Abstract
This paper presents the general synthesis of a comprehensive group of P-chiral phosphinyl derivatives with a natural coumarin-type motif. A chiral substituent was attached at the third position of the coumarin molecule via the Knoevenagel procedure using readily available P-chiral phoshinylacetic acid esters without loss of enantiomeric purity. The application of salicylaldehyde-based derivatives allowed the incorporation of substituents of different electron character into the backbone of these coumarins making them suitable for subsequent chemical modifications. As a result, we gained access to six achiral (2a–g) and a large number ((Sp)-4a–f, (Sp)-6a–e and (Rp)-8a) of new potential chiral ligand precursors, pharmaceuticals, etc. with an imbedded phosphinyl group with evidenced biological activity based on the natural coumarin backbone. The molecular structure, including absolute configuration, was determined for seven compounds.
1. Introduction
Coumarins and their derivatives as natural compounds containing a 2H-1- benzopyran-2-one core structure are a class of compounds with great application potential. Since their first synthesis in 1868 by Perkin [1], they have been extensively employed in a variety of fields. The biologically active features of coumarins, both natural and synthetic, are noteworthy to scientists. They include antibacterial, anticancer, neuroprotective, and antioxidant properties [2,3,4,5,6,7,8]. Because of their structural diversity, coumarin–metal complexes are also intriguing molecules with established biological characteristics [9]. Coumarin complexes with platinum(IV) [10] or cobalt [11] are noteworthy due to their anticancer properties, while complexes with Co(II), Ni(II), Cu(II), and Zn(II) have antimicrobial potential [12].
Coumarins’ derivatives are also extremely valuable in the field of photochemistry. Their unique properties include strong photoactivity and the ability to dimerize under UV light. Because of this, such compounds are extensively used in the production of solar cells [13], optoelectronics [14], and photoreactive polymers [15]. The use of coumarins as fluorescent probes has been the subject of equally rapid scientific expansion in recent years. Coumarin fluorescent probes can be successfully used to detect metal ions and to detect and image small active molecules in cells [16,17,18].
Organophosphorus compounds are a significant class of therapeutic agents targeting a wider range of diseases. The majority of currently approved phosphorus-containing pharmaceuticals contain a phosphoramide or phosphonate group [19]. The use of phosphine oxides as biologically active compounds is still rare; nevertheless, there is a gradual appreciation of them by scientists. Recently, the anticancer drug brigatinib [20] with a dimethylphospine oxide moiety was approved by the Food and Drug Administration (FDA). Other examples include fosenazid [21], a neurodrug with potent serotonin and epinephrine inhibitory properties, and fosazepam [22], a water-soluble diazepam derivative with sedative and anti-anxiety effects. Two years ago, researchers demonstrated that incorporation of the POMe2 substituent into prazosin increased its solubility and reduced its lipophilicity while maintaining its full biological profile [23]. In human liver cells, they saw enhanced metabolic stability linked to a longer half-life of the chemical.
The phosphinyl group is a crucial component that has the ability to control biological characteristics. Finding efficient ways of incorporating this moiety into the coumarin skeleton could provide a variety of novel compounds for application in biological research.
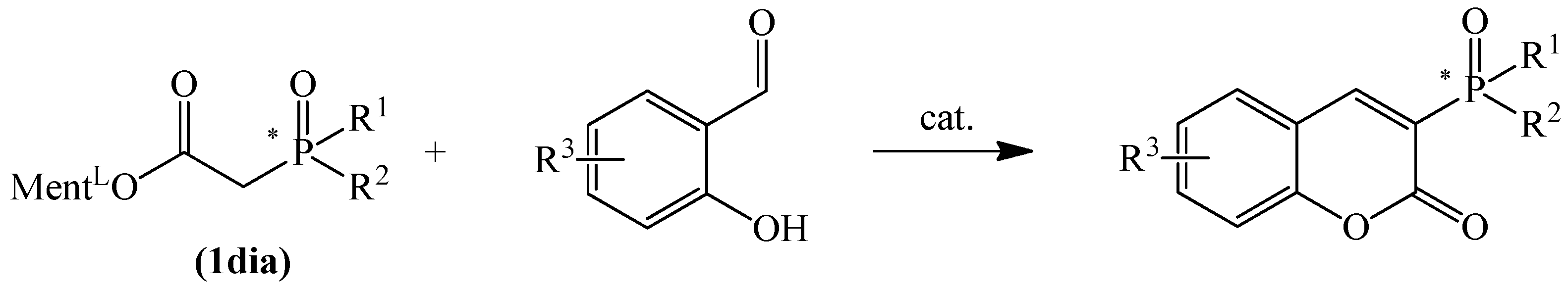
Few effective synthesis techniques have been reported to date for the preparation of 3-phosphonated or 3-phosphinylated coumarins. It is important to note that the vast majority of papers that are presently available concentrate on phosphinylation of the coumarin backbone via C-H-bond activation reactions [24,25,26], through reaction with an active P-centered radical [27,28,29,30,31], and through electrochemical phosphinylation [32,33]. In addition, there are some other studies employing Knoevenagel condensation of phosphonoacetates with aromatic aldehydes [34,35,36] and Friedel–Crafts reaction of phenols with phosphinylacrylate [37,38].
More recently, we have found that diphenylphosphinoylacetic acid esters easily react with salicylaldehydes to make coumarin derivatives in the Knoevenagel condensation reaction. To this purpose, we modified the procedure for the synthesis of such compounds proposed by Robinson [34] and Bojilova [36]. The use of a mixture of pyrrolidine and acetic acid as a catalyst in the reaction of diphenylphosphinoylacetic acid esters with salicylaldehydes allowed us to improve the yield of the formed coumarins (Scheme 1).
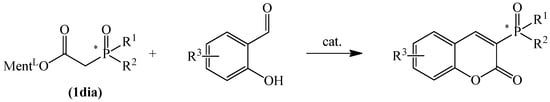
Scheme 1.
Synthesis of chiral phosphinylated coumarins.
In this article, we describe our recent efforts in the development of P-stereogenic coumarin derivatives. This is part of our research into the production of phosphinyl chromenones, in which we observe potent biologically active potential (paper in preparation). The synthesis of C-3-phosphorylated coumarins, including P-chiral ones, employing the Knoevenagel condensation reaction has not been described to date. The presented protocol for their synthesis can, therefore, make a valuable contribution to expanding the library of this class of compounds.
2. Materials and Methods
2.1. Instrumentation
2.1.1. General
All reactions were set up using standard Schlenk techniques and carried out under an argon atmosphere using anhydrous solvents, unless otherwise noted. Commercially available chemicals were obtained from Sigma-Aldrich and used as received. NMR spectra were recorded using a Bruker AV500 (1H 500 MHz, 31P 202 MHz, 13C NMR 126 MHz) spectrometer. All spectra were obtained in CDCl3 solutions, unless mentioned as otherwise, and the chemical shifts (δ) were expressed in ppm using internal reference to TMS and external reference to 85% H3PO4 in D2O for 31P. Coupling constants (J) were given in Hz. The abbreviations of signal patterns were as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad. Elemental analyses were measured on a Perkin-Elmer CHN 2400 system. Optical rotations were measured on a Perkin-Elmer 341LC digital polarimeter. Melting points were determined on a Buchi 510 apparatus and are uncorrected. Thin-layer chromatography (TLC) was completed on silica gel (Kieselgel 60, F254 on aluminum sheets, Merck (Darmstadt, Germany)) using UV light (254 nm). All column chromatographic separations and purifications were conducted using Merck silica gel 60 (230–400 mesh).
2.1.2. X-ray Crystallography
The single-crystal-diffraction data for 2d, 2e, (SP)-4a, (SP)-4d, (SP)-5, (SP)-6d, and (SP)-6e (Table S1) were collected at room temperature with a SuperNova diffractometer (Oxford Diffraction; Agilent [39]) with the graphite monochromated CuKα radiation (λ = 1.54184 Å). The CrysAlisPro program system [40] was used for data collection, cell refinement, and data reduction. The intensities were corrected for Lorentz and polarization effects, and multi-scan absorption corrections were applied. The crystal structure was solved with direct methods using the SHELXT program and refined using the full-matrix least-squares method on F2 using the SHELXL-2018/3 program [41,42]. The non-hydrogen atoms were refined with anisotropic displacement parameters; H-atoms were positioned at calculated positions and refined using the riding model. The experimental details and final atomic parameters for the analyzed crystals were deposited with the Cambridge Crystallographic Data Centre as Supplementary Material (CCDC Nos 2311368–2311374).
2.2. Procedure for Synthesis of L-menthyl Diphenylphosphinylacetate (1)
L-Menthyl chloroacetate (8.75 g, 0.04 mol) was dissolved in anhydrous THF (30 mL). The resulting solution was added dropwise to diphenylphosphine oxide (8.8 g, 0.04 mol) suspended in anhydrous THF (100 mL), and then a 60% dispersion of NaH in oil (1.6 g, 0.04 mol) was added in portions under argon in a Schlenk flask. After addition, the reaction mixture was stirred at rt for 22 h, and then evaporated and CH2Cl2 (100 mL) was added. The organic phase was washed with H2O (2 × 50 mL), dried over anhydrous MgSO4, filtered, and evaporated. The resulting yellow oil was dissolved in warm CH2Cl2 and slowly cooled to afford 13.7 g crystalline precipitate 1 with an 86% yield. 1H NMR (500 MHz, CDCl3): δ 7.90–7.72 (m, 4H), 7.60–7.34 (m, 6H), 4.58 (td, J = 10.9, 4.4 Hz, 1H), 3.51 (dd, J = 14.8, 1.4 Hz, 2H), 1.74–1.53 (m, 4H), 1.35 (tdt, J = 11.9, 6.5, 2.9 Hz, 1H), 1.23 (tt, J = 12.0, 3.2 Hz, 1H), 1.01–0.88 (m, 1H), 0.81 (dd, J = 15.6, 6.8 Hz, 6H), 0.77–0.65 (m, 2H), 0.60 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 165.67 (d, J = 5.2 Hz), 132.23 (d, J = 2.7 Hz), 131.76 (d, J = 106.3 Hz), 131.18 (d, J = 9.9 Hz), 128.63 (d, J = 12.3 Hz), 75.83, 46.52, 40.29, 39.28 (d, J = 61.2 Hz), 34.02, 31.27, 25.70, 23.01, 21.90, 20.80, 15.92. 31P NMR (202 MHz, CDCl3): δ 26.69.
General Procedure for Synthesis of Coumarins 2a–g
L-Menthyl diphenylphosphinylacetate (1 mmol), 2-hydroxybenzaldehyde (1.5 mmol, 1.5 eqv.) and 0.4 g MS (4 Å, 4–8 mesh) together with pyrrolidine (30 mol%) and acetic acid (30 mol%) were dissolved in 15 mL of acetonitrile and refluxed for times ranging from 18 to 72 h. The reaction mixture was then allowed to cool and was treated with an aqueous saturated NaHCO3 solution (25 mL). The organic layer was collected, dried, and concentrated under vacuum, and purified using column chromatography, eluting with CHCl3/ethyl acetate (24:1, v/v).
3-(Diphenylphosphinyl)-2H-chromen-2-one (2a). The reaction of 1 (399 mg, 1 mmol), salicylaldehyde (0.156 mL, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 20 h produced a white solid of 2a (306 mg, 77%). Rf = 0.4 (CHCl3/ethyl acetate 24:1). Mp. 203–205 °C. lit., ref. [30] 202.1–204.7 °C. 1H NMR (500 MHz, CDCl3): δ 8.93 (d, J = 14.0 Hz, 1H), 7.95–7.88 (m, 4H), 7.69 (dd, J = 8.0, 1.6 Hz, 1H), 7.64 (ddd, J = 8.7, 7.5, 1.6 Hz, 1H), 7.61–7.55 (m, 2H), 7.54–7.46 (m, 4H), 7.40–7.30 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 159.12 (d, J = 13.6 Hz), 155.40, 153.99 (d, J = 4.8 Hz), 134.14, 132.47 (d, J = 2.8 Hz), 132.07 (d, J = 10.6 Hz), 130.54 (d, J = 110.4 Hz), 129.47, 128.52 (d, J = 13.0 Hz), 125.00, 121.64 (d, J = 102.1 Hz), 118.53 (d, J = 10.4 Hz), 116.81. 31P NMR (202 MHz, CDCl3): δ 23.35. Anal. Calcd. for C21H15O3P C, 72.83; H, 4.37; Found C, 72.54; H, 4.51.
6-(Bromo)-3-(diphenylphosphinyl)-2H-chromen-2-one (2b). The reaction of 1 (399 mg, 1 mmol), 5-bromo-2-hydroxybenzaldehyde (302 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 18 h produced a yellow solid of 2b (366 mg, 86%). Rf = 0.42 (CHCl3/ethyl acetate 24:1). Mp. Decomposition at 256 °C. 1H NMR (500 MHz, CDCl3): δ 8.83 (d, J = 13.9 Hz, 1H), 7.94-7.89 (m, 4H), 7.80 (d, J = 2.2 Hz, 1H), 7.73 (dd, J = 11.0 and 8.8 Hz, 1H), 7.62–7.59 (m, 2H), 7.53–7.49 (m, 4H), 7.25 (d, J = 8.8 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 158.5 (d, J = 12.7 Hz), 154.2, 152.5 (d, J = 5.5 Hz), 136.8, 132.6 (d, J = 3.6 Hz), 132.1 (d, J = 10.9 Hz), 131.5, 130.1 (d, J = 110.8 Hz), 128.6 (d, J = 12.7 Hz), 123.3 (d, J = 99.9 Hz), 119.9 (d, J = 10.9 Hz), 118.5, 117.5. 31P NMR (202 MHz, CDCl3): δ 22.89 ppm. Anal. Calcd. for C21H14BrO3P C, 59.32; H, 3.32; Found C, 59.47; H, 3.21.
6,8-(Dibromo)-3-(diphenylphosphinyl)-2H-chromen-2-one (2c). The reaction of 1 (399 mg, 1 mmol), 3,5-dibromo-2-hydroxybenzaldehyde (420 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 18 h produced a yellow solid of 2c (408 mg, 81%). Rf = 0.44 (CHCl3/ethyl acetate 24:1). Mp. decomposition at 250 °C. 1H NMR (500 MHz, CDCl3): δ 8.83 (d, J = 13.6 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.93–7.89 (m, 4H), 7.77 (d, J = 2.2 Hz, 1H), 7.63-7.59 (m, 2H), 7.53-7.49 (m, 4H). 13C NMR (126 MHz, CDCl3): δ 157.6 (d, J = 12.7 Hz), 152.2 (d, J = 5.5 Hz), 151.2, 139.3, 132.7 (d, J = 2.7 Hz), 132.1 (d, J = 10.9 Hz), 130.7, 130.0 (d, J = 110.8 Hz), 128.6 (d, J = 12.7 Hz), 124.4 (d, J = 98.1 Hz), 120.7 (d, J = 10.9 Hz), 117.5, 111.4. 31P NMR (202 MHz, CDCl3): δ 22.59 ppm. Anal. Calcd. for C21H13Br2O3P C, 50.03; H, 2.60; Found C, 50.27; H, 2.54.
3-(Diphenylphosphinyl)-7-methoxy-2H-chromen-2-one (2d). The reaction of 1 (399 mg, 1 mmol), 4-methoxy-2-hydroxybenzaldehyde (342 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 48 h produced a yellow solid of 2d (210 mg, 56%). Rf = 0.39 (CHCl3/ethyl acetate 24:1). Mp 178-179 °C. Lit. [43] Mp 172.4–174.1 °C. 1H NMR (500 MHz, CDCl3): δ 8.85 (d, J = 14.2 Hz, 1H), 7.95–7.90 (m, 4H), 7.59–7.56 (m, 3H), 7.51–7.48 (m, 4H), 6.93 (dd, J = 8.8 and 2.5 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H) 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 164.8, 159.5 (d, J = 13.6 Hz), 157.7, 153.7 (d, J = 5.5 Hz), 132.3 (d, J = 2.7 Hz), 132.0 (d, J = 10.0 Hz), 131.0 (d, J = 109.9 Hz), 130.6, 128.4 (d, J = 12.7 Hz), 116.9 (d, J = 105.4 Hz), 113.4, 112.5 (d, J = 10.0 Hz), 100.6. 31P NMR (202 MHz, CDCl3): δ 23.53 ppm. Anal. Calcd. for C22H17O4P C, 70.21; H, 4.55; Found C, 70.01; H, 4.70. Crystal data for 2d: C22H16O4P, Mw = 375.32, crystal system monoclinic, space group P21/c, unit cell dimensions: a = 8.6334(4) Å, b = 16.2698(7) Å, c = 13.4284(6) Å, β = 101.916(5)°, V = 845.56(15) Å3, Z = 4, Density (calc) 1.351 g/cm3, absorption coeff. 1.535 mm−1, F(000) = 780. Collected/independent reflections 13,141/3805 [R(int) = 0.0516], data/parameters 3805/244. Goodness-of-fit on F2 1.032, final R indices [I > 2σ(I)] R1 = 0.0435, wR2 = 0.1113. CCDC No 2311368.
3-(Diphenylphosphinyl)-8-methoxy-2H-chromen-2-one (2e). The reaction of 1 (399 mg, 1 mmol), 3-methoxy-2-hydroxybenzaldehyde (342 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 18 h produced a yellow solid of 2e (331 mg, 88%). Rf = 0.40 (CHCl3/ethyl acetate 24:1). Mp 210–211 °C. 1H NMR (500 MHz, CDCl3): δ 8.90 (d, J = 14.2 Hz, 1H), 7.94–7.90 (m, 4H), 7.60–7.56 (m, 2H), 7.51–7.47 (m, 4H), 7.30–7.25 (m, 2H), 7.18 (dd, J = 7.6 and 1.9 Hz, 1H), 3.96 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 158.6 (d, J = 13.6 Hz), 154.2 (d, J = 4.5 Hz), 147.2, 145.1 (d, J = 1.8 Hz), 132.4 (d, J = 3.1 Hz), 132.1 (d, J = 11.7 Hz), 130.5 (d, J = 109.9 Hz), 128.5 (d, J = 12.7 Hz), 124.8, 121.8 (d, J = 101.7 Hz), 120.7, 119.1 (d, J = 11.8 Hz), 115.8, 56.4. 31P NMR (202 MHz, CDCl3): δ 23.40 ppm. Anal. Calcd. for C22H17O4P C, 70.21; H, 4.55; Found C, 70.11; H, 4.68. Crystal data for 2e: C22H17O4P, Mw = 376.32, crystal system triclinic, space group P1bar, unit cell dimensions: a = 9.0420(8) Å, b = 9.3558(8) Å, c = 12.4689(7) Å, α = 88.332(6)°, β = 77.755(6)°, γ = 62.401(9)°, V = 910.60(14) Å3, Z = 2, Density (calc) 1.373 g/cm3, absorption coeff. 1.556 mm−1, extinction coeff. 0.0036(6), F(000) = 392. Collected/independent reflections 6271/3675 [R(int) = 0.0365], data/parameters 3675/245. Goodness-of-fit on F2 1.047, final R indices [I > 2σ(I)] R1 = 0.0467, wR2 = 0.1197. CCDC No 2311369.
3-(Diphenylphosphinyl)-6-nitro-2H-chromen-2-one (2f). The reaction of 1 (399 mg, 1 mmol), 5-nitro-2-hydroxybenzaldehyde (376 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 20 h produced a yellow solid of 2f (294 mg, 75%). Rf = 0.35 (CHCl3/ethyl acetate 24:1). Mp. decomposition at 295 °C. 1H NMR (500 MHz, CDCl3): δ 9.01 (d, J = 13.9 Hz, 1H), 8.61 (d, J = 2.8 Hz, 1H), 8.50 (dd, J = 8.8 and 2.8 Hz, 1H), 7.94–7.90 (m, 4H), 7.64–7.60 (m, 2H), 7.55–7.50 (m, 5H). 13C NMR (126 MHz, CDCl3): δ 158.5, 157.7 (d, J = 12.7 Hz), 152.5 (d, J = 5.5 Hz), 144.3, 132.8 (d, J = 2.7 Hz), 132.0 (d, J = 10.0 Hz), 130.60 (d, J = 110.8 Hz), 128.7 (d, J = 12.7 Hz), 125.1, 125.0 (d, J = 98.1 Hz), 118.4 (d, J = 10.0 Hz), 118.1. 31P NMR (202 MHz, CDCl3): δ 22.40 ppm. Anal. Calcd. for C21H14O5PN C, 64.46; H, 3.61, N, 3.58; Found C, 64.66; H, 3.51; N, 3.78.
3-(Diphenylphosphinyl)-2H-benzo[h]chromen-2-one (2g). The reaction of 1 (399 mg, 1 mmol), 2-hydroxy-1-naphthaldehyde (258 mg, 1.5 mmol), with pyrrolidine (0.024 mL, 0.3 mmol) and acetic acid (0.017 mL, 0.3 mmol) for 72 h produced a white solid of 2g (131 mg, 34%). Rf = 0.52 (CHCl3/ethyl acetate 24:1). Mp. decomposition at 251 °C. 1H NMR (500 MHz, CDCl3): δ 9.70 (d, J = 14.2 Hz, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.09 (d, J = 9.1 Hz, 1H), 8.01–7.97 (m, 4H), 7.92 (d, J = 7.8 Hz, 1H), 7.76–7.73 (m, 1H), 7.63–7.57 (m, 3H), 7.54–7.50 (m, 4H), 7.47 (d, J = 9.1 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 159.4, 156.0, 149.5 (d, J = 5.5 Hz), 135.9, 132.5 (d, J = 2.7 Hz), 132.1 (d, J = 10.9 Hz), 130.7 (d, J = 110.8 Hz), 130.2, 129.3, 129.1 (d, J = 15.4 Hz), 128.5 (d, J = 12.7 Hz), 126.6, 122.0, 119.6 (d, J = 103.5 Hz), 116.5, 113.1 (d, J = 10.0 Hz). 31P NMR (202 MHz, CDCl3): δ 23.70 ppm. Anal. Calcd. for C25H17O3P C, 75.75; H, 4.32; Found C, 75.52; H, 4.48.
2.3. Procedure for Synthesis of (SP)-L-Menthyl (2-methoxyphenyl)phenylphosphinylacetate ((SP)-3)
The synthesis was carried out on the basis of a known procedure in the literature, ref. [44] obtaining 13.16 g (40% yield) of the diastereomerically pure (>99% de by NMR) compound (SP)-3 from 17,42 g (0.075 mol) of (2-methoxyphenyl)phenylphosphine oxide. White crystals; Mp. 98–100 °C, [α]D20 –36.81 (c 1.04, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.90–7.72 (m, 4H), 7.60–7.34 (m, 6H), 4.58 (td, J = 10.9, 4.4 Hz, 1H), 3.51 (dd, J = 14.8, 1.4 Hz, 2H), 1.74–1.53 (m, 4H), 1.35 (tdt, J = 11.9, 6.5, 2.9 Hz, 1H), 1.23 (tt, J = 12.0, 3.2 Hz, 1H), 1.01–0.88 (m, 1H), 0.82 (d, J = 6.5 Hz, 3H), 0.79 (d, J = 7.0 Hz, 3H), 0.77–0.65 (m, 2H), 0.60 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 166.04 (d, J = 6.8 Hz), 159.80 (d, J = 4.2 Hz), 134.57 (d, J = 5.9 Hz), 134.25 (d, J = 2.2 Hz), 133.14 (d, J = 106.5 Hz), 131.72 (d, J = 3.0 Hz), 130.77 (d, J = 10.3 Hz), 128.25 (d, J = 12.5 Hz), 121.17 (d, J = 11.5 Hz), 119.70 (d, J = 102.8 Hz), 110.74 (d, J = 6.9 Hz), 75.39, 55.38, 46.57, 40.29, 38.64 (d, J = 62.9 Hz), 34.09, 31.26, 25.68, 23.03, 21.93, 20.85, 15.96. 31P NMR (202 MHz, CDCl3): δ 24.97.
General Procedure for Synthesis of Coumarins (Sp)-4a–e
(SP)-L-Menthyl (2-methoxyphenyl)phenylphosphinylacetate ((SP)-3) (0.75 mmol), 2-hydroxybenzaldehyde (1.13 mmol, 1.5 eqv.) and 0.2 g MS (4 Å, 4–8 mesh), together with of pyrrolidine (30 mol%) and acetic acid (30 mol%) were dissolved in 10 mL of toluene and refluxed for times ranging from 24 to 48 h. The reaction mixture was then allowed to cool and was treated with an aqueous saturated NaHCO3 solution (25 mL). The organic layer was collected, dried, and concentrated under vacuum and purified using column chromatography, eluting with CH2Cl2/diethyl ether (19:1, v/v).
3-(SP)-(2-Methoxyphenyl)phenylphosphinyl)-2H-chromen-2-one ((SP)-(4a). The reaction of 3 (321 mg, 0.75 mmol), salicylaldehyde (0.118 mL, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4a (204 mg, 80%). Rf = 0.55 (CHCl3/ethyl acetate 24:1). [α]D20 +58.24 (c 1.08, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.73 (d, J = 14.6 Hz, 1H), 8.10–8.04 (m, 2H), 7.66 (dd, J = 7.7, 1.6 Hz, 1H), 7.60 (ddd, J = 8.7, 7.3, 1.5 Hz, 1H), 7.58–7.51 (m, 2H), 7.48 (ddd, J = 8.6, 6.8, 3.1 Hz, 2H), 7.38 (ddd, J = 15.3, 7.6, 1.7 Hz, 1H), 7.35–7.30 (m, 2H), 7.00 (tdd, J = 7.6, 2.4, 0.9 Hz, 1H), 6.95 (dd, J = 8.3, 5.5 Hz, 1H), 3.65 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.62 (d, J = 2.3 Hz), 159.15 (d, J = 13.4 Hz), 155.02, 151.65 (d, J = 5.4 Hz), 134.55 (d, J = 2.1 Hz), 133.97 (d, J = 10.0 Hz), 133.57, 132.10 (d, J = 10.5 Hz), 130.63 (d, J = 112.1 Hz), 129.33, 128.30 (d, J = 13.0 Hz), 124.82, 123.56 (d, J = 107.3 Hz), 120.85 (d, J = 13.3 Hz), 119.25 (d, J = 112.9 Hz), 118.73 (d, J = 11.0 Hz), 116.68, 111.47 (d, J = 6.6 Hz), 55.72. 31P NMR (202 MHz, CDCl3): δ 21.25. Anal. Calcd. for C22H17O4P C, 70.21; H, 4.55; Found C, 70.10; H, 4.73. Crystal data for (SP)-4a: C22H17O4P, Mw = 376.32, crystal system orthorhombic, space group P212121, unit cell dimensions: a = 9.0321(1) Å, b = 13.4636(1) Å, c = 14.8349(2) Å, V = 1803.99(3) Å3, Z = 4, Density (calc) 1.386 g/cm3, absorption coeff. 1.571 mm−1, F(000) = 784. Reflections collected/independent 13,117/3719 [R(int) = 0.0240], data/parameters 3719/244. Goodness-of-fit on F2 1.056, final R indices [I > 2σ(I)] R1 = 0.0270, wR2 = 0.0720, absolute structure parameter 0.004(9). CCDC No 2311370.
6-(Bromo)-3-(SP)-(2-methoxyphenyl)phenylphosphinyl)-2H-chromen-2-one ((SP)-4b). The reaction of (SP)-3 (321 mg, 0.75 mmol), 5-bromo-2-hydroxybenzaldehyde (227 mg, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4b (265 mg, 87%). Rf = 0.52 (CHCl3/ethyl acetate 24:1). [α]D20 +24.53 (c 1.06, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.60 (d, J = 14.5 Hz, 1H), 8.09–8.02 (m, 2H), 7.76 (d, J = 2.3 Hz, 1H), 7.68 (dd, J = 8.8, 2.4 Hz, 1H), 7.60–7.52 (m, 2H), 7.51–7.46 (m, 2H), 7.39 (ddd, J = 15.3, 7.6, 1.7 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 7.01 (tdd, J = 7.6, 2.5, 0.9 Hz, 1H), 6.95 (ddd, J = 8.4, 5.6, 0.8 Hz, 1H), 3.66 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.49 (d, J = 2.3 Hz), 158.51 (d, J = 12.9 Hz), 153.79, 149.94 (d, J = 5.6 Hz), 136.18, 134.67 (d, J = 2.1 Hz), 133.94 (d, J = 10.0 Hz), 132.30 (d, J = 2.9 Hz), 132.13 (d, J = 10.3 Hz), 131.32, 130.26 (d, J = 112.3 Hz), 128.38 (d, J = 12.9 Hz), 125.38 (d, J = 105.3 Hz), 120.94 (d, J = 13.4 Hz), 120.17 (d, J = 11.0 Hz), 119.05 (d, J = 113.2 Hz), 118.45, 117.35, 111.46 (d, J = 6.7 Hz), 55.72. 31P NMR (202 MHz, CDCl3): δ 20.72. Anal. Calcd. for C22H16O4PBr C, 58.04; H, 3.54; Found C, 58.21; H, 3.42.
6,8-(Dibromo)-3-(SP)-(2-methoxyphenyl)phenylphosphinyl)-2H-chromen-2-one ((SP)-4c). The reaction of (SP)-3 (321 mg, 0.75 mmol), 3,5-dibromo-2-hydroxybenzaldehyde (316 mg, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4c (230 mg, 75%). Rf = 0.5 (CHCl3/ethyl acetate 24:1) [α]D20 +59.46 (c 1.1, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.65 (d, J = 14.3 Hz, 1H), 8.05 (ddd, J = 12.7, 8.3, 1.4 Hz, 2H), 7.94 (d, J = 2.1 Hz, 1H), 7.75 (d, J = 2.2 Hz, 1H), 7.63–7.53 (m, 2H), 7.50 (ddd, J = 8.7, 7.0, 3.2 Hz, 2H), 7.36 (ddd, J = 15.4, 7.6, 1.7 Hz, 1H), 7.02 (tdd, J = 7.5, 2.5, 0.9 Hz, 1H), 6.98–6.93 (m, 1H), 3.68 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.62, 157.62 (d, J = 13.6 Hz), 150.82, 149.83 (d, J = 5.6 Hz), 138.76, 134.84 (d, J = 2.2 Hz), 133.93 (d, J = 10.3 Hz), 132.46 (d, J = 2.9 Hz), 132.17 (d, J = 10.3 Hz), 130.65, 129.77 (d, J = 112.7 Hz), 128.45 (d, J = 12.8 Hz), 126.20 (d, J = 104.0 Hz), 120.94 (d, J = 13.6 Hz), 118.64 (d, J = 113.6 Hz), 117.31, 111.51 (d, J = 6.5 Hz), 111.21, 55.81. 31P NMR (202 MHz, CDCl3): δ 20.73. Anal. Calcd. for C22H15O4PBr2 C, 49.47; H, 2.83; Found C, 49.59; H, 2.80.
3-(SP)-(2-Methoxyphenyl)phenylphosphinyl)-7-methoxy-2H-chromen-2-one ((SP)-4d). The reaction of (SP)-3 (321 mg, 0.75 mmol), 4-methoxy-2-hydroxybenzaldehyde (258 mg, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4d (106 mg, 39%). Rf = 0.40 (CHCl3/ethyl acetate 24:1). [α]D20 +66.54 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.56 (d, J = 14.5 Hz, 1H), 8.02–7.92 (m, 2H), 7.50–7.42 (m, 3H), 7.39 (td, J = 7.6, 3.1 Hz, 2H), 7.30 (ddd, J = 15.2, 7.6, 1.7 Hz, 1H), 6.92 (td, J = 7.5, 2.3 Hz, 1H), 6.86 (dd, J = 8.4, 5.5 Hz, 1H), 6.81 (dd, J = 8.7, 2.4 Hz, 1H), 6.74 (d, J = 2.3 Hz, 1H), 3.81 (s, 3H), 3.57 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 164.39, 161.66 (d, J = 2.3 Hz), 159.47 (d, J = 13.7 Hz), 157.24, 151.77 (d, J = 5.4 Hz), 134.43 (d, J = 2.2 Hz), 134.06 (d, J = 9.9 Hz), 131.99 (d, J = 10.4 Hz), 131.15 (d, J = 112.5 Hz), 130.44, 128.20 (d, J = 12.9 Hz), 120.79 (d, J = 13.2 Hz), 119.05 (d, J = 202.7 Hz), 119.04 (d, J = 19.3 Hz), 113.16, 112.60 (d, J = 10.9 Hz), 111.48 (d, J = 6.6 Hz), 55.96, 55.70. 31P NMR (202 MHz, CDCl3): δ 21.72. Anal. Calcd. for C23H19O5P C, 67.98; H, 4.71; Found C, 67.80; H, 4.81. Crystal data for (SP)-4d ethanol solvate: unit cell C94H82O21P4 [molecular contents: 4 × (C23H19O5P) + C2H6O], Mw = 1671.47 [4 × 406.37 + 46.07], crystal system monoclinic, space group P 21, unit cell dimensions: a = 16.3846(2) Å, b = 14.8761(2) Å, c = 17.0878(2) Å, β = 99.492(4)°, V = 4107.94(10) Å3, Z = 8, Z′ = 4. Density (calc) 1.351 g/cm3, absorption coeff. 1.480 mm−1, F(000) = 1748. Reflections collected/independent 32,156/15,333 [R(int) = 0.0246], data/restraints/parameters 15,333/4/1072. Goodness-of-fit on F2 1.017, final R indices [I > 2σ(I)] R1 = 0.0372, wR2 = 0.1001, absolute structure parameter 0.029(8). CCDC No 2311371.
3-(SP)-(2-Methoxyphenyl)phenylphosphinyl)-8-methoxy-2H-chromen-2-one ((SP)-4e). The reaction of (SP)-3 (321 mg, 0.75 mmol), 3-methoxy-2-hydroxybenzaldehyde (258 mg, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4e (232 mg, 85%). Rf = 0.39 (CHCl3/ethyl acetate 24:1). [α]D20 +95.88 (c 1.02, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.63 (d, J = 14.6 Hz, 1H), 8.03–7.91 (m, 2H), 7.50–7.41 (m, 2H), 7.38 (td, J = 7.6, 3.0 Hz, 2H), 7.26 (ddd, J = 15.3, 7.6, 1.6 Hz, 1H), 7.18–7.12 (m, 2H), 7.06 (dd, J = 7.4, 2.0 Hz, 1H), 6.90 (td, J = 7.5, 2.4 Hz, 1H), 6.85 (dd, J = 8.4, 5.5 Hz, 1H), 3.87 (s, 3H), 3.57 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.63, 158.62, 158.51, 151.70, 147.04, 144.66, 134.49, 133.98, 132.07 (d, J = 9.9 Hz), 130.75 (d, J = 112.8 Hz), 128.25 (d, J = 13.0 Hz), 124.62, 123.92 (d, J = 106.9 Hz), 120.79 (d, J = 13.4 Hz), 120.44, 119.55 (d, J = 56.1 Hz), 119.06 (d, J = 48.2 Hz), 115.16, 111.41 (d, J = 6.5 Hz), 56.30, 55.72. 31P NMR (202 MHz, CDCl3): δ 20.9. Anal. Calcd. for C23H19O5P C, 67.98; H, 4.71; Found C, 67.70; H, 4.86.
3-(SP)-(2-Methoxyphenyl)phenylphosphinyl)-6-nitro-2H-chromen-2-one ((SP)-4f). The reaction of (SP)-3 (321 mg, 0.75 mmol), 5-nitro-2-hydroxybenzaldehyde (283 mg, 1.13 mmol), with pyrrolidine (0.018 mL, 0.23 mmol) and acetic acid (0.013 mL, 0.23 mmol) for 24 h produced a white solid of (SP)-4f (213 mg, 76%). Rf = 0.35 (CHCl3/ethyl acetate 24:1). [α]D20 +18.20 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.76 (d, J = 14.4 Hz, 1H), 8.57 (d, J = 2.6 Hz, 1H), 8.45 (dd, J = 9.1, 2.7 Hz, 1H), 8.05 (ddd, J = 12.7, 8.4, 1.3 Hz, 2H), 7.64–7.54 (m, 2H), 7.55–7.48 (m, 3H), 7.41 (ddd, J = 15.4, 7.6, 1.7 Hz, 1H), 7.03 (tdd, J = 7.4, 2.5, 0.9 Hz, 1H), 6.97 (ddd, J = 8.5, 5.7, 0.9 Hz, 1H), 3.68 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 161.44 (d, J = 2.3 Hz), 158.20, 157.68 (d, J = 12.9 Hz), 149.83 (d, J = 5.7 Hz), 144.21, 134.94 (d, J = 2.2 Hz), 133.95 (d, J = 10.0 Hz), 132.57 (d, J = 2.9 Hz), 129.63 (d, J = 112.7 Hz), 128.53 (d, J = 12.9 Hz), 127.91, 126.81 (d, J = 103.8 Hz), 124.99, 121.08 (d, J = 13.6 Hz), 118.68 (d, J = 11.3 Hz), 118.51 (d, J = 113.8 Hz), 117.95, 111.51 (d, J = 6.7 Hz), 55.76. 31P NMR (202 MHz, CDCl3): δ 20.63. Anal. Calcd. for C22H16O6PN C, 62.71; H, 3.83, N, 3.32; Found C, 62.66; H, 3.81; N, 3.58
2.4. Procedure for Synthesis of (SP)-L-menthyl phenylvinylphosphinylacetate ((SP)-5)
The synthesis was carried out on the basis of a known procedure in the literature, ref. [45] obtaining 25.0 g (32% yield) of the diastereomerically pure (>99% de by NMR) (SP)-5. White crystals; Mp. 150–151 °C, [α]D20 –62.3 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.84–7.70 (m, 2H), 7.60–7.52 (m, 1H), 7.49 (tdd, J = 7.1, 3.1, 1.7 Hz, 2H), 6.69 (ddd, J = 28.2, 18.7, 12.6 Hz, 1H), 6.43 (ddd, J = 22.8, 18.7, 1.6 Hz, 1H), 6.33 (ddd, J = 42.1, 12.6, 1.6 Hz, 1H), 4.61 (td, J = 10.9, 4.4 Hz, 1H), 3.25 (d, J = 15.8 Hz, 2H), 2.00–1.79 (m, 1H), 1.63 (ddq, J = 13.9, 9.8, 3.2 Hz, 3H), 1.41 (dddd, J = 12.0, 8.6, 6.5, 3.3 Hz, 1H), 1.28 (ddt, J = 14.5, 10.8, 3.1 Hz, 1H), 1.03–0.89 (m, 2H), 0.87 (d, J = 6.5 Hz, 3H), 0.85–0.81 (m, 1H), 0.79 (d, J = 7.0 Hz, 3H), 0.60 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.79 (d, J = 4.4 Hz), 135.50, 132.31 (d, J = 2.9 Hz), 131.13 (d, J = 108.4 Hz), 130.66 (d, J = 10.0 Hz), 129.81 (d, J = 97.5 Hz), 128.71 (d, J = 12.5 Hz), 75.83, 46.58, 40.56, 39.41 (d, J = 62.7 Hz), 34.03, 31.34, 25.78, 23.08, 21.94, 20.74, 15.99. 31P NMR (202 MHz, CDCl3): δ 22.22. Crystal data for (SP)-5: C20H29O3P, Mw = 348.40, crystal system monoclinic, space group P21, unit cell dimensions: a = 9.2836(3) Å, b = 9.8728(3) Å, c = 10.9586(3) Å, β = 95.256(2)°, V = 1000.19(5) Å3, Z = 2, Density (calc) 1.157 g/cm3, absorption coeff. 1.322 mm−1, F(000) = 376. Reflections collected/independent 6730/3592 [R(int) = 0.0238], data/restraints/parameters 3592/1/217. Goodness-of-fit on F2 1.030, final R indices [I > 2σ(I)] R1 = 0.0343, wR2 = 0.0894, absolute structure parameter 0.017(15). CCDC No 2311372.
General Procedure for Synthesis of Coumarins (SP)-6a–e
(SP)-L-Menthyl 2-(phenyl(vinyl)phosphinyl)acetate ((SP)-5) (1.43 mmol), 2-hydroxyaldehyde (2.15 mmol, 1.5 eqv.) and 0.2 g MS (4 Å, 4–8 mesh), together with of pyrrolidine (30 mol%) and acetic acid (30 mol%) were dissolved in 10 mL of toluene and refluxed for times ranging from 24 to 48 h. The reaction mixture was then allowed to cool and was treated with an aqueous saturated NaHCO3 solution (25 mL). The organic layer was collected, dried, concentrated under a vacuum, and purified using column chromatography, eluting with hexane/propanol (15:1, v/v).
3-(SP)-(Phenyl(vinyl)phosphinyl)-2H-chromen-2-one ((SP)-6a). The reaction of (SP)-5 (500 mg, 1.43 mmol), salicylaldehyde (0,178 mL, 2.14 mmol), with pyrrolidine (0.035 mL, 0.43 mmol) and acetic acid (0.024 mL, 0.43 mmol) for 48 h produced a white solid of (SP)-6a (250 mg, 59%). Rf = 0.45 (hexane/propanol 5:1). [α]D20 +32.0 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.77 (d, J = 8.7 Hz, 1H), 7.96–7.92 (m, 2H), 7.69–7.64 (m, 2H), 7.58–7.48 (m, 3H), 7.39–7.36 (m, 2H), 7.16 (ddd, J = 29.2, 18.8, 12.7 Hz, 1H), 6.60 (ddd, J = 23.4, 18.9, 1.5 Hz, 1H), 6.42 (ddd, J = 43.1, 14.9, 1.5 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 159.26 (d, J = 14.5 Hz), 159.17, 152.14 (d, J = 4.5 Hz), 135.96, 133.99, 132.34 (d, J = 3.6 Hz), 131.58 (d, J = 28.6 Hz), 131.03, 130.99 (d, J = 9.9 Hz), 129.36, 129.12, 128.58 (d, J = 13.6 Hz), 128.32, 125.05, 122.43 (d, J = 103.5 Hz), 118.48 (d, J = 9.9 Hz), 116.87. 31P NMR (202 MHz, CDCl3): δ 17.27. Anal. Calcd. for C17H13O3P C, 68.92; H, 4.42; Found C, 68.70; H, 4.56.
6-(Bromo)-3-(SP)-(phenyl(vinyl)phosphinyl)-2H-chromen-2-one ((SP)-6b). The reaction of (SP)-5 (500 mg, 1.43 mmol), 5-bromo-2-hydroxybenzaldehyde (430 mg, 2.14 mmol), with pyrrolidine (0.035 mL, 0.43 mmol) and acetic acid (0.024 mL, 0.43 mmol) for 48 h produced a white solid of (SP)-6b (334 mg, 62%). Rf = 0.50 (hexane/propanol 5:1). [α]D20 +28.0 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.68 (d, J = 13.5 Hz, 1H), 7.94–7.89 (m, 2H), 7.78 (d, J = 2.2 Hz, 1H), 7.72 (dd, J = 8.3, 2.5, Hz, 1H), 7.57– 7.47 (m, 3H), 7.58–7.48 (m, 3H), 7.25 (d, J = 8.3 1H), 7.12 (ddd, J = 29.3, 18.6, 12.6 Hz, 1H), 6.62 (ddd, J = 23.3, 18.6, 1.6 Hz, 1H), 6.44 (ddd, J = 43.1, 14.9, 1.6 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 158.75 (d, J = 13.5 Hz), 153.96, 150.67 (d, J = 4.5 Hz), 136.38 (d, J = 36.3 Hz), 132.47, 131.46 (d, J = 127.16 Hz), 131,41, 131.02 (d, J = 9.9 Hz), 130.60, 128.87 (d, J = 102.6 Hz), 124.19 (d, J = 100.8 Hz), 119.90 (d, J = 10.9 Hz), 118.57, 117.61. 31P NMR (202 MHz, CDCl3): δ 16.96. Anal. Calcd. for C17H12O3PBr C, 54.43; H, 3.22; Found C, 54.60; H, 3.28.
3-(SP)-(Phenyl(vinyl)phosphinyl)-7-methoxy-2H-chromen-2-one ((SP)-6c). The reaction of (SP)-5 (500 mg, 1.43 mmol), 4-methoxy-2-hydroxybenzaldehyde (326 mg, 2.14 mmol), with pyrrolidine (0.035 mL, 0.43 mmol) and acetic acid (0.024 mL, 0.43 mmol) for 48 h produced a white solid of (SP)-6c (1187 mg, 40%). Rf = 0.51 (hexane/propanol 5:1). [α]D20 +26.8 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.67 (d, J = 13.5 Hz, 1H), 7.94–7.89 (m, 2H), 7.56–7.45 (m, 4H), 7.05 (ddd, J = 28.7, 18.6, 1.6 Hz, 1H), 6.92 (dd, J = 8.8, 2.5 Hz, 1H), 6.83 (d, J = 2.2 Hz, 1H), 6.62 (ddd, J = 23.3, 18.6, 12.6 Hz, 1H), 6.32 (ddd, J = 42.8, 12.9, 1.9 Hz, 1H), 3.90 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 164.68, 159.71, 157.41, 153.96, 151.98 (d, J = 4.5 Hz), 135.36, 132.16 (d, J = 2.7 Hz), 131.43 (d, J = 110.8 Hz), 130.91 (d, J = 9.9 Hz), 130.46, 128.77 (d, J = 101.7 Hz), 128.63, 128.52, 116.77 (d, J = 106.2 Hz), 113.43, 112. 38, 100.64, 56.00. 31P NMR (202 MHz, CDCl3): δ 17.64. Anal. Calcd. for C18H15O4P C, 66.24; H, 4.63; Found C, 66.42; H, 4.73.
3-(SP)-(Phenyl(vinyl)phosphinyl)-8-methoxy-2H-chromen-2-one ((SP)-6d). The reaction of (SP)-5 (500 mg, 1.43 mmol), 3-methoxy-2-hydroxybenzaldehyde (326 mg, 2.14 mmol), with pyrrolidine (0.035 mL, 0.43 mmol) and acetic acid (0.024 mL, 0.43 mmol) for 48 h produced a white solid of (SP)-6d (299 mg, 64%). Rf = 0.42 (hexane/propanol 5:1). [α]D20 +9.3 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.74 (d, J = 14.9 Hz, 1H), 7.94–7.89 (m, 2H), 7.55–7.45 (m, 3H), 7.29–7.08 (m, 4H), 6.55 (ddd, J = 20.4, 18.9, 1.6 Hz, 1H), 6.34 (ddd, J = 8.8, 2.5, 1.9 Hz, 1H), 6.83 (d, J = 2.2 Hz, 1H), 6.62 (ddd, J = 23.3, 18.6, 12.6 Hz, 1H), 6.32 (ddd, J = 43.9, 12.3, 1.9 Hz, 1H), 3.96 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 158.71(d, J = 14.5 Hz), 152.20 (d, J = 4.5 Hz), 147.19, 144.89, 135.80, 132.28 (d, J = 2.7 Hz), 130.99 (d, J = 119.9 Hz), 128.37 (d, J = 101.7 Hz), 131.43 (d, J = 110.8 Hz), 130.91 (d, J = 9.9 Hz), 130.46, 128.57 (d, J = 12.7 Hz), 128.77 (d, J = 101.7 Hz), 124.86, 121.92 (d, J = 102.6 Hz), 120.51, 119.04 (d, J = 9.9 Hz), 115.59, 56.37. 31P NMR (202 MHz, CDCl3): δ 17.28. Anal. Calcd. for C18H15O4P C, 66.24; H, 4.63; Found C, 66.13; H, 4.61. Crystal data for (SP)-6d: C18H15O4P, Mw = 326.27, crystal system monoclinic, space group P21, unit cell dimensions: a = 8.5085(2) Å, b = 5.9492(1) Å, c = 30.6549(7) Å, β = 91.769(2)°, V = 1550.97(6) Å3, Z = 4, Z′ = 2, Density (calc) 1.397 g/cm3, absorption coeff. 1.734 mm−1, F(000) = 680. Reflections collected/independent 10,647/5095 [R(int) = 0.0490], data/restraints/parameters 5095/1/415. Goodness-of-fit on F2 1.084, final R indices [I > 2σ(I)] R1 = 0.0995, wR2 = 0.2814, absolute structure parameter 0.07(2). CCDC No 2311373.
3-(SP)-(Phenyl(vinyl)phosphinyl)-6-nitro-2H-chromen-2-one ((SP)-6e). The reaction of (SP)-5 (500 mg, 1.43 mmol), 5-nitro-2-hydroxybenzaldehyde (357 mg, 2.14 mmol), with pyrrolidine (0.035 mL, 0.43 mmol) and acetic acid (0.024 mL, 0.43 mmol) for 48 h produced a yellow solid of (SP)-6e (230 mg, 47%). Rf = 0.4 (hexane/propanol 5:1). [α]D20 +14.8 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.82 (d, J = 13.5 Hz, 1H), 8.59 (d, J = 2.5 Hz, 1H), 8.47 (dd, J = 8.8, 2.5 Hz, 1H), 7.94–7.90 (m, 2H), 7.60–7.49 (m, 4H), 6.55 (ddd, J = 29.6, 18.6, 12.6 Hz, 1H), 6.60 (ddd, J = 20.4, 18.9, 1.6 Hz, 1H), 6.40 (ddd, J = 43.5, 12.9, 1.6 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 158.31, 157.79, 150.58 (d, J = 4.5 Hz), 144.33, 136.92, 132.72 (d, J = 2.7 Hz), 131.07, 130.99 (d, J = 109.9 Hz), 128.80 (d, J = 12.7 Hz), 128.25, 118.44 (d, J = 9.9 Hz), 118.13. 31P NMR (202 MHz, CDCl3): δ 16.41. Anal. Calcd. for C17H12O5PN C, 59.83; H, 3.54, N, 4.10; Found C, 59.01; H, 3.49; N, 4.16. Crystal data for (SP)-6e: C17H12NO5P, Mw = 341.25, crystal system monoclinic, space group C2, unit cell dimensions: a = 8.6060(2) Å, b = 28.2190(7) Å, c = 13.1554(3) Å, β = 96.077(2)°, V = 3176.87(13) Å3, Z = 8, Z′ = 2, Density (calc) 1.427 g/cm3, absorption coeff. 1.790 mm−1, F(000) = 1408. Reflections collected/independent 6835/5040 [R(int) = 0.0271], data/restraints/parameters 5040/1/433. Goodness-of-fit on F2 1.083, final R indices [I > 2σ(I)], R1 = 0.0379, wR2 = 0.105, absolute structure parameter 0.002(18). CCDC No 2311374.
2.5. Procedure for Synthesis of (RP)-tert-butylphenylphosphinyl Acetic Acid Menthyl Ester ((RP)-7)
The synthesis was carried out on the basis of a known procedure in the literature, ref. [46] obtaining diastereomerically pure (RP)-7 (>98% de by 1H NMR) compound. [α]D20 –80.8 (c 1.0, MeOH). 1H NMR (500 MHz, CDCl3): δ 7.82–7.78 (m, 2H), 7.53–7.50 (m, 3H), 4.61 (dt, J = 4.4 and 11.0 Hz, 1H), 3.32–3.21 (m, 2H), 1.74–0.76 (m, 15H), 1.18 (d, J = 15.5 Hz, 9H), 0.67 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 166.7 (d, J = 7.0 Hz), 132.0 (d, J = 8.6 Hz), 131.8 (d, J = 2.6 Hz), 129.5 (d, J = 92.3 Hz), 128.1 (d, J = 12.0 Hz), 75.6, 46.5, 40.2, 34.2, 34.0 (d, J = 62.8 Hz), 33.3 (d, J = 50.8 Hz), 31.3, 25.6, 24.4, 23.0, 22.0, 20.0, 15.8. 31P NMR (202 MHz, CDCl3): δ 43.83. Anal. Calcd. for C22H35O3P C, 69.81; H, 9.32; Found C, 69.86; H, 9.13.
Synthesis of 3-(RP)-(tert-butylphenylphosphinyl)-2H-chromen-2-one ((RP)-8a)
The reaction of (RP)-tert-butylphenylphospinoyl acetic acid menthyl ester (100 mg, 0.264 mmol), salicylaldehyde (80 mg, 0.0661 mmol), with pyrrolidine (0.0056 mL, 0.08 mmol) and acetic acid (0.005 mL, 0.08 mmol) for 144 h produced a yellow pale solidified oil of (RP)-8a (23 mg, 27%). Rf = 0.5 (hexane/propanol 5:1). [α]D20 +84.9 (c 0.55, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.93 (d, J = 13.6 Hz, 1H), 8.26–8.22 (m, 2H), 7.65–7.62 (m, 2H), 7.54–7.56 (m, 1H), 7.51–7.48 (m, 2H), 7.39–7.32 (m, 2H), 1.34 (d, J = 16.4 Hz, 9H). 13C NMR (126 MHz, CDCl3): δ 160.1, 155.6 (d, J = 3.7 Hz), 155.1, 134.0, 132.4 (d, J = 9.3 Hz), 132.1 (d, J = 3.1 Hz), 129.5 (d, J = 94.5 Hz), 129.4, 128.2, 124.9, 121.5, 118.5 (d, J = 9.1 Hz), 36.0 (d, J = 72.7 Hz), 25.5. 31P NMR (202 MHz, CDCl3): δ 40.26. Anal. Calcd. for C19H19O3P C, 69.93; H, 5.87; Found C, 69.86; H, 6.03.
3. Results and Discussion
3.1. Synthesis of Phosphinylated Coumarins
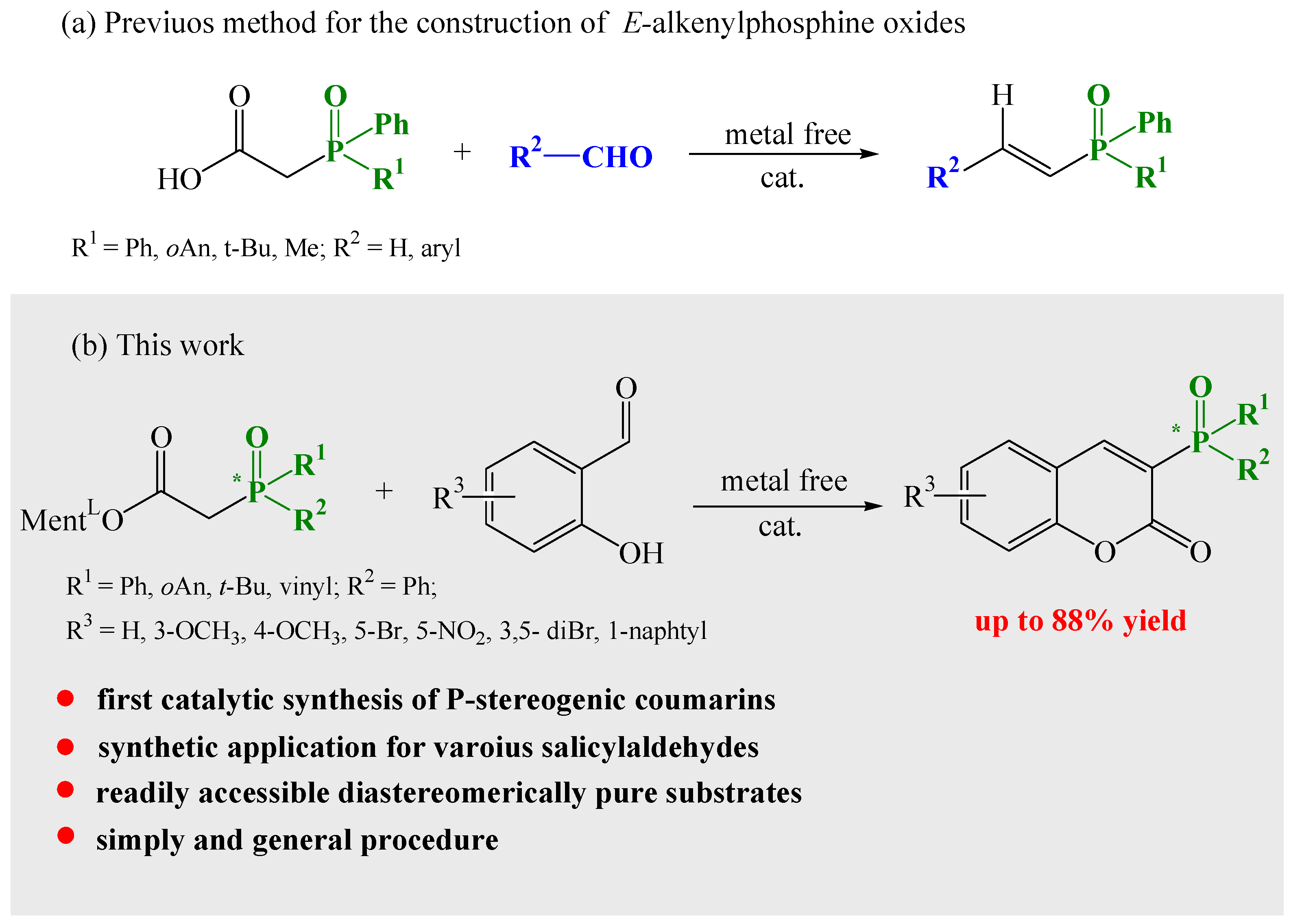
Owing to our interest in tertiary phosphine oxides, we have developed a simple and efficient method for the construction of α,β-unsaturated E-alkenylphosphine oxides using Knoevenagel condensation reactions (Scheme 2a) [47].
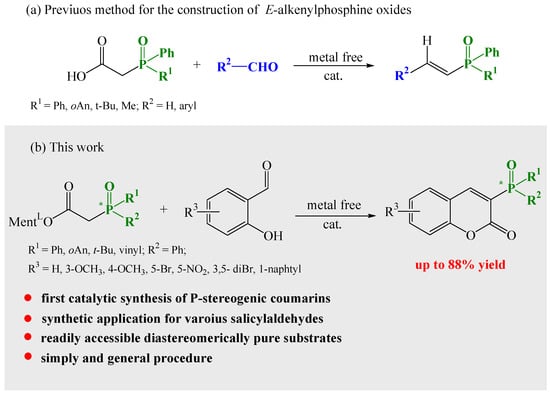
Scheme 2.
Deployment of condensation reactions for the synthesis of phosphine oxide derivatives.
Investigating further the broad utility potential of the condensation reaction, we noted that salicylaldehyde and its derivatives, in the presence of catalytic amounts of amine and acid, readily undergo reactions with L-menthyl diphenylphosphinylacetate. Bojilova and Nikolova [36,48] reported the reaction of salicylaldehyde with triethylphosphonoacetate in the presence of piperidine in refluxing toluene. A Dean–Stark water separator was used to remove the resulting water from the reaction mixture. For our synthesis of 3-diphenylphosphoryl coumarins to improve the yield of the products, we modified the catalyst, and we noticed that the addition of molecular sieves (MS 4 Å, 4–8 mesh) was enough to remove the water from the reaction. A deeper understanding of the parameters governing this reaction allowed us to develop an efficient method for the preparation of coumarins bearing the P-chiral moiety.
3.1.1. Synthesis of 3-Diphenylphosphoryl Coumarins
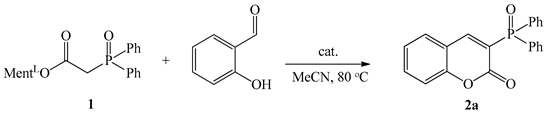
Our initial study commenced with the reaction of diphenylphosphinoylacetic acid menthyl ester with commercially available salicylaldehyde in the presence of 30 mol% piperidine and 30 mol% acetic acid. The experimental conditions used were the same as those previously described by our group in the synthesis of E-alkenylphosphine oxides [48]. To our delight, the condensation product 2a was obtained in acetonitrile at 80 °C with 100% conversion and 77% yield (Table 1). It was possible to use pyridine and piperidine in synthesis alongside selected acid such as acetic acid (AcOH), trichloroacetic acid (TCA), or p-toluenosulfonic acid (TSA). However, if pure piperidine or pyrrolidine (30 mol%) was exploited as a catalyst, product 2a was not afforded.

Table 1.
Optimization reaction of phosphinoylacetic acids menthyl ester with salicylaldehyde.
With the optimized conditions on hand, i.e., pyrrolidine, and acetic acid as catalysts at 30 mol%, a reaction temperature of 80 °C, and acetonitrile as solvent, the scope of salicylaldehydes was investigated. As demonstrated in Scheme 3, salicylaldehydes bearing electron-donating groups and electron-withdrawing groups were all efficiently coupled with 1 under these reaction conditions to generate the responding 3-(diphenylphosphoryl)-2H-chromen-2-one derivatives in moderate to good yields that demonstrated the generality of this method. Thus, the functionalities, such as bromo, dibromo, nitro, methoxyl, and 1-naphtyl were all well tolerated for this method.
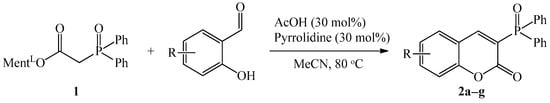
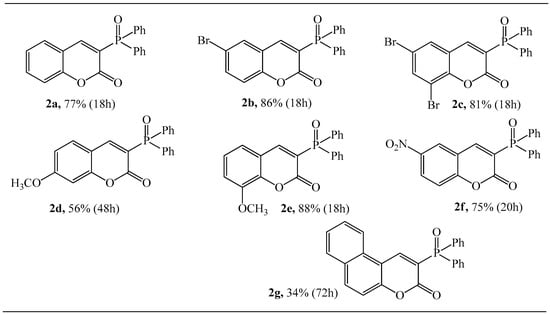
Scheme 3.
Reaction of 1 with substituted salicylaldehydes. Reaction conditions: 1 (1.0 mmol), aldehyde (1.5 equiv.), pyrrolidine (30 mol%), AcOH (30 mol%),CH3CN (15 mL), 80 °C, reaction time 18–72 h. The isolated yield is given.
At 80 °C and after 18 h, the highest reaction yield of 88% was found for reaction 1 with 3-methoxy-2-hydroxybenzaldehyde. It is worth noting that with 4-methoxy-2-hydroxybenzaldehyde, the corresponding product 2d was obtained with a much lower yield of 56%. The substituents such as Br, diBr, and NO2 of salicylaldehedes were compatible with the developed reaction conditions in the transformation, furnishing the corresponding products 2b, 2c, and 2f in good yields (86%, 81%, and 75%, respectively). In contrast, the lower yield (34%) was observed only in the cases of 2-hydroxy-1-naphthaldehyde.
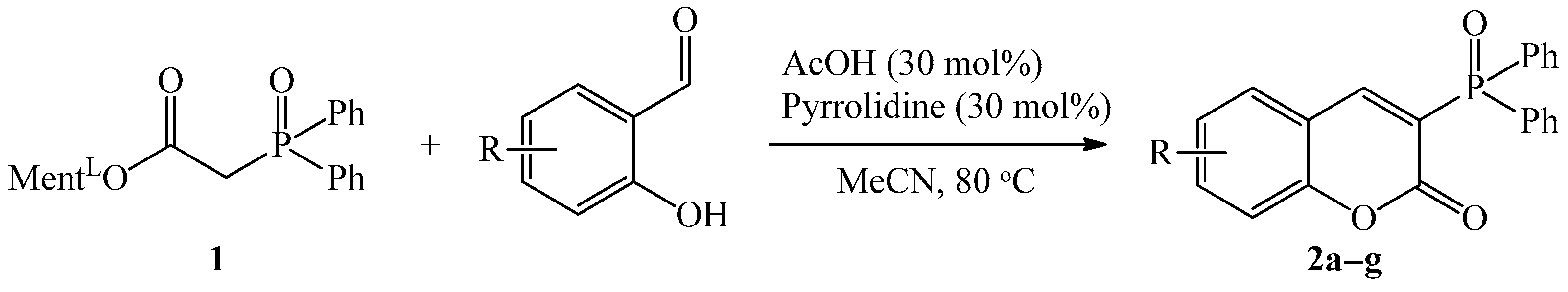
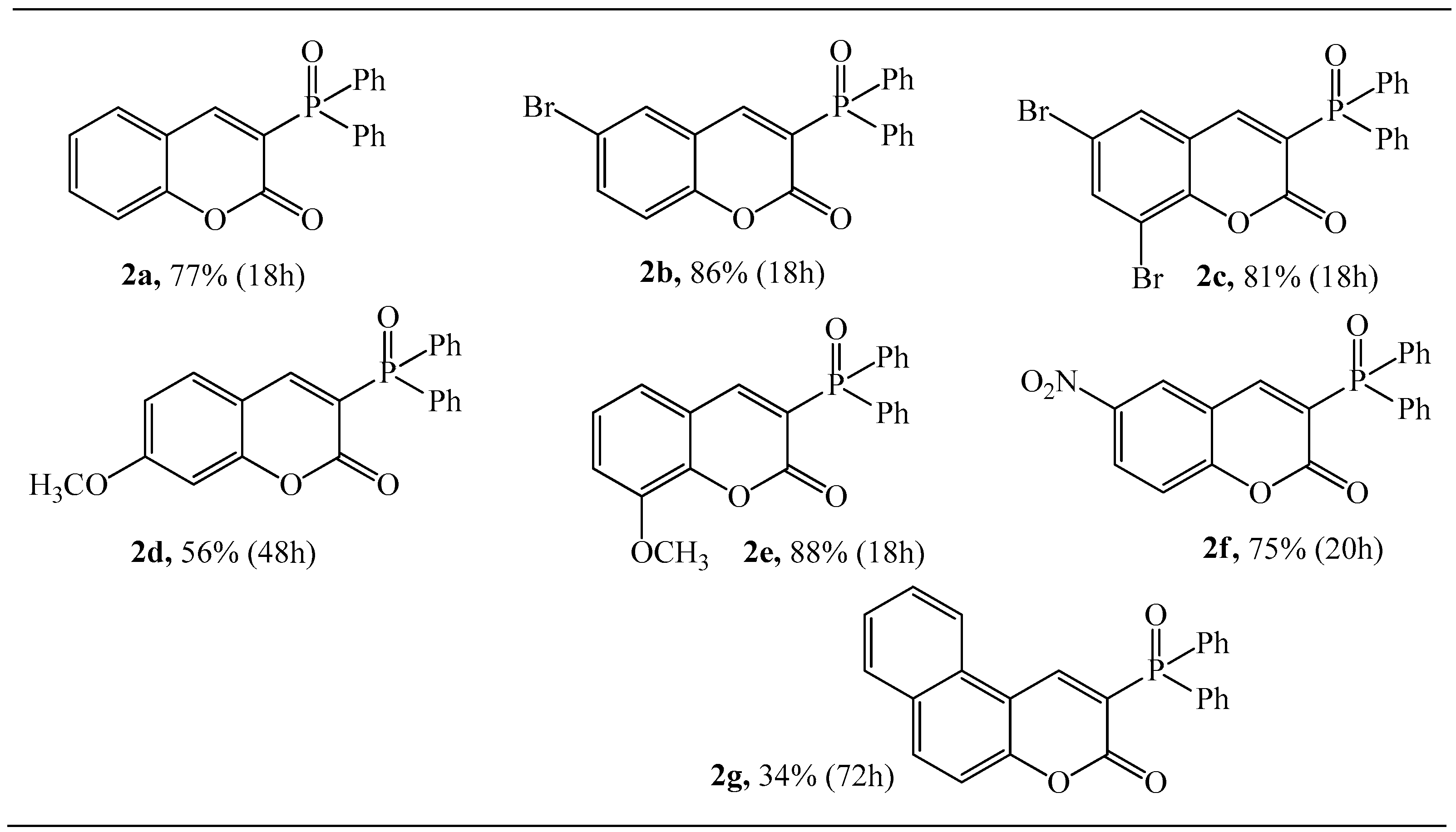
The molecular structure for coumarins 2d and 2e was determined by means of an X-ray crystallography (Figure 1 and Figure 2).
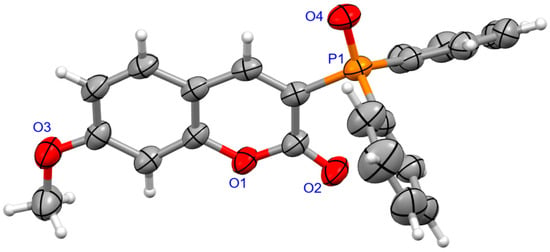
Figure 1.
The molecular structure of 2d.
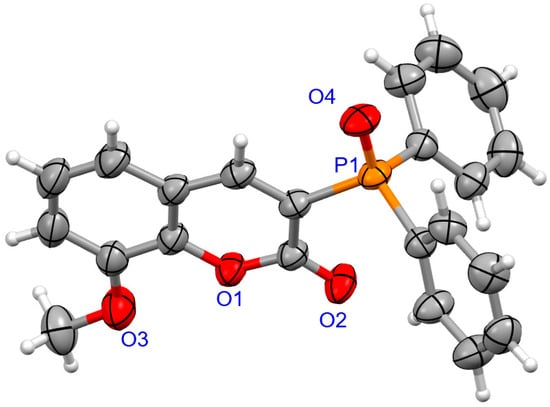
Figure 2.
The molecular structure of 2e.
3.1.2. Synthesis of Coumarins with P-stereogenic Moiety
After exploring the scope of Knoevenagel condensation of mentyl esters of phosphinoacetic acids with salicylic aldehydes, we designed a series of simple enantiomerically pure phosphine oxides that, owning chirality on the phosphorus atom, could become very attractive as ligands for asymmetric transition metal catalysis or asymmetric organocatalysis, and they could also show biological activity. Following our convenient and efficient synthetic pathway, we subjected diastereomerically pure menthyl phosphinoacetates to a condensation reaction with a series of salicylaldehydes.
As shown in Scheme 4, when ester (Sp)-3 was utilized to react with salicylaldehyde in the presence of pyrrolidine (30 mol%) and AcOH (30 mol%) in MeCN at 80 °C the reaction offered the product (Sp)-4a in 52% yield. It was necessary to raise the reaction temperature to 110 °C and change the solvent to toluene. The salicyladehydes containing various electron-donor and electron-withdrawing substituents were well tolerated in the reaction with 3 and provided the desired products (4b–f) in good yields (up to 87%). In particular, the course of the reaction was significantly affected by the use of 4-methoxysalicylaldehyde as a substrate, and the product 4d could be obtained in yields as low as 39%.
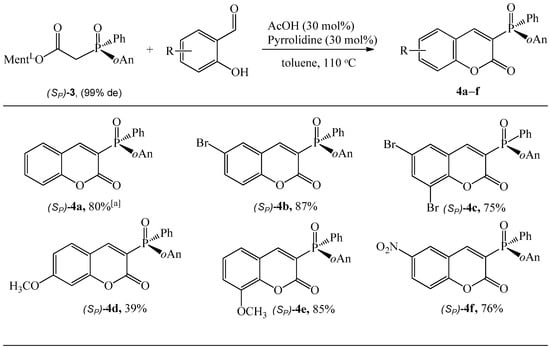
Scheme 4.
Reaction of (Sp)-3 with substituted salicylaldehydes. Reaction conditions: (Sp)-3 (0.75 mmol), aldehyde (1.5 equiv.), pyrrolidine (30 mol%), AcOH (30 mol%), toluene (10 mL), 110 °C, reaction time 48 h. The isolated yield is shown; [a] in MeCN and 80 °C, after 48 h the yield was 52%.
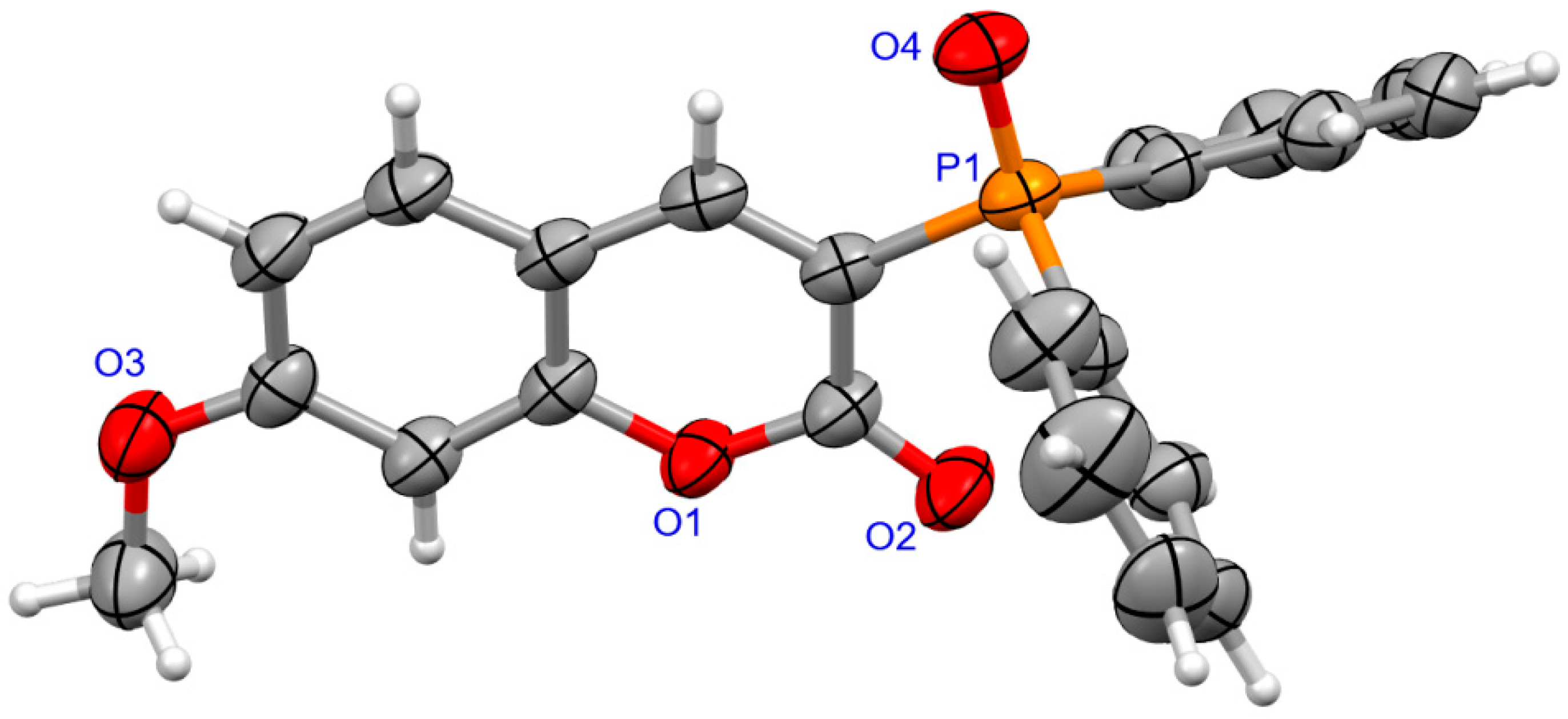
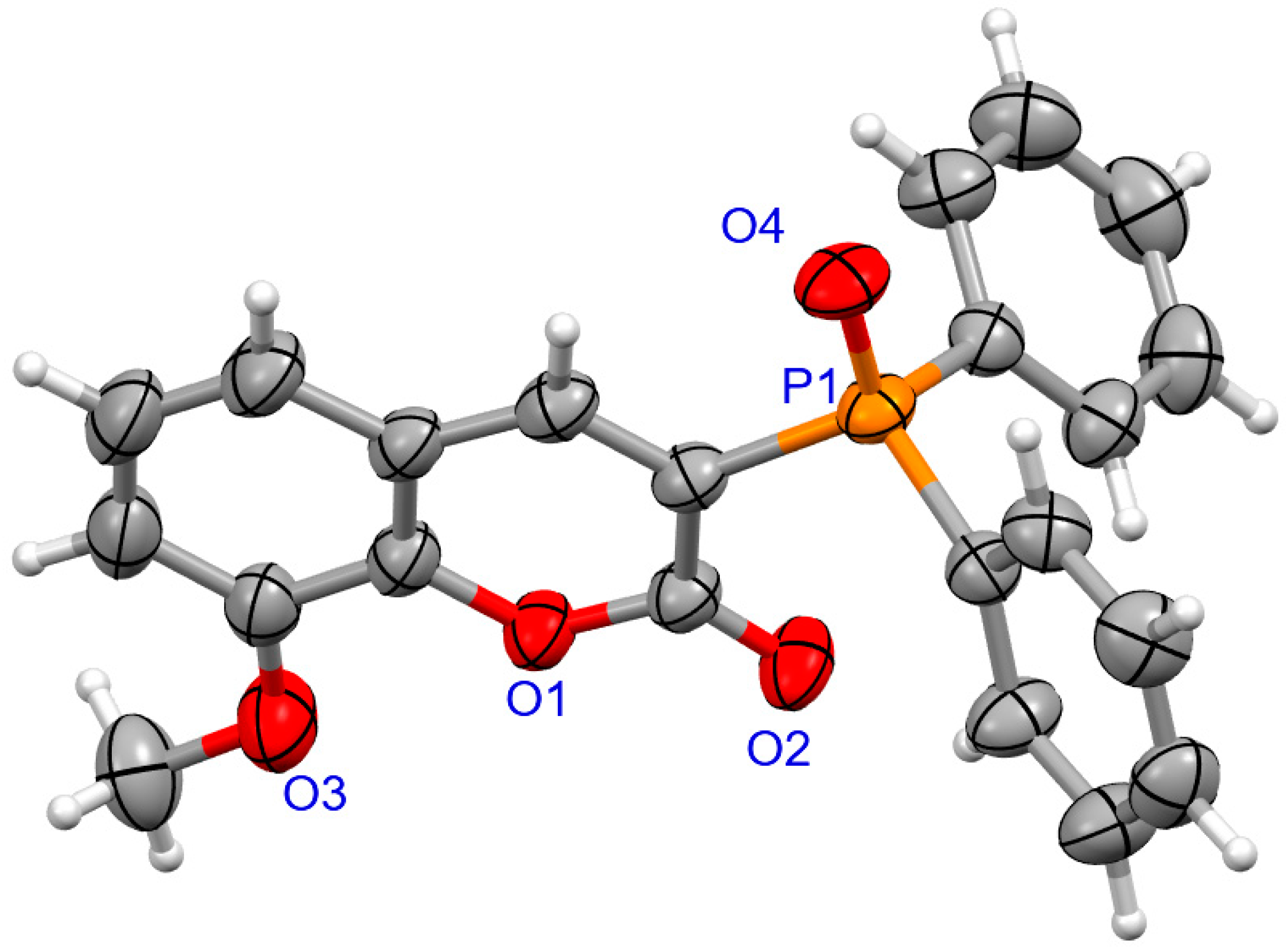
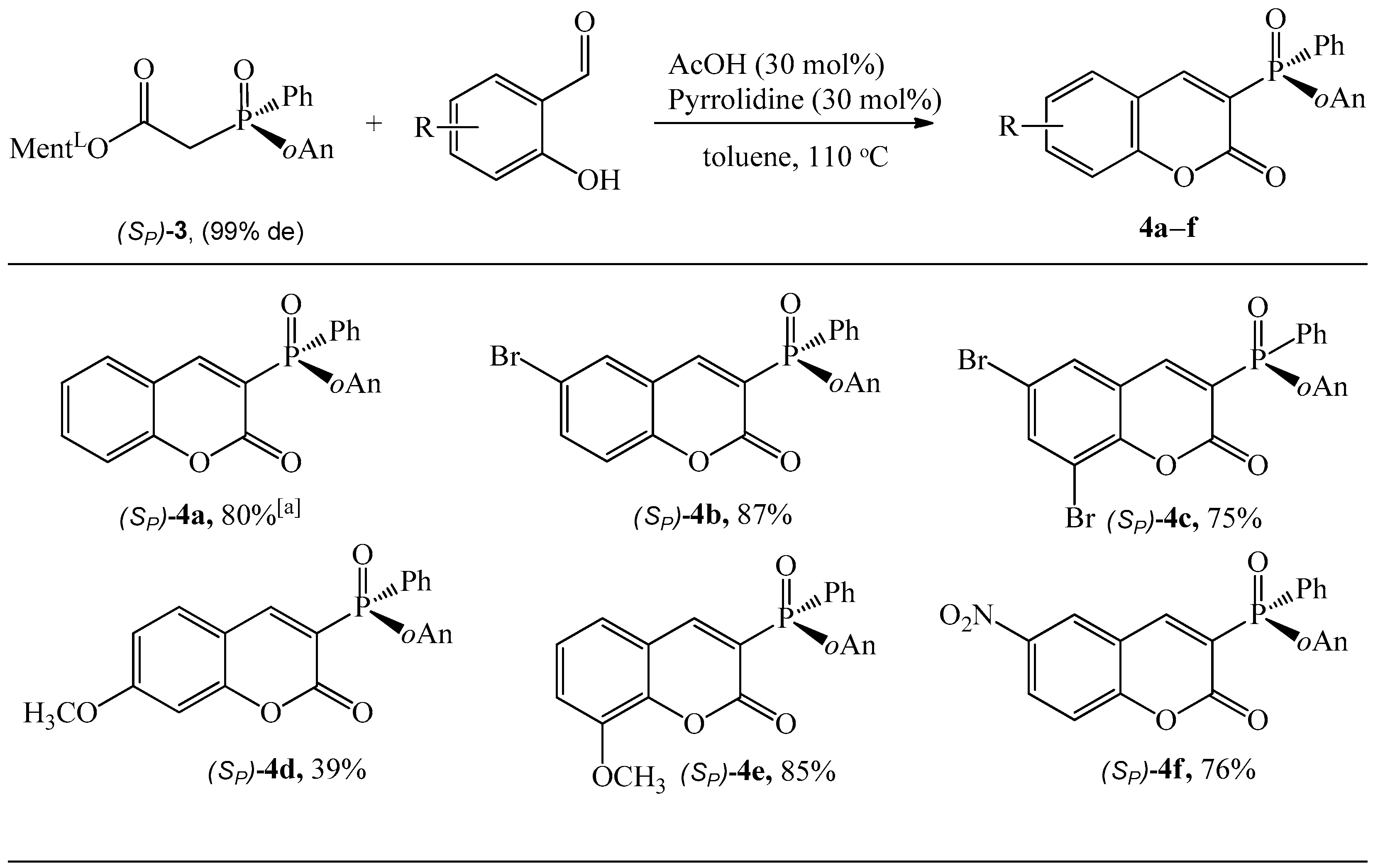
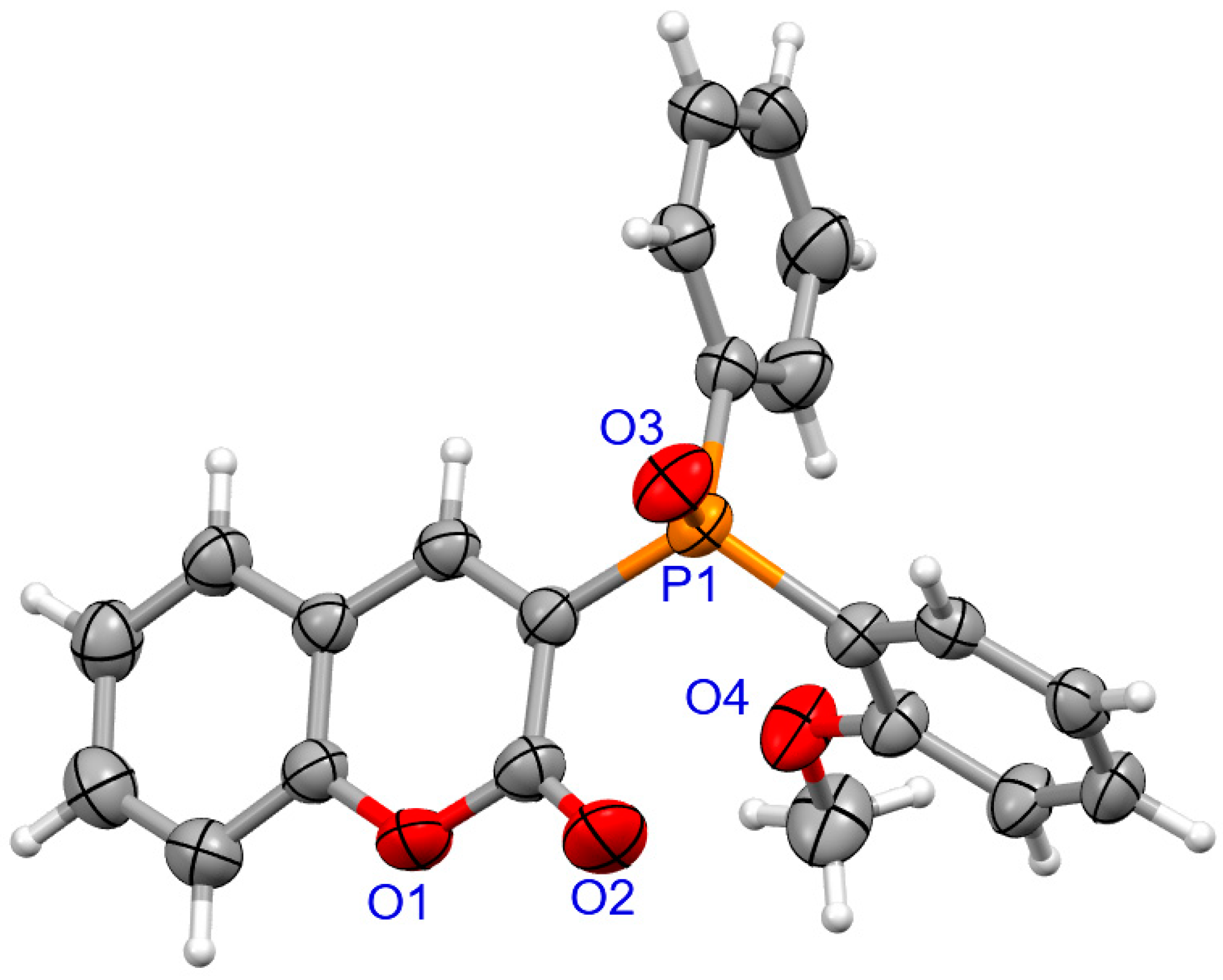
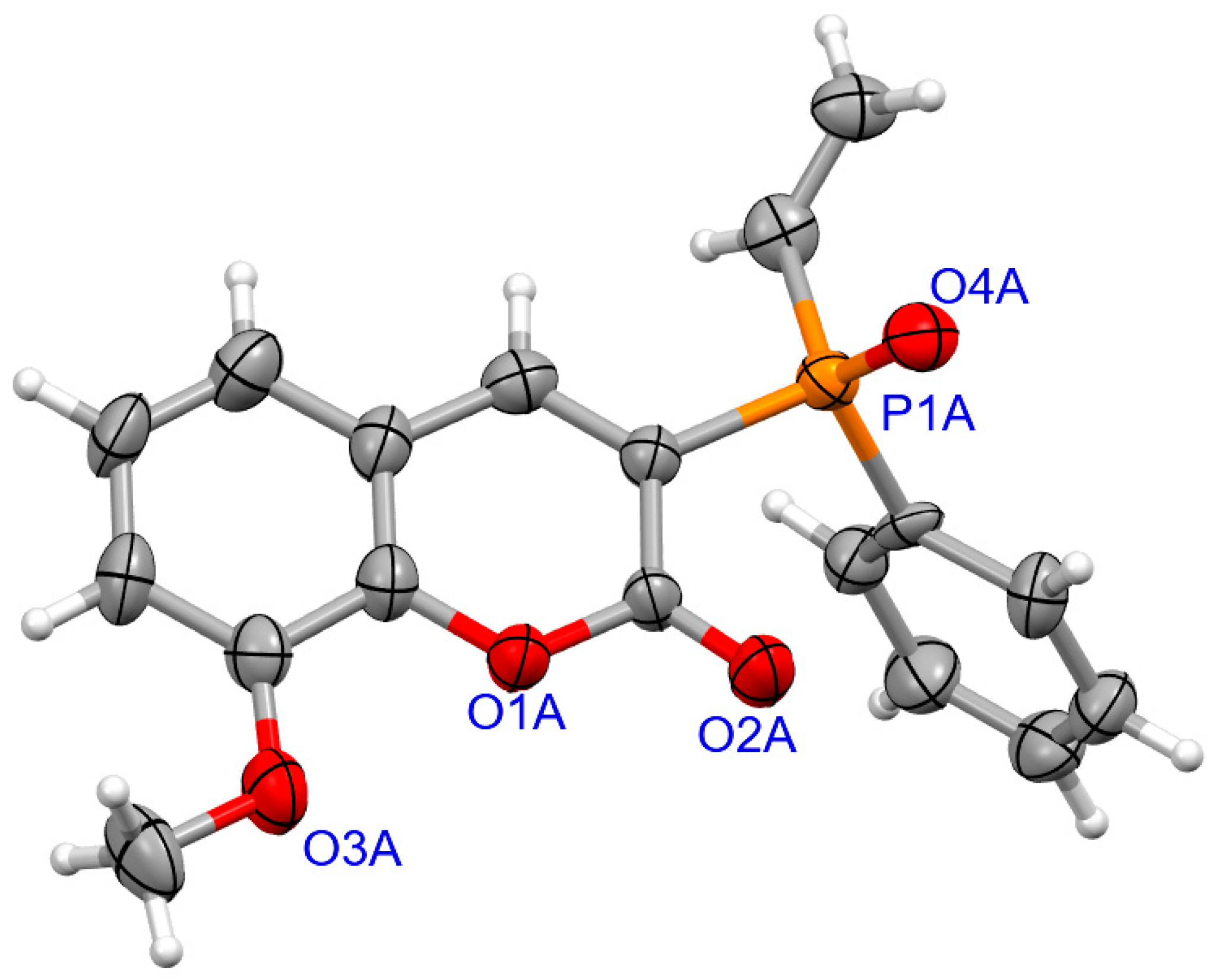
The determination of molecular structure and assignment of configuration for (SP)-4a and (SP)-4d was performed using an X-ray crystallography (Figure 3 and Figure 4). For the presented reactions, the configuration of the phosphorus atom has not changed.
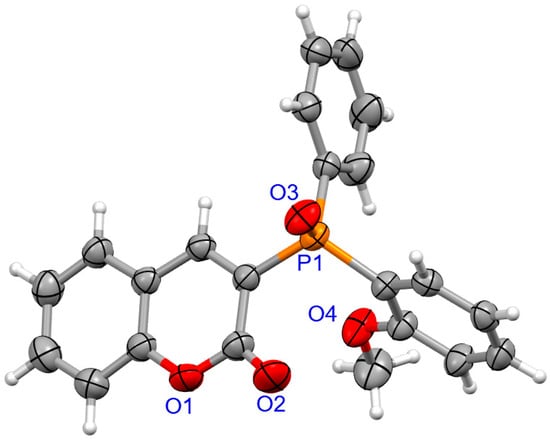
Figure 3.
The molecular structure of (SP)-4a.
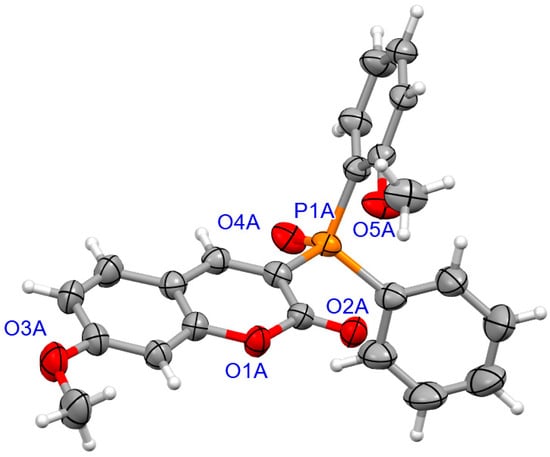
Figure 4.
The molecular structure of (SP)-4d.
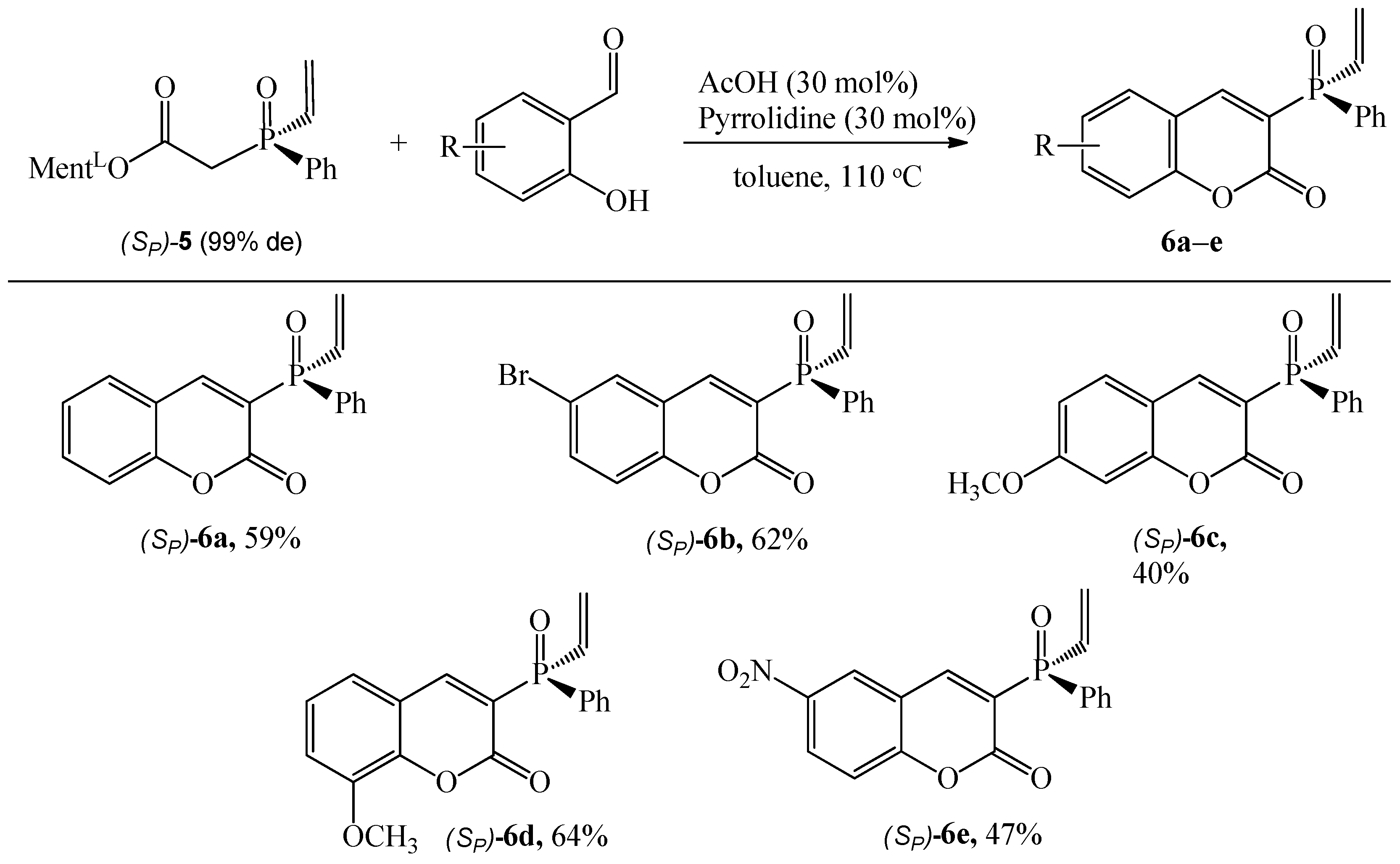
Finally, from the reaction of diastereomerically pure L-menthyl 2-(phenyl(vinyl)phosphinyl)acetate (SP)-5 with salicylaldehydes, the coumarins (SP)-6 were isolated with a satisfactory yield (40–64%), Scheme 5. However, the reactions required extended reaction times of up to 48 h. The result showed that 3-methoxysalicylaldehyde as a substrate is more suitable for this process than 4-methoxysalicylaldehyde (yielding 64% and 40%, respectively).
Scheme 5.
Reaction of (Sp)-5 with substituted salicylaldehydes. Reaction conditions: (Sp)-5 (1.43 mmol), aldehyde (1.5 equiv.), pyrrolidine (30 mol%), AcOH (30 mol%), toluene (10 mL), 110 °C, reaction time 48 h. The isolated yield is shown.
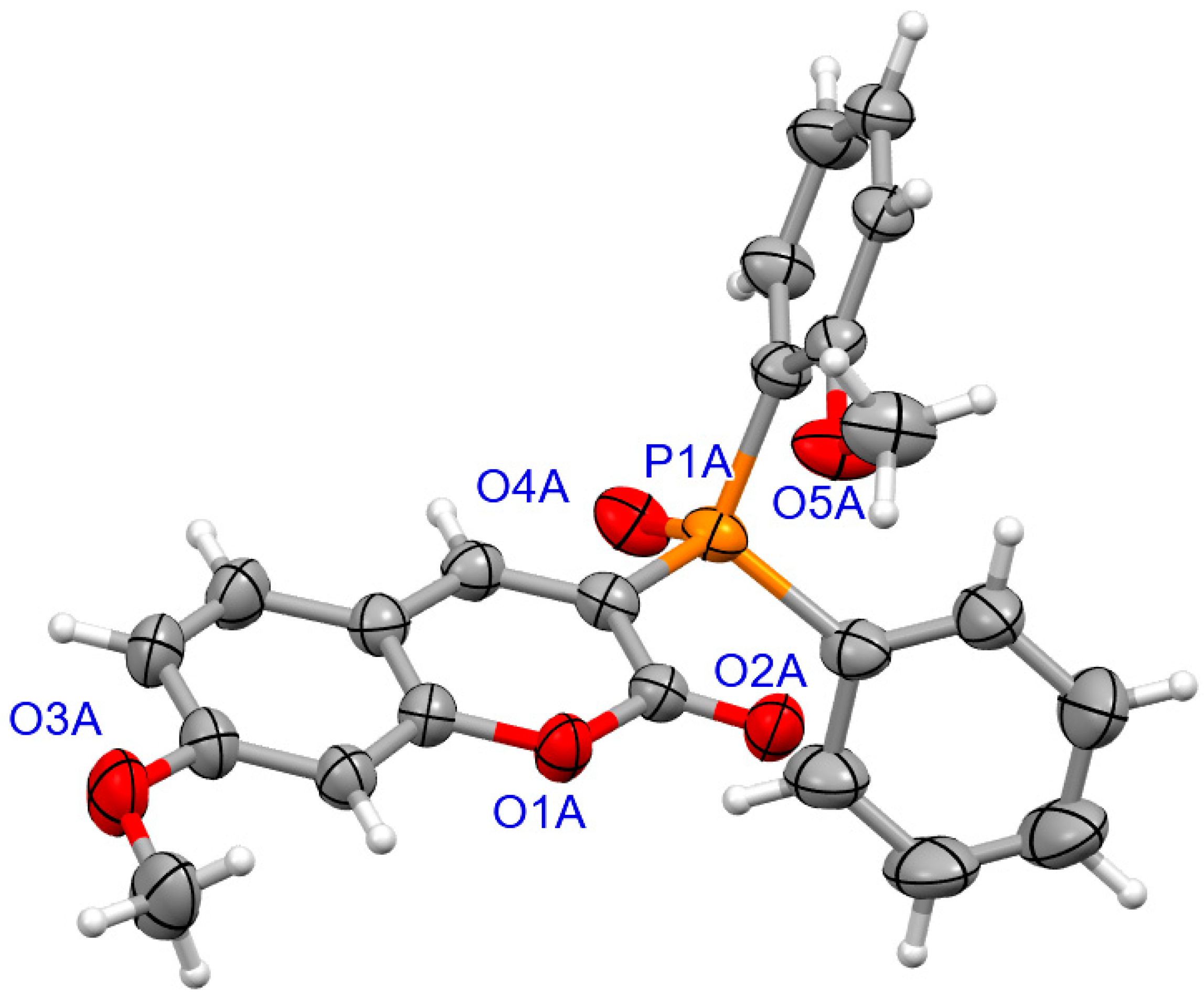
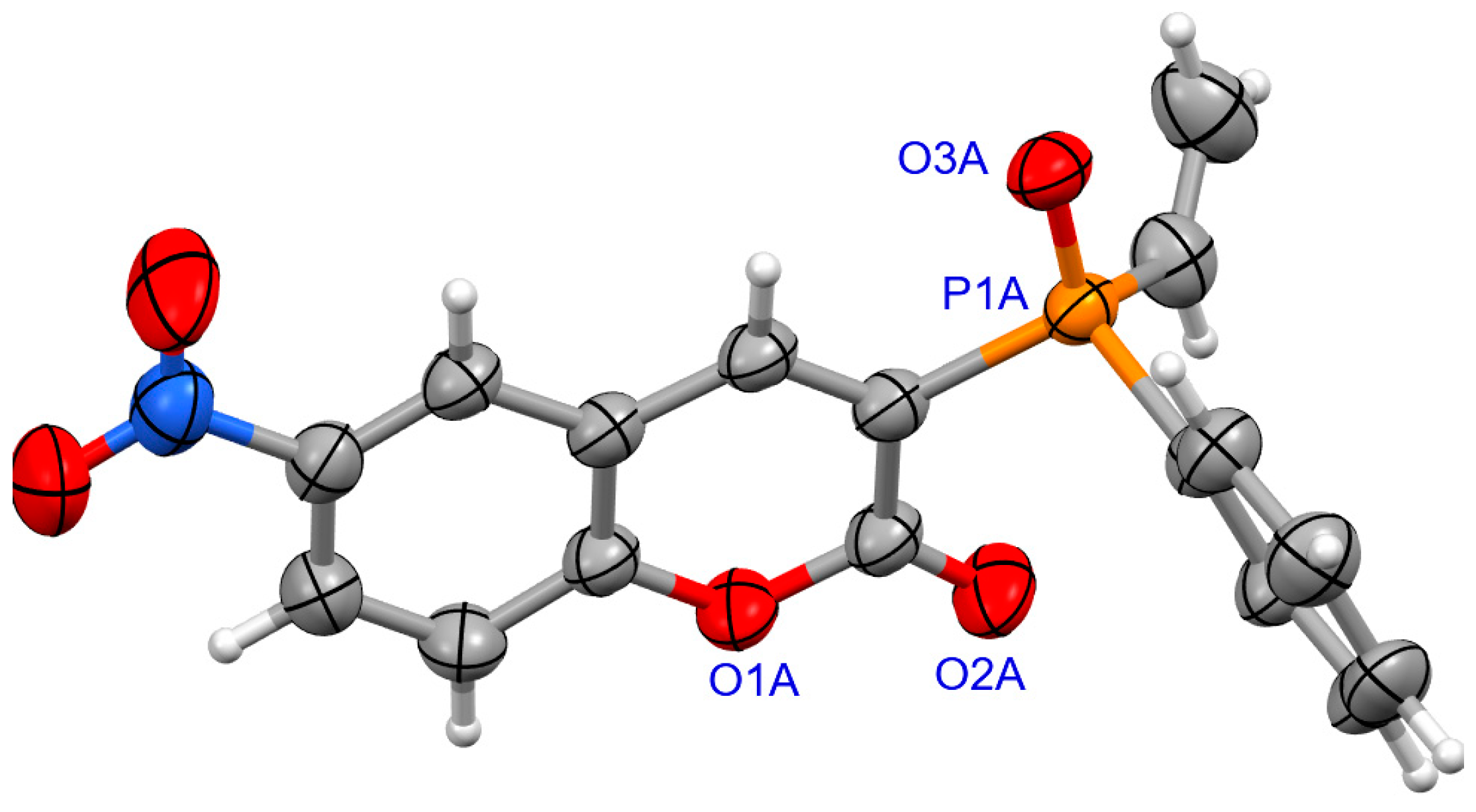
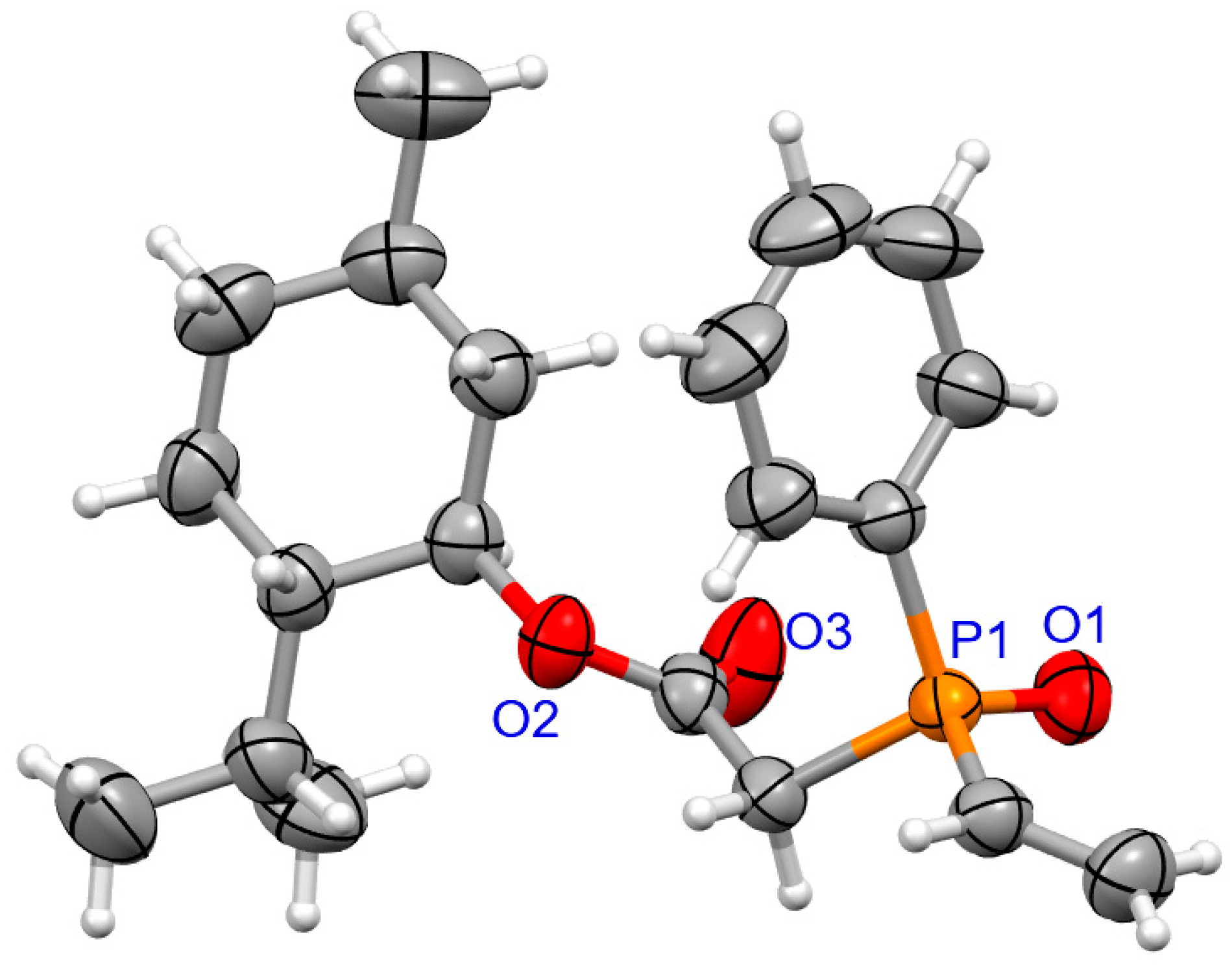
For coumarins 6d and 6e (Figure 5 and Figure 6), the absolute configuration at P was determined to be (Sp) which is in accord with the configuration of the starting substrate (Sp)-5 (Figure 7).
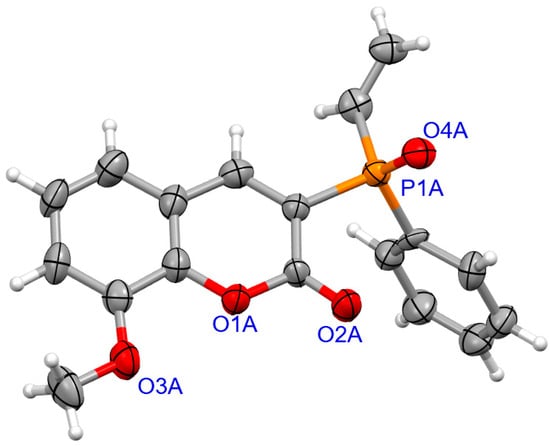
Figure 5.
The molecular structure of (SP)-6d.
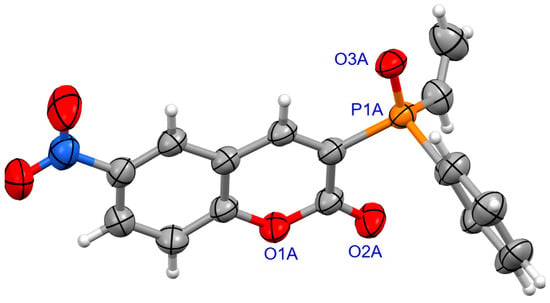
Figure 6.
The molecular structure of (SP)-6e.
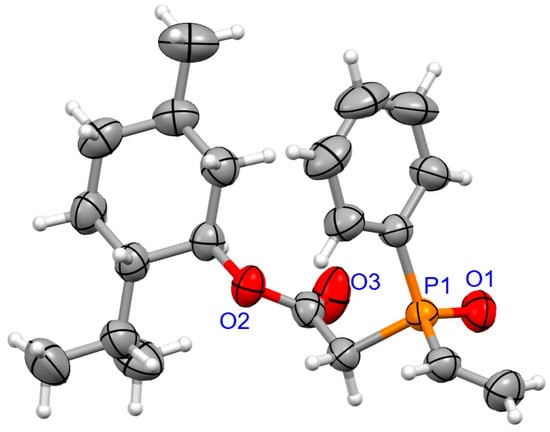
Figure 7.
The molecular structure of (SP)-5.
As we suspected, the presence of a vinyl substituent at the phosphorus atom significantly affected the conversion of the process. Even stronger substituent effects on reaction efficiencies were observed in the synthesis of chiral tert-butylphenylphosphinoyl coumarin 8a (Scheme 6). The reaction of ester (Rp)-7 with salicylaldehyde was carried out in toluene at 110 °C for 144 h. Probably due to the steric hindrance around the phosphorus, the conversion of the reaction was low (40%), and the pure product was isolated in 27% yield.
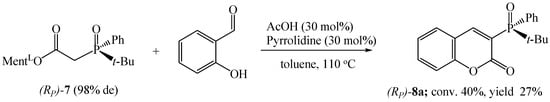
Scheme 6.
Synthesis of tert-butylphenylphosphinoyl coumarin 8a.
To summarize, it was found that phoshinylacetic acid L-menthyl esters can react with diverse 2-hydroxybenzaldehydes. The formation of 3-phosphinylated coumarins occurred in refluxing acetonitrile or toluene in the presence of pyrrolidine and acetic acid. With the developed protocol, structurally diverse phosphorus atom coumarins were smoothly obtained in high yields of up to 88%.
The structures of the isolated derivatives were supported by using elemental analysis, 1HNMR, 13C-NMR, and 31PNMR. The confirmation of structure of 2d and 2e, and assignment of configuration for the enantiopure (SP)-L-menthyl phenylvinylphosphinyl- acetate (SP)-5 as well as for the enantiopure coumarins (SP)-4a, (SP)-4d, (SP)-6d, (SP)-6e were obtained by means of an X-ray crystallography (see Supplementary Materials).
4. Conclusions
We found that the diastereomeric pure phoshinylacetic acid L-menthyl esters react with salicylaldehydes in the Knoevenagel condensation condition to yield 3-phosphinylated coumarins in one straightforward synthetic step. The salicylaldehydes bearing electron-donating groups and electron-withdrawing groups were efficiently coupled with esters to generate the corresponding 3-(diphenylphosphoryl)- 2H-chromen-2-one derivatives in moderate to very good yields. Importantly, these new enantiomerically pure compounds combine a coumarin backbone and a phosphinyl group with an asymmetric center at the phosphorus atom in a single molecule. With consideration of this, they could serve as potential chiral precursors for ligands or pharmaceuticals.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/sym16010073/s1. Supplementary material includes 1HNMR, 31PNMR, 13CNMR spectra of isolated compounds, and Table S1-llustration of molecular conformers.
Author Contributions
Conceptualization, K.F.D. and K.S.; investigation, K.F.D., K.S., S.F. and A.E.K.; methodology, K.F.D. and K.S.; writing—original draft, K.S., K.F.D., S.F. and A.E.K.; writing—review and editing, K.S., K.F.D., S.F. and A.E.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Crystallographic data for the structures presented in this paper have been deposited in the Cambridge Crystallographic Data Center as a Supplementary Publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Perkin, W.H. On the artificial production of coumarin and formation of its homologues. J. Chem Soc. 1868, 21, 53–63. [Google Scholar] [CrossRef]
- Patel, G.; Banerjee, S. Review on Synthesis of Bio-Active Coumarins Fused Heterocyclic Molecules. Curr. Org. Chem. 2020, 24, 2566–2587. [Google Scholar] [CrossRef]
- Todorov, L.; Saso, L.; Kostova, I. Antioxidant Activity of Coumarins and Their Metal Complexes. Pharmaceuticals 2023, 16, 651. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, V.; Villasana-Ruíz, A.P.; Garza-Veloz, I.; González-Delgado, S.; Martinez-Fierro, M.L. Therapeutic Effects of Coumarins with Different Substitution Patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef] [PubMed]
- Cubukcu, B.; Bray, D.H.; Warhurst, D.C.; Mericli, A.H.; Ozhatay, N.; Sariyar, G. In vitro antimarlarial activity of crude extracts and compounds from Artemisia abrotanum L. Phytother. Res. 1990, 4, 203–204. [Google Scholar] [CrossRef]
- Song, X.F.; Fan, J.; Liou, L.; Liu, X.F.; Gao, F. Coumarin derivatives with anticancer activities: An update. Arch. Pharm. 2020, 353, 2000025. [Google Scholar] [CrossRef]
- Stasi, L.C. Natural Coumarin Derivatives Activating Nrf2 Signaling Pathway as Lead Compounds for the Design and Synthesis of Intestinal Anti-Inflammatory Drugs. Pharmaceuticals 2023, 16, 511. [Google Scholar] [CrossRef]
- Berzina, L.; Mierina, I. Antiradical and Antioxidant Activity of Compounds Containing 1,3-Dicarbonyl Moiety: An Overview. Molecules 2023, 28, 6203. [Google Scholar] [CrossRef]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A Mini-Review: Recent Advances in Coumarin-Metal Complexes With Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Li, G.; Liu, Z.; Ma, J.; Liu, M.; Li, D.; Han, J.; Wang, B. Synthesis and Evaluation of Bi-functional 7-hydroxycoumarin Platinum(IV) Complexes as Antitumor Agents. Bioorg. Med. Chem. 2019, 27, 2112–2121. [Google Scholar] [CrossRef]
- Sarkar, T.; Kumar, A.; Sahoo, S.; Hussain, A. Mixed-ligand Cobalt(III) Complexes of a Naturally Occurring Coumarin and Phenanthroline Bases as Mitochondria-Targeted Dual-Purpose Photochemotherapeutics. Inorg. Chem. 2021, 60, 6649–6662. [Google Scholar] [CrossRef] [PubMed]
- Yernale, N.G.; Bennikallu Hire Mathada, M. Preparation of Octahedral Cu(II), Co(II), Ni(II) and Zn(II) Complexes Derived from 8- Formyl-7-Hydroxy-4-Methylcoumarin: Synthesis, Characterization and Biological Study. J. Mol. Struct. 2020, 1220, 128659. [Google Scholar] [CrossRef]
- Stojanović, M.; Flores-Diaz, N.; Ren, Y.; Vlachopoulos, N.; Pfeifer, L.; Shen, Z.; Liu, Y.; Zakeeruddin, S.M.; Milić, J.V.; Hagfeldt, A. The Rise of Dye-Sensitized Solar Cells: From Molecular Photovoltaics to Emerging Solid-State Photovoltaic Technologies. Helv. Chim. Acta 2021, 104, e2000230. [Google Scholar] [CrossRef]
- Kumar, A.; Baccoli, R.; Fais, A.; Cincotti, A.; Pilia, L.; Gatto, G. Substitution Effects on the Optoelectronic Properties of Coumarin Derivatives. Appl. Sci. 2019, 10, 144. [Google Scholar] [CrossRef]
- Safavi-Mirmahalleh, S.A.; Golshan, M.; Gheitarani, B.; Hosseini, M.S.; Salami-Kalajahi, M. A review on applications of coumarin and its derivatives in preparation of photo-responsive polymers. Eur. Polym. J. 2023, 199, 112430. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Sun, J.; Wang, X. Synthesis and application of coumarin fluorescence probes. RSC Adv. 2020, 10, 10826–10847. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Liu, Z.; Verwilst, P.; Koo, S.; Jangjili, P.; Kim, J.S.; Lin, W. Coumarin-Based Small-Molecule Fluorescent Chemosensors. Chem. Rev. 2019, 119, 10403–10519. [Google Scholar] [CrossRef]
- Szwaczko, K. Fluorescent Coumarin-based Probe for Detection of Biological Thiols. Curr. Org. Chem. 2023, 15, 1329–1335. [Google Scholar] [CrossRef]
- Yu, H.; Yang, H.; Shi, E.; Tang, W. Development and Clinical Application of Phosphorus-Containing Drugs. Med. Drug Discov. 2020, 8, 100063. [Google Scholar] [CrossRef]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948. [Google Scholar] [CrossRef]
- Val’dman, A.; Kozlovskaia, M.M.; Zaikonnikova, I.V.; Bravkov, M.F.; Rzhevskaia, G.F. Vliianie gidifena na povedenie i gemodinamicheskie proiavleniia émotsional’no-stressovoĭ reaktsii [Effect of gidifen on behavior and hemodynamic signs of an emotionally stressful reaction]. Biull. Eksp. Biol. Med. 1980, 89, 310–312. [Google Scholar] [PubMed]
- Nicholson, A.; Wright, C. Activity of fosazepam, a soluble analogue of diazepam. Br. J. Clin. Pharmacol. 1977, 4, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Stambirskyi, M.V.; Kostiuk, T.; Sirobaba, S.I.; Rudnichenko, A.; Titikaiev, D.L.; Dmytriv, Y.V.; Kuznietsova, H.; Pishel, I.; Borysko, P.; Mykhailiuk, P.K. Phosphine Oxides (-POMe2) for Medicinal Chemistry: Synthesis, Properties, and Applications. J. Org. Chem. 2021, 86, 12783–12801. [Google Scholar] [CrossRef] [PubMed]
- Szwaczko, K. Coumarins Synthesis and Transformation via C–H Bond Activation—A Review. Inorganics 2022, 10, 23. [Google Scholar] [CrossRef]
- Mi, X.; Huang, M.; Zhang, J.; Wang, C.; Wu, Y. Regioselective palladium-catalyzed phosphonation of coumarins with dialkyl H-phosphonates via C-H functionalization. Org. Lett. 2013, 15, 6266–6269. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Yuan, J.; Xiao, Y.; Mao, P. Catalytic activity of chelating N-heterocyclic carbene palladium complexes towards selective phosphorylation of coumarins. J. Organomet. Chem. 2016, 818, 179. [Google Scholar] [CrossRef]
- Yuan, J.; Li, Y.; Yang, L.; Mai, W.; Mao, P.; Xiao, Y.; Qu, L. Silver-catalyzed direct Csp2-H radical phosphorylation of coumarins with H-phosphites. Tetrahedron 2015, 71, 8178–8186. [Google Scholar] [CrossRef]
- Mi, X.; Wang, C.; Huang, M.; Zhang, J.; Wu, Y.; Wu, Y. Silver-Catalyzed Synthesis of 3-Phosphorated Coumarins via Radical Cyclization of Alkynoates and Dialkyl H-Phosphonates. Org. Lett. 2014, 12, 3356–3359. [Google Scholar] [CrossRef]
- Zhou, P.; Jiang, Y.J.; Zou, J.P.; Zhang, W. Manganese(III) Acetate Mediated Free-Radical Phosphonylation of Flavones and Coumarins. Synthesis 2012, 7, 1043. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, X.; Li, Y.; Huang, M.; Kim, J.K.; Wu, Y. Regioselective phosphinylation of coumarins under green LED irradiation and its mechanism. Org. Biomol. Chem. 2017, 15, 9775. [Google Scholar] [CrossRef]
- Liu, D.; Chen, J.; Wang, X.; Xu, P. Metal-Free, Visible-Light-Promoted Synthesis of 3-Phosphorylated Coumarins via Radical C−P/C−C Bond Formation. ASC Commun. 2017, 359, 2773–2777. [Google Scholar] [CrossRef]
- Grinenko, V.; Khrizanforov, M.; Strekalova, S.; Khrizanforova, V.; Kholin, K.; Gryaznova, T.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Kadirov, M.; Gryaznova, T.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Robinson, C.N.; Addison, J.F. Condensation of Triethyl Phosphonoacetate with Aromatic Aldehydes. J. Org. Chem. 1966, 31, 4325–4326. [Google Scholar] [CrossRef]
- Bouyssou, P.; Chenault, J. Phosphonates and phosphine oxides as reagents in a one-pot synthesis of coumarins. Tetrahedron Lett. 1991, 32, 5341–5344. [Google Scholar] [CrossRef]
- Bojilova, A.; Nikolova, R.; Ivanov, C.; Rodios, N.; Terzis, A.; Raptopoulou, C. A comparative study of the interaction of salicylaldehydes with phosphonoacetates under Knoevenagel reaction conditions. Synthesis of 1,2-benzoxaphosphorines and their dimers. Tetrahedron 1996, 52, 12597–12612. [Google Scholar] [CrossRef]
- Janecki, T.; Albrecht, A.; Koszuk, J.; Mondranka, J.; Slowak, D. A simple and effective synthesis of activated vinylphosphonates from 3-methoxy-2-diethoxyphosphorylacrylate. Tetrahedron Lett. 2010, 51, 2274–2276. [Google Scholar] [CrossRef]
- Mondranka, J.; Albrecht, A.; Janecki, T. A Convenient Entry to 3-Methylidenechroman-2-ones and 2-Methylidene- dihydrobenzochromen-3-ones. Synlett 2010, 19, 2867–2870. [Google Scholar] [CrossRef]
- SuperNova Diffractometer (Oxford Diffraction); Agilent Technologies Inc.: Yarnton, UK, 2014.
- CrysAlisPro, 1.171.42.79a; Rigaku Oxford Diffraction: Tokyo, Japan, 2022.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Kim, I.; Min, M.; Kang, D.; Kim, K.; Hong, S. Direct Phosphonation of Quinolinones and Coumarins Driven by the Photochemical Activity of Substrates and Products. Org. Lett. 2017, 19, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Lubańska, M.; Dziuba, K.F.; Pietrusiewicz, K.M. Enantiodivergent Synthesis of Both PAMPO Enantiomers Using L-Menthyl Chloroacetate and Stereomutation at P in Classical Quaternisation Reactions. Synthesis 2020, 52, 909–916. [Google Scholar] [CrossRef]
- Bodalski, R.; Rutkowska-Olma, E.; Pietrusiewicz, K.M. Optically active Phosphine oxides: Synthesis and absolute configuration of (menthoxycarbonylmethyl) phenylvinyl phosphine oxide. Tetrahedron 1980, 36, 2353–2355. [Google Scholar] [CrossRef]
- Cheng, X.; Horton, P.N.; Hursthouse, M.B.; Hii, K.K. Aminohydroxy phosphine oxide ligands in ruthenium catalysed asymmetric transfer hydrogenation reactions. Tetrahedron Asymmetry 2004, 15, 2141–2246. [Google Scholar] [CrossRef]
- Dziuba, K.; Szwaczko, K.; Frynas, S. Knoevenagel Condensation of Phosphinoylacetic Acids with Aldehydes: An Efficient One-Pot Strategy for the Synthesis of P-Functionalized Alkenyl Compounds. Synthesis 2021, 53, 2142–2154. [Google Scholar] [CrossRef]
- Koleva, A.I.; Petkova-Yankova, N.I.; Nikolova, R.D. Synthesis and Chemical Properties of 3-Phosphono-coumarins and 1,2-Benzoxaphosphorins as Precursors for Bioactive Compounds. Molecules 2019, 24, 2030. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).