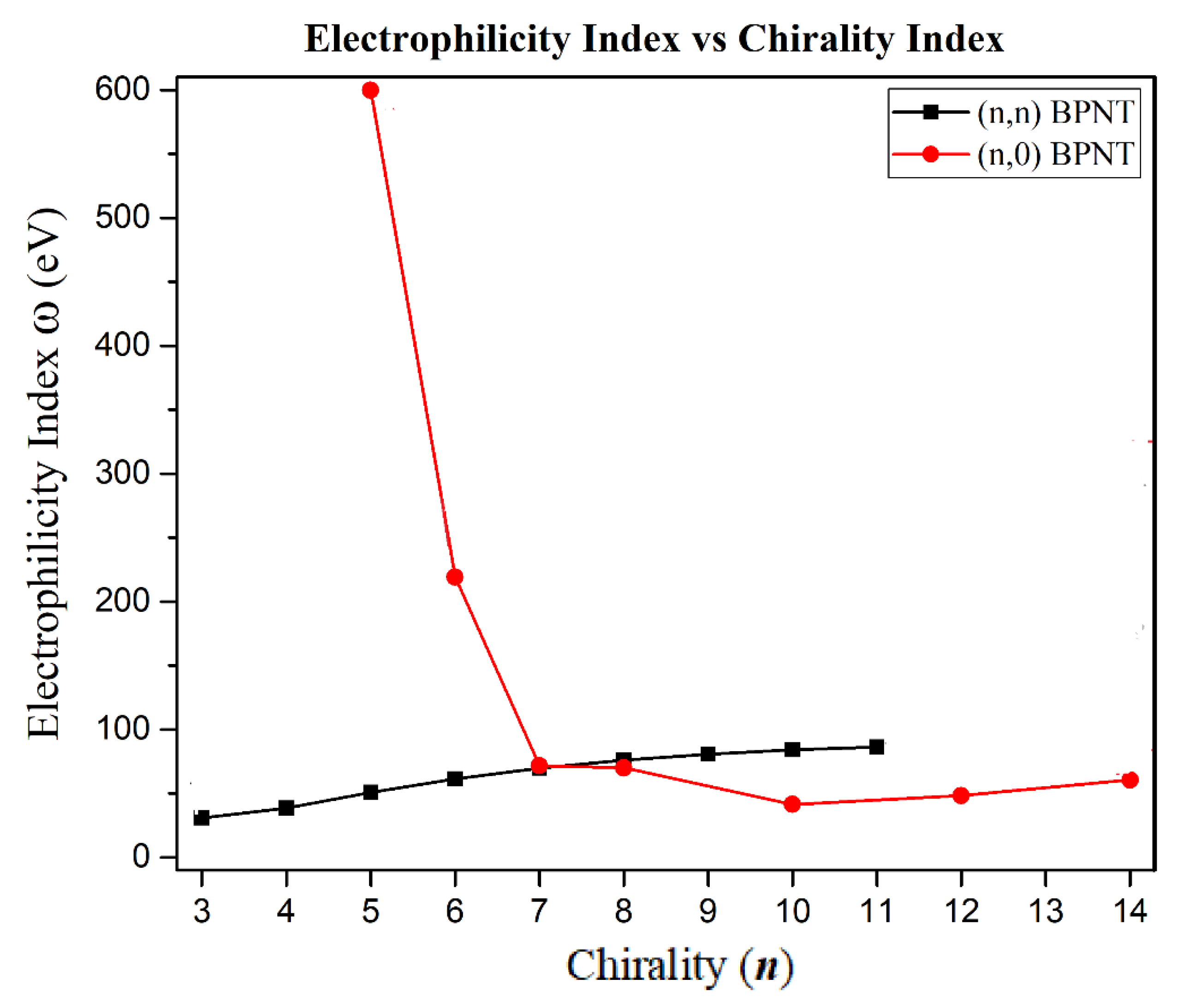
Regarding the electrophilicity index, the (5,0) BPNT exhibits a large value provided that
(599.59 eV) is large. However, the electrophilic character of the zigzag BPNTs drops abruptly with increasing chirality, while in the armchair type, this parameter increases gradually (see
Figure 9). The ionization potential (
) measures the tendency to yield electrons, while electron affinity (
) measures the tendency to accept electrons. Thus, high ionization potential values indicate that large energy is required to yield electrons from the HOMO orbital, while large values of electron affinity indicate high electron accepting capacity. Both the armchair- and the zigzag-type chiralities exhibit large energy values of
and
, therefore, BPNTs are systems that easily accept electrons. This justifies their great electrophilic character, as indicated by the
index.
The dipole moment
is a quantitative measure of the dipole moment magnitude and allows differentiating between polar and non-polar molecules. It is considered to a certain extent an index of reactivity, which is very important to define some biological properties [
49]. The dipole moment of a polyatomic system is the vector sum of the bond dipoles. Our results show that the (
n,
n) BPNTs exhibit nearly zero dipole moment values (i.e., non-polar systems), as induced by the structural symmetry of the armchair-type nanotubes (see
Figure 10). Nearly-zero polarity is independent of the type of atoms that make up the nanotube, since other nanotubes such as GaNNT, BNNT and AlNNT also display null dipole moment values of armchair-type chirality [
43,
50]. On the other hand, zigzag-type nanotubes are polar structures, with the (14,0) BPNT being the one with the largest value
, although with no dependence of polarity (
) on the chiral index (
). This suggests that zigzag-type boron phosphide nanotubes are more soluble than armchair-type structures in polar solvents such as water, which in turn indicates that they are suitable for applications in biological systems. This characteristic of zigzag-type nanotubes of having a higher dipole moment, and therefore greater solubility, compared to armchair type, has also been reported for BNNTs [
51,
52]. These results make it clear that the zigzag-type nanotubes have a higher degree of solubility, since polarity is a key property for high solubility. We can validate this through the solvation energy values calculated using Equation (7). The results show that, in general, the solvation energy in the zigzag type is higher (more negative) than in armchair nanotubes, as expected. It may be thought that
Figure 11 contradicts this last assertion since, for example, the armchair-type (11,11) BPNT has higher
values than most of the zigzag-type nanotubes. However, this is not an appropriate comparison, since the (11,11) BPNT contains a much larger number of atoms that promotes intermolecular interactions which in turn favors solvation. Then, it is necessary to compare the
values between nanotubes with a similar number of atoms and having almost the same diameters, for example the (5,5) BPNT (−12.08 kcal/mol) vs. (8,0) BPNT (−17.27 kcal/mol) or the (6,6) BPNT (−13.49 kcal/mol) vs. (10,0) BPNT (−19.83 kcal/mol). There are several recent investigations that show the important role that the dipole moment plays on the solubility of some pristine and/or functionalized nanotubes [
51,
52,
53,
54,
55,
56].


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}