
Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes



Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. General Considerations
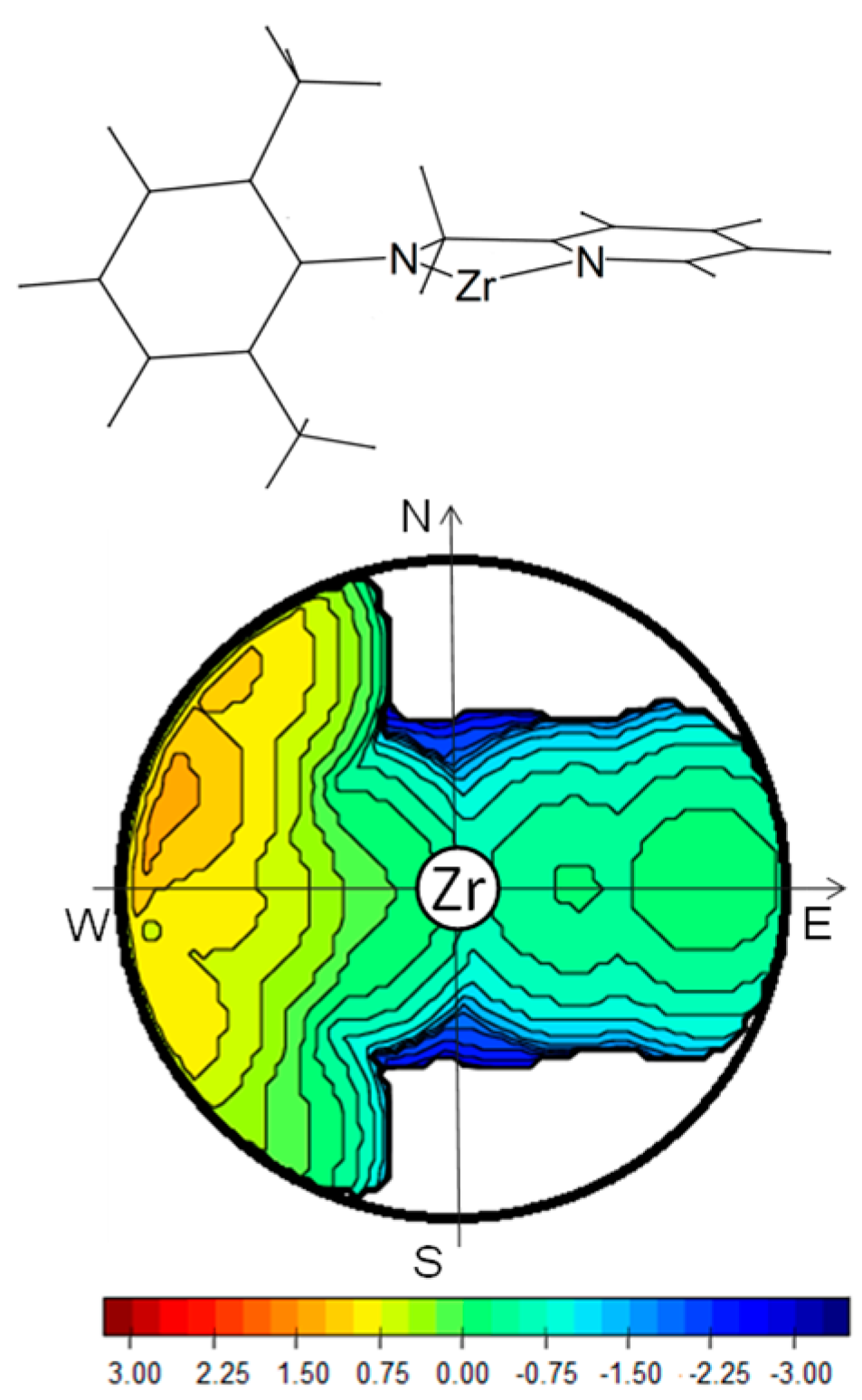
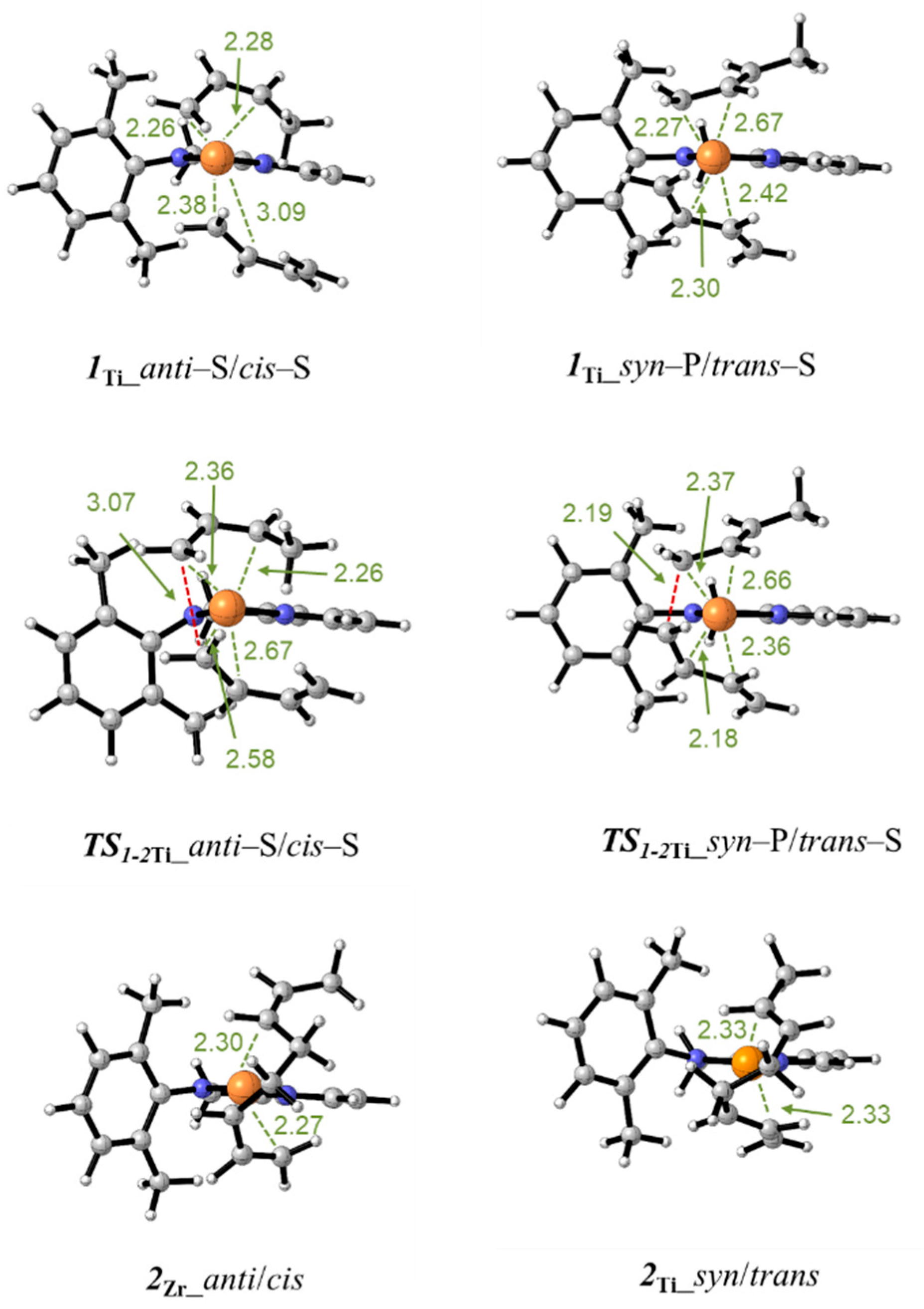
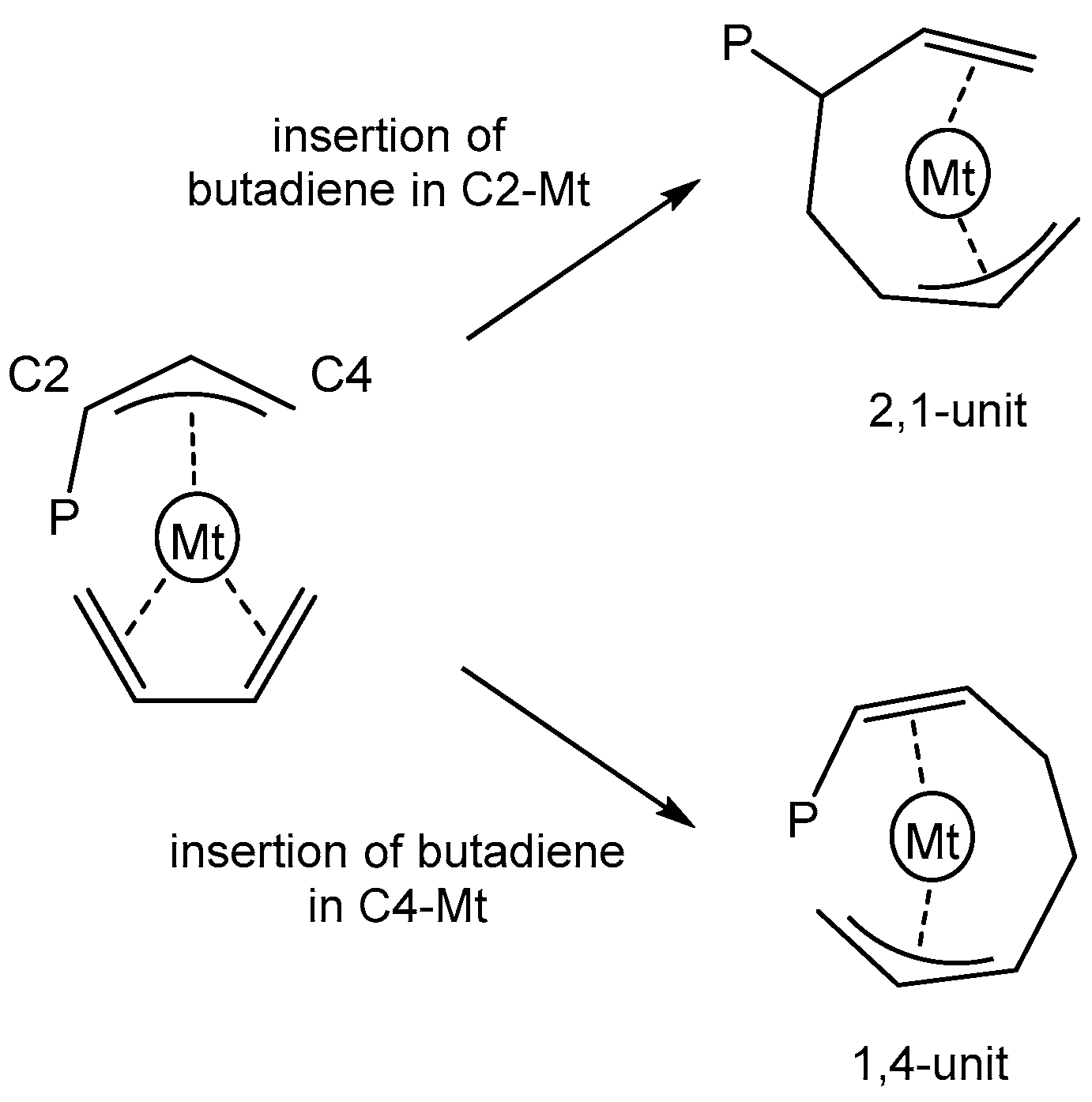
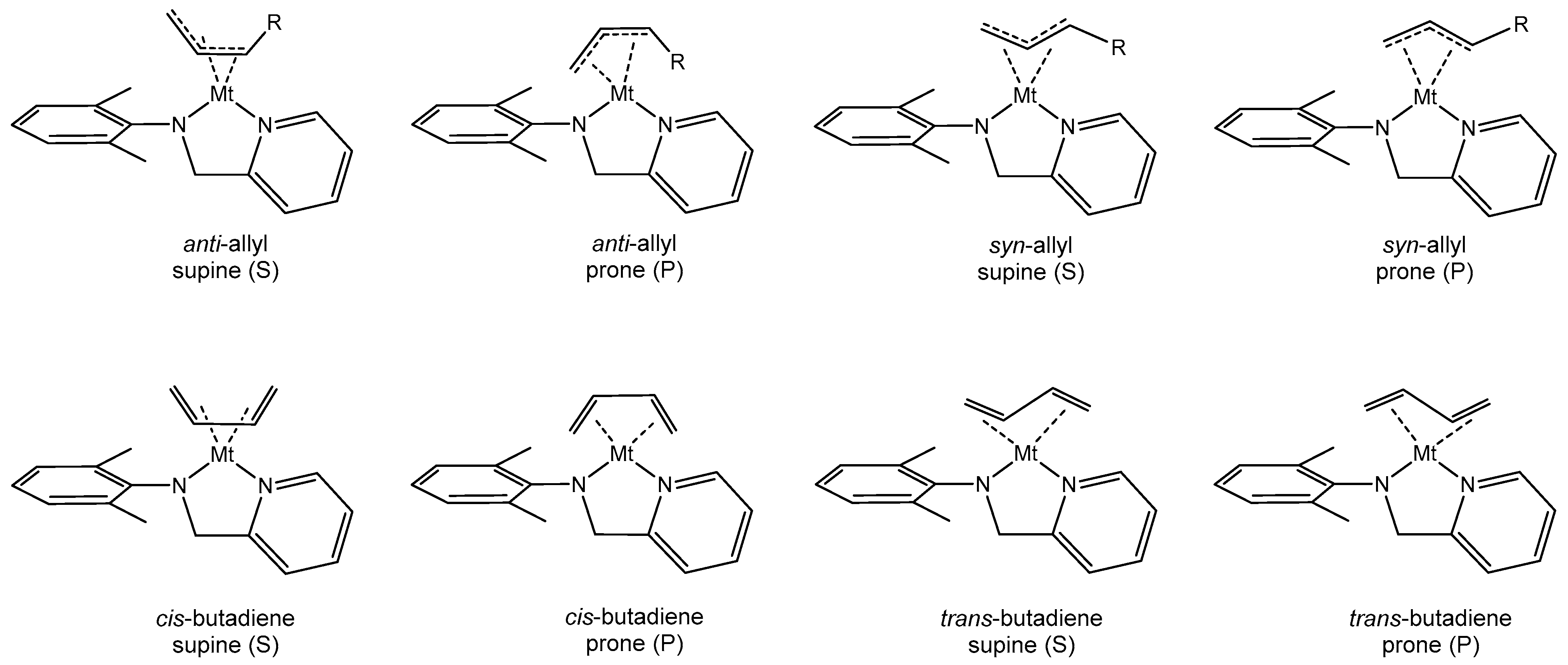
3.2. Model of the Active Species
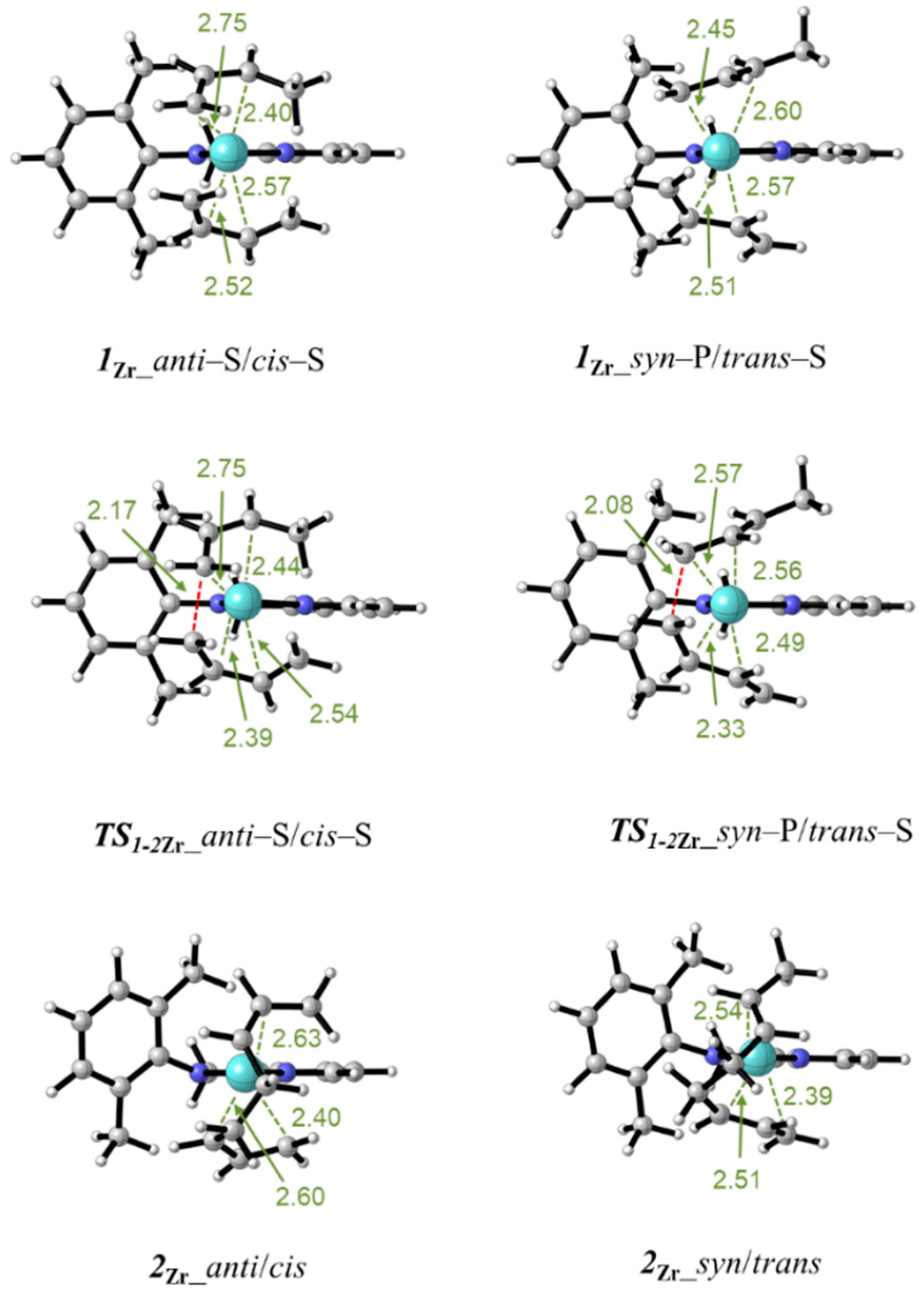
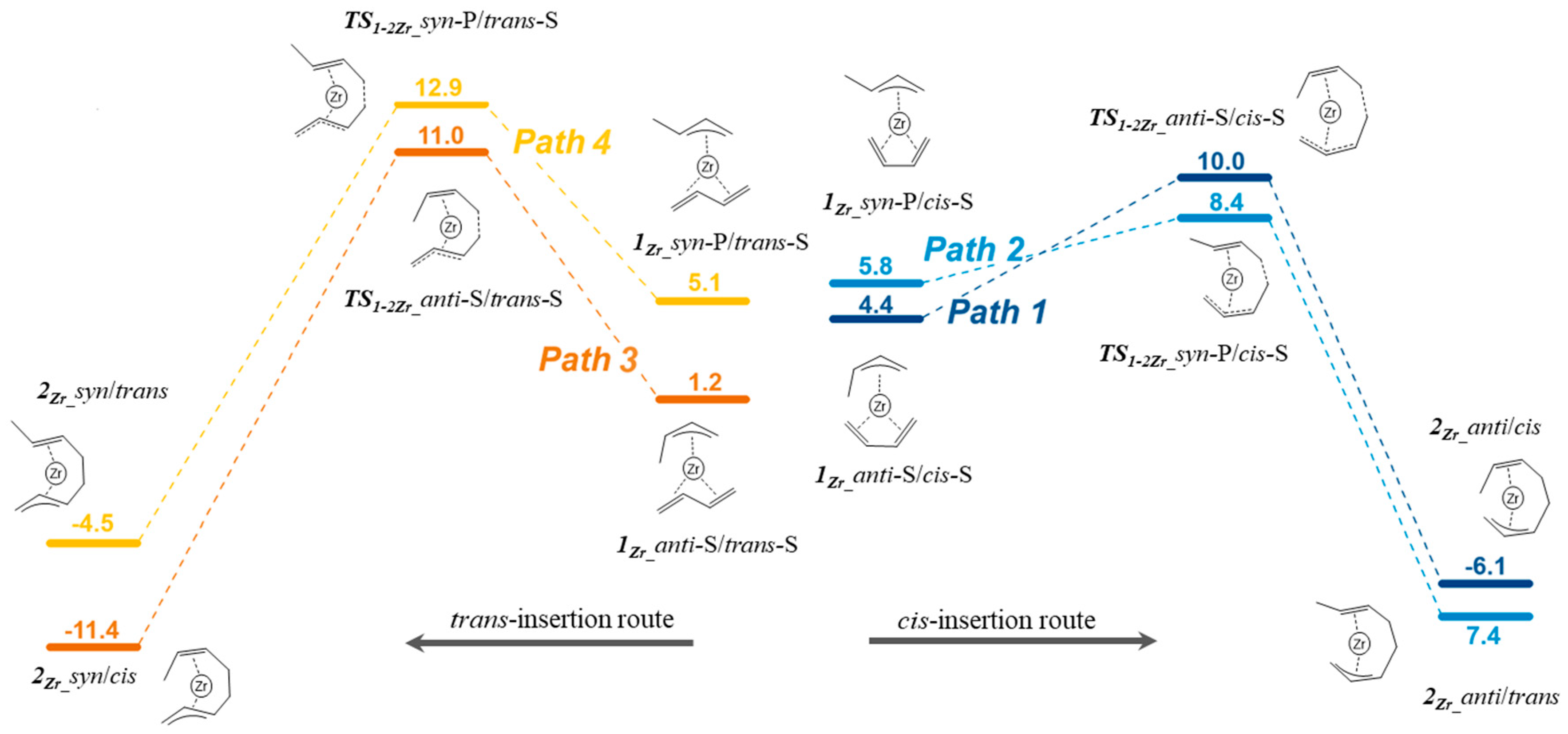
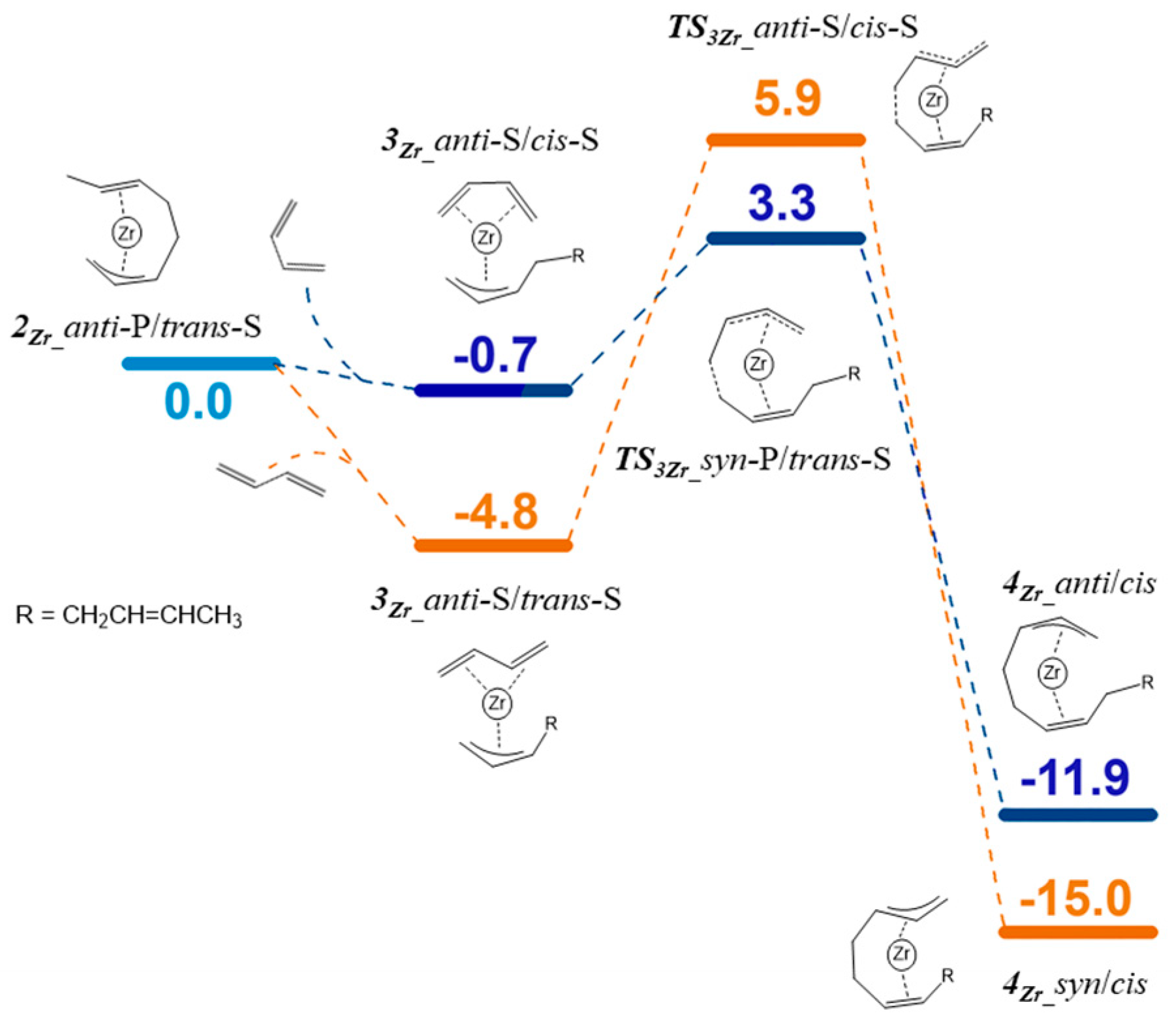
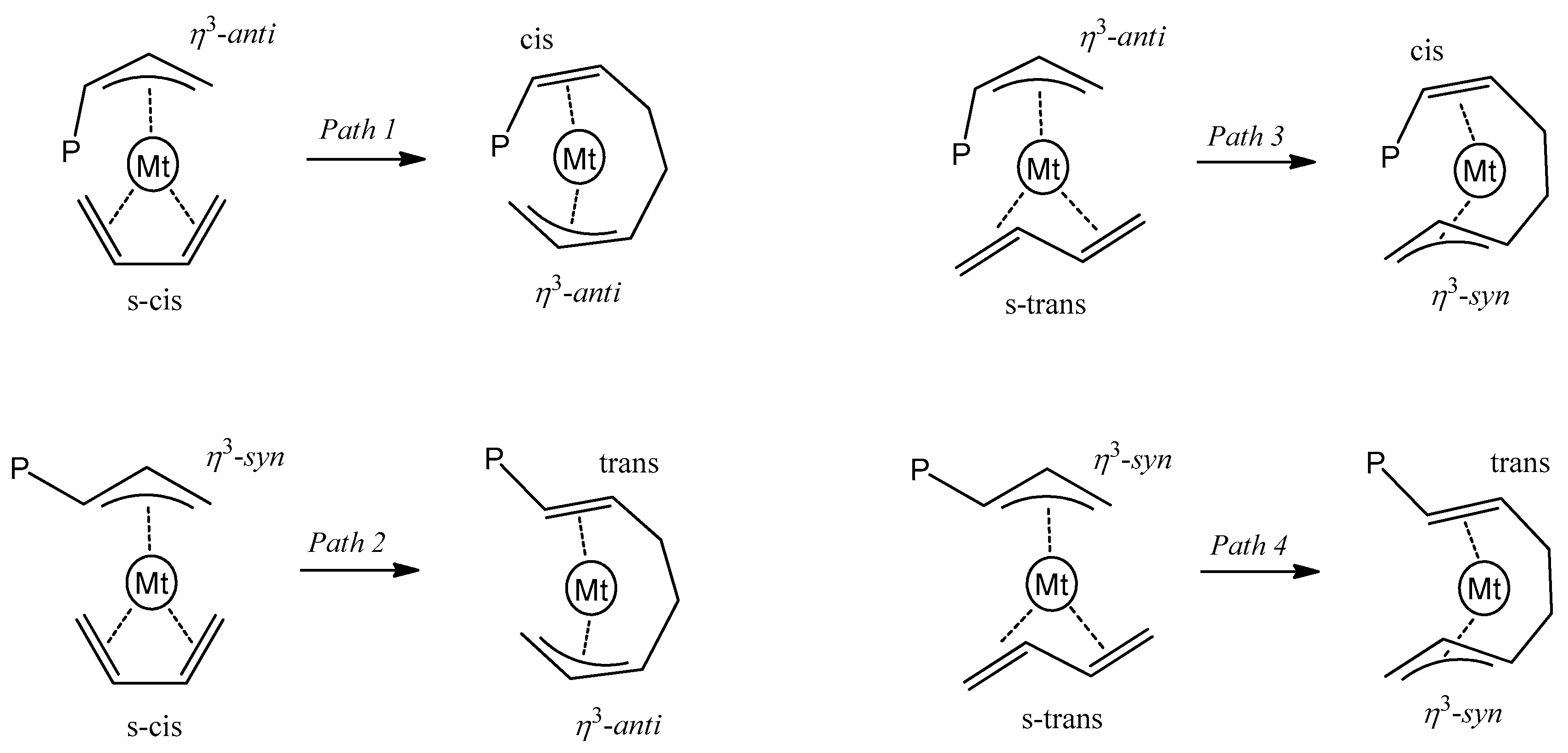
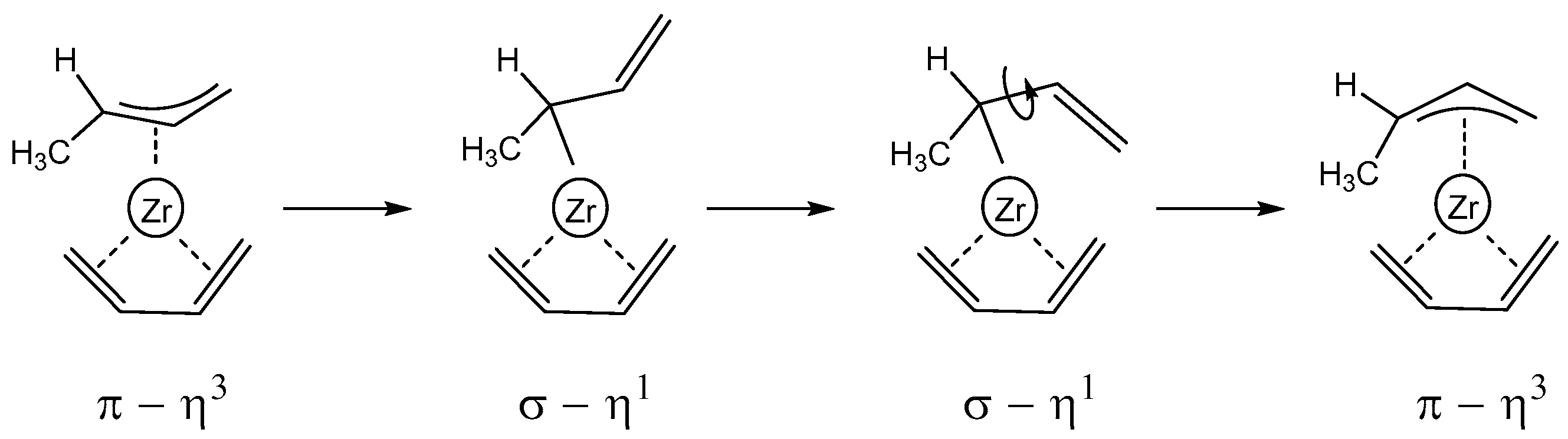
3.3. Polymerization Paths Promoted by Using the Zirconium-Based Catalyst
3.4. Polymerization Paths Promoted by Using the Titanium-Based Catalyst
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, A.; Mohanty, S.; Gupta, V.R. Butadiene Rubber: Synthesis, Microstructure, and Role of Catalysts. Rubber Chem. Technol. 2021, 94, 393–409. [Google Scholar] [CrossRef]
- Porri, L.; Giarrusso, A. Conjugated Diene Polymerization. In Comprehensive Polymer Science; Eastmon, G.C., Ledwith, A., Russo, S., Sigwalt, P., Eds.; Elsevier: Oxford, UK, 1989; Volume 4, pp. 53–108. [Google Scholar]
- Takeuchi, D. Stereoselective polymerization of conjugated dienes. In Encyclopedia of Polymer Science and Technology, 4th ed.; Mark, H.F., Ed.; Wiley: New York, NY, USA, 2014; Volume 13, pp. 126–150. [Google Scholar]
- Ricci, G.; Leone, G. Polymerization of 1,3-butadiene with organometallic complexes-based catalysts. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, 3rd ed.; Cornils, B., Hermann, W.A., Beller, M., Paciello, R., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2017; Volume 1, p. 251. [Google Scholar]
- Ricci, G.; Pampaloni, G.; Sommazzi, A.; Masi, F. Dienes Polymerization: Where We Are and What Lies Ahead. Macromolecules 2021, 54, 5879–5914. [Google Scholar] [CrossRef]
- Ricci, G.; Leone, G. Recent advances in the polymerization of butadiene over the last decade. Polyolefins J. 2014, 1, 43–60. [Google Scholar]
- Pino, P.; Giannini, U.; Porri, L. Insertion Polymerization. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bikales, N.M., Overberger, C.G., Menges, G., Eds.; Wiley: New York, NY, USA, 1987; Volume 8, pp. 147–220. [Google Scholar]
- Porri, L.; Giarrusso, A.; Ricci, G. On the Mechanism of Formation of Isotactic and Syndiotactic Polydiolefins. Macromol. Symp. 2002, 178, 55–68. [Google Scholar] [CrossRef]
- Porri, L.; Ricci, G.; Shubin, N. Polymerization of 1,3-dienes with neodymium catalysts. Macromol. Symp. 1998, 128, 53–61. [Google Scholar] [CrossRef]
- Porri, L.; Giarrusso, A.; Ricci, G. Recent views on the mechanism of diolefin polymerization with transition metal initiator systems. Prog. Polym. Sci. 1991, 16, 405–441. [Google Scholar] [CrossRef]
- Pragliola, S.; Botta, A.; Longo, P. Solvent effect in 1,3-butadiene polymerization by cyclopentadienyl titanium trichloride (CpTiCl3)/methylaluminoxane (MAO) and pentamethylcyclopentadienyl titanium trichloride (Cp*TiCl3)/MAO catalysts. Eur. Polym. J. 2019, 111, 20–27. [Google Scholar] [CrossRef]
- Pragliola, S.; Costabile, C.; Venditto, V. Ethylene/1,3-butadiene cyclocopolymerization catalyzed by zirconocene systems. Eur. Polym. J. 2014, 58, 157–163. [Google Scholar] [CrossRef]
- Nath, D.C.D.; Shiono, T.; Ikeda, T. Copolymerization of 1,3-butadiene and isoprene with cobalt dichloride/methylaluminoxane in the presence of triphenylphosphine. J. Polym. Sci. A Polym. Chem. 2002, 40, 3086–3092. [Google Scholar] [CrossRef]
- Kaita, S.; Takeguchi, Y.; Hou, Z.; Nishiura, M.; Doi, Y.; Wakatsuki, Y. Pronounced Enhancement Brought in by Substituents on the Cyclopentadienyl Ligand: Catalyst System (C5Me4R)2Sm(THF)x/MMAO (R = Et, iPr, nBu, TMS; MMAO = Modified Methylaluminoxane) for 1,4-Cis Stereospecific Polymerization of 1,3-Butadiene in Cyclohexane Solvent. Macromolecules 2003, 36, 7923–7926. [Google Scholar]
- Maiwald, S.; Sommer, C.; Müller, G.; Taube, R. Highly Active Single-Site Catalysts for the 1,4-cis Polymerization of Butadiene from Allylneodymium(III) Chlorides and Trialkylaluminiums–A Contribution to the Activation of Tris(allyl)neodymium(III) and the Further Elucidation of the Structure-Activity Relationship. Macromol. Chem. Phys. 2002, 203, 1029–1039. [Google Scholar]
- Milione, S.; Cuomo, C.; Capacchione, C.; Zannoni, C.; Grassi, A.; Proto, A. Stereoselective Polymerization of Conjugated Dienes and Styrene−Butadiene Copolymerization Promoted by Octahedral Titanium Catalyst. Macromolecules 2007, 40, 5638–5643. [Google Scholar] [CrossRef]
- Pires, N.M.T.; Ferreira, A.A.; de Lira, C.H.; Coutinho, P.L.A.; Nicolini, L.F.; Soares, B.G.; Coutinho, F.M.B. Performance Evaluation of High-cis 1,4-Polybutadienes. J. Appl. Polym. Sci. 2006, 99, 88–99. [Google Scholar] [CrossRef]
- Pires, N.M.T.; Coutinho, F.M.B.; Costa, M.A.S. Synthesis and characterization of high cis-polybutadiene: Influence of monomer concentration and reaction temperature. Eur. Polym. J. 2004, 40, 2599–2603. [Google Scholar] [CrossRef]
- Gargani, L.; Bruzzone, M. Fatigue Resistance of Polybutadienes and Effect of Microstructure. In Advances in Elastomers and Rubber Elasticity; Lal, J., Mark, J.E., Eds.; Springer US: Boston, MA, USA, 1986; pp. 233–251. [Google Scholar]
- Ricci, G.; Sommazzi, A.; Masi, F.; Forni, A. Polymerization of 1,3-Butadiene with Catalysts Based on Cobalt Dichloride Complexes with Aminophosphines: Switching the Regioselectivity by Varying the MAO/Co Molar Ratio. Chin. J. Polym. Sci. 2023, in press. [Google Scholar] [CrossRef]
- Cass, P.; Pratt, K.; Mann, T.; Laslett, B.; Rizzardo, E.; Burford, R. Investigation of methylaluminoxane as a cocatalyst for the polymerization of 1,3-butadiene to high cis-1,4-polybutadiene. J. Polym. Sci. A Polym. Chem. 1999, 37, 3277–3284. [Google Scholar] [CrossRef]
- Endo, K.; Hatakeyama, N. Stereospecific and molecular weight-controlled polymerization of 1,3-butadiene with Co (acac) 3-MAO catalyst. J. Polym Sci. Part A Polym. Chem. 2001, 39, 2793–2798. [Google Scholar] [CrossRef]
- Sivaram, S.; Upadhyay, V.K. Synthesis of High–cis–Polybutadiene Using Cobalt (II)-2-Ethylhexoate Modified Triethylaluminum Catalyst. J. Macromol. Sci. Pure Appl. Chem. 1992, 29, 13–19. [Google Scholar] [CrossRef]
- Wang, B.; Liu, H.; Tang, T.; Zhang, T. cis-1,4 Selective Coordination Polymerization of 1,3-Butadiene and Copolymerization with Polar 2-(4-Methoxyphenyl)-1,3-butadiene by Acenaphthene-Based α-Diimine Cobalt Complexes Featuring Intra-Ligand π-π Stacking Interactions. Polymers 2021, 13, 3329. [Google Scholar] [CrossRef]
- Gong, D.; Jia, X.; Wang, B.; Zhang, X.; Jiang, L. Synthesis, characterization, and butadiene polymerization of iron(III), iron(II) and cobalt(II) chlorides bearing 2,6-bis(2-benzimidazolyl)pyridyl or 2,6-bis(pyrazol)pyridine ligand. J. Organomet. Chem. 2011, 702, 10–18. [Google Scholar] [CrossRef]
- Liu, W.; Pan, W.; Wang, P.; Li, W.; Mu, J.; Mu, J.; Weng, G.; Jia, X.; Gong, D.; Huang, K.W. Synthesis of mixed-ligand cobalt complexes and their applications in high cis-1,4-selective butadiene polymerization. Inorganica Chim. Acta 2015, 436, 132–138. [Google Scholar] [CrossRef][Green Version]
- Gong, D.; Liu, W.; Pan, W.; Chen, T.; Jia, X.; Huang, K.W.; Zhang, X. Tunable regioselectivity in 1,3-butadiene polymerization by using 2,6-bis(dimethyl-2-oxazolin-2-yl)pyridine incorporated transition metal (Cr, Fe and Co) catalysts. J. Mol. Catal. A Chem. 2015, 406, 78–84. [Google Scholar] [CrossRef]
- Liu, J.; Fan, X.; Min, X.; Zhu, X.; Zhao, N.; Wang, Z. Synthesis of high cis-1,4 polybutadiene with narrow molecular weight distribution via a neodymium-based binary catalyst. RSC Adv. 2018, 8, 21926–21932. [Google Scholar] [CrossRef] [PubMed]
- Friebe, L.; Nuyken, O.; Obrecht, W. Ziegler/Natta Catalysts and their Application in Diene Polymerization. In Neodymium Based Ziegler Catalysts–Fundamental Chemistry. Advances in Polymer Science; Nuyken, O., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 204, pp. 5–7. [Google Scholar]
- Annunziata, L.; Pragliola, S.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. New (Anilidomethyl)pyridine Titanium(IV) and Zirconium(IV) Catalyst Precursors for the Highly Chemo- and Stereoselective cis-1,4-Polymerization of 1,3-Butadiene. Macromolecules 2011, 44, 1934–1941. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef]
- Taube, R.; Schmidt, U.; Gehrke, J.-P.; Böhme, P.; Langlotz, J.; Wache, S. New mechanistic aspects and structure activity relationships in the allyl nickel complex catalysed butadiene polymerization. Makromol. Chem. Makromol. Symp. 1993, 66, 245–260. [Google Scholar] [CrossRef]
- Taube, R.; Windisch, H.; Maiwald, S. The catalysis of the stereospecific butadiene polymerization by allyl nickel and allyl lanthanide complexes–A mechanistic comparison. Macromol. Symp. 1995, 89, 393–409. [Google Scholar] [CrossRef]
- Tobisch, S.; Bögel, H.; Taube, R. Mechanistic studies of the 1,4-polymerization of butadiene according to the π-allyl-insertion mechanism. 2. Density functional study of the C-C bond formation reaction in cationic and neutral (η3-crotyl)(η2-/η4-butadiene)nickel(II) complexes [Ni(C4H7)(C4H6)]+, [Ni(C4H7)]. Organometallics 1996, 15, 3563–3571. [Google Scholar] [CrossRef]
- Taube, R.; Sylvester, G. Applied Homogeneous Catalysis with Organometallic Complexes; Cornils, B., Herrmann, W.A., Eds.; VCH: Weinheim, Germany, 1996; pp. 280–317. [Google Scholar]
- Peluso, A.; Improta, R.; Zambelli, A. Mechanism of Isoprene and Butadiene Polymerization in the Presence of CpTiCl3−MAO Initiator: A Theoretical Study. Macromolecules 1997, 30, 2219–2227. [Google Scholar] [CrossRef]
- Guerra, G.; Cavallo, L.; Corradini, P.; Fusco, R. Molecular mechanics and stereospecificity in Ziegler-Natta 1,2 and cis-1,4 polymerizations of conjugated dienes. Macromolecules 1997, 30, 677–684. [Google Scholar] [CrossRef]
- Tobisch, S.; Taube, R. Mechanistic studies of the 1,4-polymerization of butadiene according to the π-allyl-insertion mechanism. 3. Density functional study of the C-C bond formation reaction in cationic “ligand-free” (η3:η2-heptadienyl) (η2-/η4-butadiene)nickel(II) complexes [Ni(C7H11)(C4H6)]+. Organometallics 1999, 18, 5204–5218. [Google Scholar]
- Costabile, C.; Milano, G.; Cavallo, L.; Guerra, G. Stereoselectivity and Chemoselectivity in Ziegler-Natta Polymerizations of Conjugated Dienes. 1. Monomers with Low-Energy s-Cis h4 Coordination. Macromolecules 2001, 34, 7952–7960. [Google Scholar] [CrossRef]
- Taube, R.; Sylvester, G. Stereospecific polymerization of butadiene or isoprene. In Applied Homogeneous Catalysis with Organometallic Compounds, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002; Volume 1, pp. 285–315. [Google Scholar]
- Tobisch, S. Reaction Mechanism in the Stereospecific 1,4-Polymerization of Butadiene with Ziegler-Natta Type Catalysts of Early Transition Metals: Comprehensive Density Functional Investigation for the Cationic [TiIIICp(polybutadienyl)(butadiene)]+ Active catalyst. Organometallics 2003, 22, 2729–2740. [Google Scholar] [CrossRef]
- Tobisch, S. The stereospecific polymerization of 1,3-butadiene mediated by early and late transition-metal catalysts. Towards a deeper understanding of the catalytic structure–reactivity relationships from computational-mechanistic studies. J. Mol. Struct. Theochem 2006, 771, 171–179. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Path 1 | Path 3 | ||||||
|---|---|---|---|---|---|---|---|
| 1Zr | TS1-2Zr | 2Zr | 1Zr | TS1-2Zr | 2Zr | ||
| anti–S/cis–S | 4.4 | 10.0 | −6.1 | anti–S/trans–P | 0.0 | 21.4 | −5.8 |
| anti–S/cis–P | 5.2 | 22.9 | −5.6 | anti–S/trans–S | 1.2 | 11.0 | −8.6 |
| anti–P/cis–S | 9.7 | 12.3 | −3.9 | anti–P/trans–P | 4.5 | 23.0 | −0.5 |
| anti–P/cis–P | 6.6 | 25.0 | −4.0 | anti–P/trans–S | 4.3 | 16.3 | −0.3 |
| Path 2 | Path 4 | ||||||
| 1Zr | TS1-2Zr | 2Zr | 1Zr | TS1-2Zr | 2Zr | ||
| syn–S/cis–S | 6.2 | 11.0 | −7.2 | syn–S/trans–P | 2.5 | 22.6 | −4.3 |
| syn–S/cis–P | 5.5 | 21.1 | −5.8 | syn–S/trans–S | 2.8 | 13.7 | −7.1 |
| syn–P/cis–S | 5.8 | 8.4 | −7.4 | syn–P/trans–P | 4.8 | 20.9 | −3.9 |
| syn–P/cis–P | 5.3 | 21.9 | −5.3 | syn–P/trans–S | 5.1 | 12.9 | −4.5 |
| Path 1 | Path 3 | ||||||
|---|---|---|---|---|---|---|---|
| 1Ti | TS1-2Ti | 2Ti | 1Ti | TS1-2Ti | 2Ti | ||
| anti–S/cis–S | 3.2 | 5.4 | −12.4 | anti–S/trans–P | 0.2 | 15.4 | −12.1 |
| anti–S/cis–P | 4.8 | 15.5 | −11.1 | anti–S/trans–S | 0.8 | 8.0 | −13.2 |
| anti–P/cis–S | 8.6 | 8.7 | −11.7 | anti–P/trans–P | 3.5 | 15.9 | −5.2 |
| anti–P/cis–P | 6.1 | 18.6 | −4.3 | anti–P/trans–S | 3.2 | 8.6 | −8.9 |
| Path 2 | Path 4 | ||||||
| 1Ti | TS1-2Ti | 2Ti | 1Ti | TS1-2Ti | 2Ti | ||
| syn–S/cis–S | 5.2 | - | −13.0 | syn–S/trans–P | 1.0 | 15.8 | −9.7 |
| syn–S/cis–P | 4.2 | 14.2 | −3.7 | syn–S/trans–S | 0.0 | 7.0 | −11.1 |
| syn–P/cis–S | 4.5 | 5.1 | −14.9 | syn–P/trans–P | 2.3 | 13.5 | −7.8 |
| syn–P/cis–P | 4.4 | 15.3 | −5.5 | syn–P/trans–S | 2.5 | 5.4 | −9.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milione, S.; Pragliola, S. Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes. Symmetry 2024, 16, 18. https://doi.org/10.3390/sym16010018
Milione S, Pragliola S. Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes. Symmetry. 2024; 16(1):18. https://doi.org/10.3390/sym16010018
Chicago/Turabian StyleMilione, Stefano, and Stefania Pragliola. 2024. "Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes" Symmetry 16, no. 1: 18. https://doi.org/10.3390/sym16010018
APA StyleMilione, S., & Pragliola, S. (2024). Stereoselectivity in Butadiene Polymerization Promoted by Using Ziegler–Natta Catalysts Based on (Anilidomethyl)pyridine Group (IV) Complexes. Symmetry, 16(1), 18. https://doi.org/10.3390/sym16010018