Abstract
The hydrolase-catalyzed kinetic resolution of fluorinated racemates of 3-arylcarboxylic acids is described. Hydrolysis of ethyl esters of fluorinated acids by esterases and hydrolases in all cases resulted in the formation of hydrolyzed (S)-carboxylic acids and unreacted (R)-esters in high yields and high enantiomeric purity. The influence of separation conditions on the efficiency and enantioselectivity of biocatalytic conversion was also studied. The reactions were carried out under normal conditions (stirring with a magnetic stirrer at room temperature) and microwave irradiation in the presence of hydrolases. Amano PS showed excellent selectivity and good yields in the hydrolysis of fluorinated aromatic compounds. The absolute configuration of the resulting compounds was based on biokinetic studies and the use of chiral HPLC. A molecular modeling of the kinetic resolution of carboxylic acid esters was carried out.
1. Introduction
Symmetry is an integral property of living organisms. Asymmetric carbon atoms create chirality in molecules—the possibility of the existence of two forms of mirror images. Almost all chiral molecules in living organisms occur in only one form: sugars are exclusively right-handed, amino acids are levorotatory, and DNA folds into right-handed helices. The presence of carbon atoms with four chemically different substituents results in the formation of chiral molecules with non-movable mirror-image forms known as stereoisomers. Symmetry is a fundamental aspect of nature, and many important problems in science depend on understanding the reasons for the presence or absence of symmetry.
In recent years, the trend towards the use of chiral pharmaceuticals has increased significantly.
Requirements on this issue were published by the US Food and Drug Administration (FDA) back in 1992 in a document entitled “Development of new stereoisomeric drugs” [1,2]. Based on these requirements, there has been a significant shift towards the development of enantiomerically pure pharmaceuticals. As a result, the demand for enantiomerically pure drugs is steadily growing by 13–15% per year [3,4]. As a result, a significant amount of fluorine- and phosphorus-containing synthetic blocks used in the production of pharmaceuticals were obtained in enantiomerically pure form [5,6]. Of particular interest is the enzymatic deracemization of synthetic blocks of biologically active substances in order to achieve “chiral switching” of racemic pharmaceuticals [6]. Enzymatic resolution offers a simple and fruitful route for the synthesis of enantiomerically pure pharmaceuticals. Crude, purified, or immobilized lipases can be used directly in water, organic solvents, or ionic liquids [7].
Enantiomerically pure 3-arylalkanoic acids are used as chiral synthons in the asymmetric synthesis of antibacterial agents such as (-)-malingolide, curcumene and curcuphenol, biologically important bisabolene sesquiterpenes, as well as in the synthesis of the amino acids p-methyl phenylalanine and p-methyltyrosine. The predominance of this structural motif in pharmaceuticals has led to methods for the preparation of 3-arylalkanoic acids becoming an important topic in organic chemistry and drug discovery. Most commonly, these compounds are prepared by asymmetric metal complex hydrogenation of unsaturated carboxylic acids. Lipase-catalyzed cleavage of racemic secondary alcohols and esters by esterification and trans-esterification in non-aqueous media is the most suitable approach to obtain bisabolene–sesquiterpenes. The availability of lipases from sources such as plants, bacteria, and fungi makes them attractive biocatalysts. Lipases are widely used as biocatalysts for organic synthesis due to their stability in non-aqueous media, their ability to accept a wide range of substances, and their use of mild reaction conditions [6,7,8,9].
Desymmetrization reactions starting from symmetric compounds are among the most cost-effective transformations in synthetic organic chemistry, which are advantageous for the total synthesis of complex natural products or pharmaceuticals because the starting materials, symmetric compounds, are usually produced at a large scale from inexpensive sources or commercially available at a low cost. Among numerous solvents, water is one of the most environmentally friendly and inexpensive solvents, so organic reactions that occur with water are generally considered “green chemistry”. Combining these concepts is expected to result in water-mediated desymmetrization of symmetrical organic compounds providing remarkable synthetic advantages. Isolating identical functional groups existing in symmetrical compounds is considered a difficult task. Organic compounds generally do not dissolve in water in significant quantities due to their hydrophobic properties. As a result, studies of desymmetrization reactions of symmetrical organic compounds under the influence of water are very limited. Enzymatic reactions are a relatively common approach to accomplish desymmetrization in aqueous media. Well-known examples of enzymatic desymmetrization reactions include the monohydrolysis of symmetric diesters and the monoesterification of symmetric diols. The conditions for enzymatic reactions are usually mild and environmentally friendly, which is why such approaches are considered green chemistry [9].
2. Materials and Methods
2.1. Preparation of Racemic Esters of Arylcarboxylic Acids
In this work, we obtained enantiomerically pure 3-aryl-2-methylpropanoic and 3-arylbutanoic acids using enzymatic deracemization. As biocatalysts, we chose some of the most effective esterases and hydrolases (lipases from Burkholderia cepacia, Pseudomonas cepacia, and Candida antarctica B), which combine broad substrate specificity with high regio- and enantioselectivity, which allows them to be used for the separation of enantiomers with a high degree of efficiency and selectivity. These properties make biocatalysts especially attractive for the pharmaceutical and agrochemical industries, where there is growing interest in enantiomerically pure and specifically functionalized compounds. Traditional aqueous hydrolysis of esters catalyzed by Burkholderia cepacia has been described for the resolution of 3-phenylbutanoic acid (E > 50); however, this work has not been extended to the acidic substrates, including more sterically hindered substituents in the stereogenic center. Racemic -aryl-2-methylpropanoic and 3-arylbutanoic acid esters 2a–e and 4a–f were synthesized according to the two-step Figure 1 below.
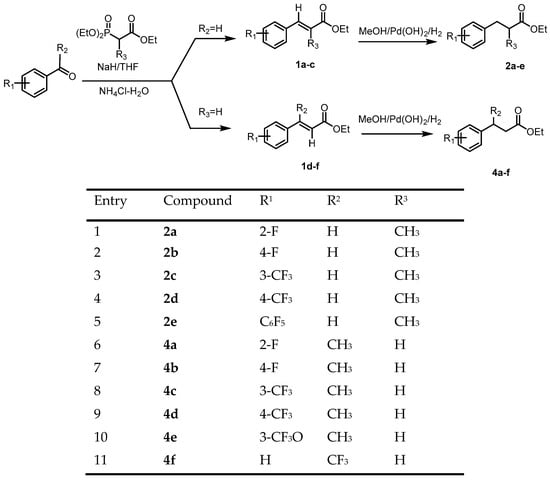
Figure 1.
Synthesis of racemic ethyl 3-aryl-2-methylpropanoates 2a–e and ethyl 3-arylbutanoates 4a–f.
At the first stage of the synthesis, esters of unsaturated arylcarboxylic acids were obtained using the Horner–Wittig reaction. In the next stage, hydrogenation was carried out with hydrogen into methanol in the presence of a palladium catalyst. As a result, racemic ethyl (E)-3-arylbuta-2-noates were obtained in 88–90% yield, which was used in the following steps: 5.0 g of palladium hydroxide was added to ethyl (E)-3-phenylbut-2-noate in a methanol solution. Then, a low pressure hydrogen source (1 atm) was connected. The reaction mixture was stirred for 16 h at room temperature and filtered under an argon atmosphere, the precipitate was washed with methanol. The filtrate was evaporated in a vacuum. As a result, 3-arylcarboxylic acids 2a–e and 4a–f were obtained in good yields, which were used without additional purification. The structure and chemical purity of the products were confirmed by data from NMR spectra, HPLC, and LC–MS (Figure 2).
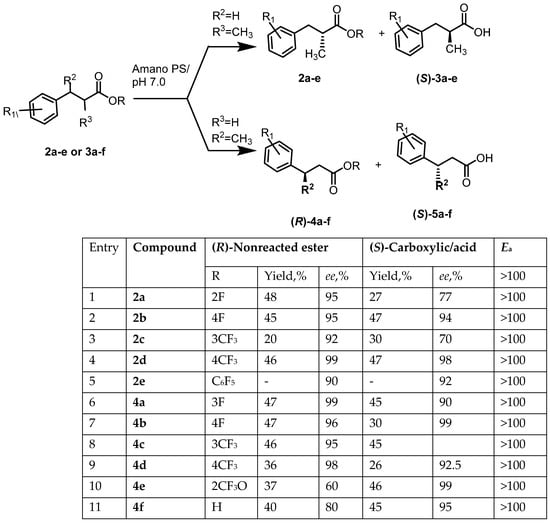
Figure 2.
Enzymatic resolution of enantiomeric racemic arylcarboxylic acids esters 3a–e.
2.2. Enzymative Deracemization of Arylcarboxylic Acids
Enzymes act in solution, at ambient temperature and neutral pH. They demonstrate a high level of reaction stereoselectivity, as well as high chemo-, regio-, and enantioselectivity. Hydrolases are enzymes that catalyze hydrolysis reactions directly in water, through aqueous buffers, or even using two-phase organic solvent/water systems. The category of hydrolases is very broad, and there is much variation among hydrolases. For example, hydrolase activity typically requires dissolution of the substrate in the reaction medium, which does not apply to lipases. Indeed, the latter demonstrate low catalytic activity towards substrates dissolved in an aqueous solution. However, lipase activity increases significantly when the substrate reaches a critical micelle concentration, where it tends to form a second phase. This activation of the water/lipid interface is attributed to the presence of the oligopeptide. (helical “lid”), which blocks access to the active site of the enzyme. When interaction with a hydrophobic interface occurs, the enzyme rearranges itself into its active conformation. This phenomenon is specific to lipases and does not occur in other hydrolases. Hydrolysis uses esterases, proteases, and some lipases. The chiral or prochiral moiety of esters performs an acidic function. The substrates presented in this article are racemic esters. Cleavage of racemic esters by hydrolase-catalyzed enantioselective hydrolysis is an efficient and practical method for the preparation of pharmaceutical intermediates (Figure 2). Transesterification in an organic medium is an equilibrium reaction. Therefore, it is important to shift the balance in the right direction to obtain the desired result. For example, during enzymatic digestion by Burchholderia cepacia lipase, an aryl carboxylic acid ester can be transesterified in the presence of vinyl acetate. The vinyl alcohol thus formed tautomerizes into an aldehyde during the reaction, making it irreversible.
Enantioselective esterification of racemic acid with Burkholderia cepacia lipase in hexane can be carried out, provided that the water formed during the reaction is removed. Enzymatic hydrolysis of the racemic aryl carboxylic acid ester was carried out in phosphate buffer pH 7.0 in the presence of Amano PS lipase. The mixture was stirred for 16 h at room temperature, then filtered; the filtrate was acidified with 2 M HCl to pH 2 and extracted with MTBE. Carboxylic acid esters 3a–e were obtained in 50–80% yield and 93–100% ee enantioselectivity. The aqueous solution after extraction with an organic solvent was acidified with 2M HCl to pH 2 and washed with MTBE. The resulting extract was washed with a solution of sodium chloride, dried with sodium sulfate, and evaporated in a vacuum. As a result, arylcarboxylic acids 3a–e or 5a–e were obtained in 52–60% yield and 77–99% enantiomeric purity. To increase the enantiomeric purity of arylcarboxylic acids, additional (double) enzymatic deracemization was used (Figure 3).
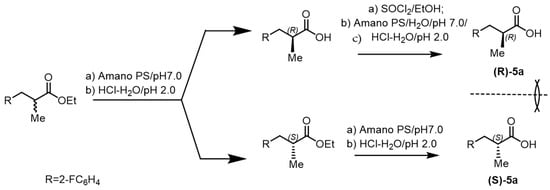
Figure 3.
Additional deracemization of 2-methylarylcarboxylic acid 5a.
To a solution of methyl aryl carboxylic acid in ethanol solution at 0 °C, 1.6 equiv of thionyl chloride was added dropwise over 10 min. The mixture was stirred for 16 h at room temperature, then evaporated in a vacuum. The residue was dissolved in water and neutralized with an aqueous solution of potash to pH > 9. The resulting ester was added to a suspension of Amano PS in a solution of MTBE with 5 eq. water. The reaction mixture was stirred for 16 h at 40 °C, then filtered, washed with MTBE, and the filtrate was evaporated in a vacuum. The residue was dissolved by heating in MTBE, then cooled and left in the refrigerator for 10 h. In total, several lipases and esterases were tested for the resolution of racemic 3-phenylbutanoic acid ester (±)-5a. All hydrolases tested resulted in the hydrolysis of ethyl 3-phenylbutanoate (±)-3a, and the screening results are summarized in Table 1. Pseudomonas cepacia and Burcholderia cepacia showed excellent enantioselectivity for hydrolysis of the substrate (±)-3a. It was previously reported that hydrolysis of Burholderia cepacia (±)-3a methyl ester (E > 50) provides access to (S)-3a-acid with 89% ee. Here, these enzymes produce (S)-3a-acid with a high enantioselectivity of 97%. Unreacted (R)-3a was isolated at 98% ee, allowing access to both enantiomeric series in a single resolution. The presence of a bulky trifluoromethyl group in ethyl (S)-4,4,4-trifluoro-3-phenylbutanoate 4f significantly increased the reaction time, which made it difficult to bring the reaction to the optimal 50% hydrolysis (Figure 4). Therefore, we resorted to initiating the reaction using microwave radiation, which allowed the reaction to be completed within an optimal term. The reaction in toluene at 40 °C proceeded at a constantly high ee. Kinetic resolution occurred under the influence of microwave irradiation, and the conversion and ee remained stable throughout the entire resolution process (Table 1). In recent years, the trend towards the use of chiral pharmaceuticals has been steadily increasing, although requirements on this issue were published by the US Food and Drug Administration (FDA) back in 1992 in a document entitled “Development of new stereoisomeric drugs” [1,2].

Table 1.
3-phenylbutanoic acids and methyl-3-phenylpropanoic acids.
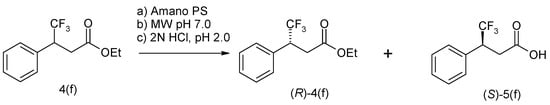
Figure 4.
Enzymatic resolution by Amano PS of ethyl (R)-4,4,4-trifluoro-3-phenylbutanoate 5f under microwave irradiation.
General procedure for the resolution of fluorinated arylcarboxylic acids by lipase. A solution of 0.25 mol of racemic aryl carboxylic acid ester 2(a–e) in 1500 mL of phosphate buffer pH 7.0. Add 20 g of Amano PS lipase. The mixture was stirred for 16 h at room temperature, and then filtered, the filtrate was acidified with 2 M HCl to pH 2 and extracted with MTBE (3 × 300 mL). The organic extract was washed with 0.2 M aqueous potassium carbonate solution (2 × 300 mL), dried with sodium sulfate, and evaporated in a vacuum. Carboxylic acid esters 2(a–e) or 4(a–e) were obtained. Yield 50–80%; 93–100% ee. After extraction, the aqueous solution was acidified with 2 M HCl to pH 2 with an organic solvent and extracted with MTBE (3 × 500 mL). The extract was washed with sodium chloride solution, dried with sodium sulfate, and evaporated in a vacuum. Arylcarboxylic acids 3(a–e) or 5(a–e) were obtained. Yield 52–60%, 77–99% ee. To an enantiomerically enriched solution of 55 g (0.335 mol; 1 eq.) of aryl carboxylic acid in 500 mL of ethanol at 0 °C, 29.1 mL (0.4 mol; 1.2 eq.), thionyl chloride was added dropwise over a period of 10 min. The mixture was stirred for 16 h at room temperature, then concentrated in vacuo. The residue was dissolved in 500 mL of MTBE and washed twice with a 5% solution of sodium bicarbonate in water (2 × 300 mL). The organic layer was dried over Na2SO4 and evaporated in a vacuum. The resulting ester was added to a suspension of 5.5 g Amano PS (10 weight percent) in 550 mL phosphate buffer pH 7.0 and stirred for 16 h at room temperature. The mixture was filtered and washed with MTBE (2 × 200 mL); the filtrate was acidified to pH 2 with 2M HCl and extracted three times with MTBE (3 × 200 mL). The organic layer was washed twice with 0.5 M (2 × 300 mL) potassium carbonate solution and disposed of. The aqueous layer was acidified to pH 2 with 2M HCl and extracted three times with MTBE (3 × 500 mL). The organic layer was washed twice with saturated sodium chloride solution (2 × 500 mL), dried over sulfate, and evaporated in a vacuum. We obtained 41.5 g (252.7 mmol; ee = 99; yield 75.5%) of substance 4a in the form of a pale yellow liquid.
(R)-3-(2-Fluorophenyl)-2-methylpropanoic acid ((R)-3a). Pale yellow oil (97% yield). [α]D20 = −25.22 (C = 0.5, CH3OH), ee 95%. 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.18 (d, J = 6.8 Hz, 3H, CH3); 2.75 m (2H, CH2); 3.1 (1H, CHCH3); 6.96–7.21 m (4H, C6H4). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −118.34; LCMS: M 182.19 (M 182.09) (R); HPLC: 10.177 min (R); 9.674 min (S); Column: Chiralpak AD-H (250 × 4.6 mm, 5 mkm)—ADH0CE-WG064—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 90:10; Flow Rate: 0.6 mL/min.
(S)-3-(2-Fluorophenyl)-2-methylpropanoic acid ((S)-3a). Pale yellow oil (97% yield), [α]D20 = +25.34 (C = 0.5, CH3OH), 97.5% ee. 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.18 (d, J = 6.8 Hz, 3H, CH3); 2.75 m (2H, CH2) 3.1 (1H, CHCH3); 6.96–7.21 m (4H, C6H4). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −118.34; LCMS: M 182.19 (M 182.09) (S); HPLC: 10.081 min (R); 9.656 min (S), Column: Chiralpak AD-H (250 × 4.6 mm, 5 mkm)—ADH0CE-WG064—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 90:10; Flow Rate: 0.6 mL/min.
(R)-3-(4-fluorophenyl)-2-methylpropanoic acid ((R)-3b). Pale yellow oil (97% yield). [α]D20 =−27.1 (C = 0.5, CH3OH) ee 99%. 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.19 (d, J = 6.7 Hz, 3H, CH3); 2.73 m (2H, CH2) 3.02 (1H, CHCH3); 6.97–7.14 m (4H, C6H4). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −117.24; LCMS: M 182.19 (M 182.09) (R); HPLC 7.944 min (R); 8.958 min (S), Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—3; Mobile Phase: Hexane/IPA, 95:5; Flow Rate: 1.0 mL/min.
(S)-3-(4-fluorophenyl)-2-methylpropanoic acid ((S)-3b). Pale yellow oil (97% yield). [α]D20 = +27.2 (C = 0.5, CH3OH), ee 94% (S). 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.19 (d, J = 6.7 Hz, 3H, CH3); 2.73 m (2H, CH2) 3.02 (1H, CHCH3); 6.97–7.14 m (4H, C6H4). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −117.67. LCMS: M 182.19 (M 182.09);HPLC 8.125 min (R); 9.148 min (S), Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—3; Mobile Phase: Hexane/IPA, 95:5; Flow Rate: 1.0 mL/min.
(R)-2-methyl-3-(3-(trifluoromethyl)phenyl)propanoic acid ((R)-3c). Oil; [α]D20 = −25.04 (C = 0.5, CH3OH), (S); 97% ee. 1H NMR (500 MHz, ppm, J, Hz, DMSO) δ 1.04 (d, J = 6.8 Hz, CH3); 2.70 m (2H, CH2) 2.90 (1H, CHCH3); 7.5 d (4H, C6H4); 12.19 br (1H, OH). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 16.5 s; 38.95 s; 41.1 s; 123.42 q; 125.7 q; 128.87 s; 131 q; 132.4 s; 139.9 s; 182.2 s; J 120.08; 122.78; 125.49; 128.19. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.47; LCMS: M 232.2 (M 232.09); HPLC 7.24 min (R); 7.67 min (S), Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 97:3.
(S)-2-methyl-3-(3-(trifluoro-methyl)phenyl) propanoic acid ((S)-3c). Oil; [α]D20 = +25.27 (C = 0.5, CH3OH)(S); 99% ee 1H NMR (500 MHz, ppm, J, Hz, DMSO) δ 1.04 (d, J = 6.8 Hz, CH3); 2.70 m (2H, CH2) 2.90 (1H, CHCH3); 7.5 d (4H, C6H4); 12.19 br (1H, OH), 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 16.5 s; 38.95 s; 41.1 s; 123.42 q; 125.7 q; 128.87 s; 131 q; 132.4 s; 139.9 s; 182.2 s; JCF 120.08; 122.78; 125.49; 128.19. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.47; LCMS: M 232.2 (M 232.09); HPLC 7.24 min (R); 7.67 min (S), Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 97:3.
(R)-2-methyl-3-(4-(trifluoromethyl)phenyl)propanoic acid ((R)-3d). Mp 61 °C; [α]D20 = −24.16 (C = 0.5, CH3OH)(R); ee 98%, 1H NMR (500 MHz, ppm, J, Hz, DMSO); δ 1.04 (d, J = 6.7 Hz, 3H, CH3); 2.70 m (2H, CH2); 2.93 (1H, CHCH3); 7.40–7.63 m (4H, C6H4); 12.19 br (1H, OH). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 16.07s; 38.41 s; 40.45 s; 122.62 s; 124.8 5q, JCF 128.7; 142.57 s; 181.58 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.23; LCMS: M 232.2 (M 232.09); HPLC 9.89 min (R); 9.275 min (S), Column: Chiralpak AD-H (250 × 4.6 mm, 5 mkm)-3; Mobile Phase: Hexane (0.1% TFA)/IPA, 97:3 Flow Rate: 1.0 mL/min.
(S)-2-methyl-3-(4-(trifluoromethyl)phenyl)propanoic acid ((S)-3d). Mp 61 °C; [α]D20 = +22.5 (C = 0.5, CH3OH)(S); ee 98%. 1H NMR (500 MHz, ppm, J, Hz, DMSO) δ 1.04 (d, J = 6.7 Hz, 3H, CH3); 2.70 m (2H, CH2); 2.93 (1H, CHCH3); 7.40–7.63 m (4H, C6H4); 12.19 br (1H, OH). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 16.07 s; 38.41 s; 40.45 s; 122.62 s; 124.8 5q, JCF 128.7; 142.57 s; 181.58 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.23; LCMS: M 232.2 (M 232.09); HPLC 9.89 min (R); 9.275 min (S); Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)-3; Mobile Phase: Hexane/IPA, 98:2 Flow Rate: 0.6 mL/min.
(S)-2-methyl-3-(perfluorophenyl)propanoic acid ((S)-3e). [α]D20 = +16 (C = 0.5, CH3OH)(S); ee 99%. 1H NMR (400 MHz, ppm, J, Hz, CDCl3) δ 1.21 (d, J = 7 Hz); 2.81 m (2H, CH2); 3.1 m (1H, CHCH3). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −143 dd; −156.62 t; −162.8 dt. LCMS: M 254.04 (M 233.0 Neg [-HF]); HPLC 8.65 min (R); 9.14 min (S); Column: Chiralpak IB (250 × 4.6 mm, 5 mkm)—IB00CE-BN025—14; Mobile Phase: Hexane (0.1% TFA)/IPA, 97:3; Flow Rate: 0.6 mL/min.
(R)-2-methyl-3-(perfluorophenyl)propanoic acid ((R)-3e). [α]D20 = −16 (C = 0.5, CH3OH)(S); ee 49%. 1H NMR (400 MHz, ppm, J, Hz, CDCl3) δ 1.21 (d, J = 7 Hz); 2.81 m (2H, CH2); 3.1 m (1H, CHCH3). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −143 dd; −156.62 t; −162.8 dt. LCMS: M 254.04 (M 233.0 Neg [-HF]); HPLC 8.75 min (R); 9.25 min (S); Column: Chiralpak IB (250 × 4.6 mm, 5 mkm)—IB00CE-BN025—14; Mobile Phase: Hexane (0.1% TFA)/IPA, 97:3; Flow Rate: 0.6 mL/min.
(S)-3-(2-fluorophenyl)butanoic acid ((S)-5a). Mp 36 °C; [α]D20 = +29 (C = 0.5, CH2Cl2), ee 99%. 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.3 d (CH3); 2.55–2.8 m (2H, CH2) 3.5 (1H, CHCH3); 6.9–7.25 m (4H, C6H4)). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 19.9 s; 29.6 s; 40.4 s; 115.1 d; 123.7 d, 124.45 d; 124.51 d; 131.5 d; 159.2–161.5 d, 178.35s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −118.6; LCMS: M 182.19 (M 182.09) (S); HPLC: 12.07 min (R); 10.45 min (S), Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 90:10; Flow Rate: 0.6 mL/min.
(R)-3-(2-fluorophenyl)butanoic acid ((R)-5a). Mp 36 °C; [α]D20 = −25.95 (C = 0.5, CH2Cl2) ee 99% 1H NMR (CDCl3, 500 MHz, 25 °C): δH 1.3 d (CH3); 2.55–2.8 m (2H, CH2) 3.5 (1H, CHCH3); 6.9–7.25 m (4H, C6H4). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 19.9 s; 29.6 s; 40.4 s; 115.1 d; 123.7 d, 124.45 d; 124.51 d; 131.5 d; 159.2–161.5 d, 178.35 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −118.6; LCMS: M 182.19 (M 182.09) (R); HPLC: 11.94 min (R); 10.34 min (S), Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 90:10; Flow Rate: 0.6 mL/min.
(S)-3-(4-fluorophenyl)butanoic acid ((S)-5b). Oil; [α]D20 = +43.09 (C = 0.5, CHCl3) ee 99%. 1H NMR (DMSO, 500 MHz, 25 °C): δH 1.17 d (CH3); 2.45 d (2H, CH2) 3.13 m (1H, CHCH3); 7.09 t, 7.27 d (4H, C6H4), 12.04 br (1H, OH). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −117.7; LCMS: M 182.19 (M 182.09) (S); HPLC: 10.12 min (R); 8.88 min (S), Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 80:20; Flow Rate: 0.6 mL/min.
(R)-3-(4-fluorophenyl)butanoic acid ((R)-5b). Oil; [α]D20 = −39 (C = 0.5, CHCl3) ee 99%. 1H NMR (DMSO, 500 MHz, 25 °C): δH 1.17 d (CH3); 2.45 d (2H, CH2) 3.13 m (1H, CHCH3); 7.09 t, 7.27 d (4H, C6H4), 12.04 br (1H, OH). 19F NMR (CDCl3, 376 MHz, 25 °C): δF −117.7; LCMS: M 182.19 (M 182.09) (R); HPLC: 14.9 min (R); 11.95 min (S), Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—OJH0CE-UF008—3; Mobile Phase: Hexane/IPA, 90:10; Flow Rate: 0.6 mL/min.
(S)-3-(3-(trifluoromethyl)phenyl)butanoic acid ((S)-5c). Oil; [α]D20 = +22.5 (C = 0.5, MeOH) ee 99%. 1H NMR (DMSO, 500 MHz, 25 °C): δH 1.14 d (CH3); 2.55 d (2H, CH2); 3.16 m (1H, CHCH3); 7.4–7.6m (4H, C6H4), 12.04 br (1H, OH). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 21.13 s; 35.48; 41.8 s; JF 120.4, 122.56, 124.73, 125.9; 122.92 d; 123.05 d; 128.5 s; 129.67 s; 130.33 q; 145.79; 177.72. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.38; LCMS: M 232.2 (M 232.09) (S); HPLC: 9.45 min (R); 8.48 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 90:10; Flow Rate: 0.6 mL/min.
(R)-3-(3-(trifluoromethyl)phenyl)butanoic acid ((R)-5c). Oil; [α]D20 = −22.5 (C = 0.5, MeOH) ee 98%. 1H NMR (DMSO, 500 MHz, 25 °C): δH 1.14 d (CH3); 2.55 d (2H, CH2); 3.16 m (1H, CHCH3); 7.4–7.6 m (4H, C6H4), 12.04 br (1H, OH). 13C NMR (CDCl3, 273.8 MHz, 25 °C): δC = 21.13 s; 35.48; 41.8 s; JF 120.4, 122.56, 124.73, 125.9; 122.92 d; 123.05 d; 128.5 s; 129.67 s; 130.33 q; 145.79; 177.72. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −61.38;LCMS: M 232.2 (M 232.09) (S); HPLC: 9.12 min (R); 8.22 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 90:10; Flow Rate: 0.6 mL/min.
(S)-3-(4-(trifluoromethyl)phenyl)butanoic acid ((S)-5d). Mp 56 °C; [α]D20 = +35.0 (C = 1.1, CHCl3); ee 99%. 1H NMR (500 MHz, ppm, J, Hz, CDCl3) δ 1.3 d (CH3); 2.63 m (2H, CH2); 3.32 m (1H, CHCH3); 7.32 d, 7.54d, 4H (C6H4). 13C NMR (237.8 MHz, CDCl3) δC = 21.7 s; 35.98 s; 42.08 s; 122.5 s, 125.5 q; 127.1 s; 130 q; 149.3 s; 177.96 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −62.94; LCMS Mol 232.2 (M 232.09) (S). HPLC: 13.057 min (R); 10.79 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3.
(R)-3-(4-(trifluoromethyl)phenyl)butanoic acid ((R)-5d). Mp 56 °C; [α]D20 = −23.7 (C = 0.5, MeOH); ee 99%. 1H NMR (500 MHz, ppm, J, Hz, CDCl3) δ 1.3 d (CH3); 2.63 m (2H, CH2); 3.32 m (1H, CHCH3); 7.32d, 7.54d, 4H (C6H4). 13C NMR (237.8 MHz, CDCl3) δC = 21.7 s; 35.98 s; 42.08 s; 122.5 s, 125.5 q; 127.1 s; 130 q; 149.3 s; 177.96 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −62.94; LCMS Mol 232.2 (M 232.09) (S). HPLC: 13.057 min (R); 10.76 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 95:5; Flow Rate: 0.6 mL/min.
(S)-3-(3-(trifluoromethoxy)phenyl)butanoic acid ((S)-5e). Oil; [α]D20 = +32.0 (C = 0.5, CHCl3); ee 99%. 1H NMR (500 MHz, ppm, J, Hz, CDCl3) δH 1.31 d (J 10, 3H, CH3); 2.64 m (2H, CH2) 3.32 (1H, CHCH3); 7.05–7.35 m, 4H (C6H4). 13C NMR (237.8 MHz, CDCl3) δC = 21.1 s; 35.38 s; 41.77 s; 118.4 s, 118.9 s; 121 s; 124.6 s; 129.3 s; 147.2 s; 148.9 s; 177.49 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −62.94; LCMS Mol 248.2 (M 248.08) (S) HPLC: 22.87 min (R); 20.95 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 99:1; Flow Rate: 0.6 mL/min.
(R)-3-(3-(trifluoromethoxy) phenyl)butanoic acid ((R)-5e). Oil; [α]D20 = −20 (C = 0.5, CHCl3); ee 96%. 1H NMR (500 MHz, ppm, J, Hz, CDCl3) δH 1.31 d (J 10, 3H, CH3); 2.64 m (2H, CH2) 3.32 (1H, CHCH3); 7.05–7.35 m, 4H (C6H4). 13C NMR (237.8 MHz, CDCl3) δC = 21.1 s; 35.38 s; 41.77 s; 118.4 s, 118.9 s; 121 s; 124.6 s; 129.3 s; 147.2 s; 148.9 s; 177.49 s. 19F NMR (CDCl3, 376 MHz, 25 °C): δF −62.94; LCMS Mol 248.2 (M 248.08) (S) HPLC: 23.05 min (R); 20.99 min (S), Column: Chiralpak AS-H (250 × 4.6 mm, 5 mkm)—203VB002-DB181—3; Mobile Phase: Hexane (0.1% TFA)/IPA, 99:1; Flow Rate: 0.6 mL/min.
Lipase-catalyzed microwave-assisted enzymatic separation of (S)-4,4,4-trifluoro-3-phenylbutanoic acid (S)-5(f). Lipase-catalyzed hydrolysis of ethyl 4,4,4-trifluoro-3-phenylbutanoate under microwave irradiation was carried out in SEM Discover. Model Explorer SP 48. Power 100 W (CEM Corporation, Matthews, North Carolina, USA). Phosphate buffer (10 mL pH 7.0), lipase (160 mg Amano Lipase PS), and aryl carboxylic acid ester 2f, (0.5 g, 0,2 mmol). The entire reaction solution was placed in a microwave apparatus and irradiated at a frequency of 2.45 GHz, and at 200 m. The formation of the hydrolyzed product was monitored by TLC and NMR. The reaction mixture was filtered, and evaporated in a vacuum, the remaining ether was purified by chromatography on columns with silica gel and hexane-ethyl acetate eluent (95:53:1). The separated 4,4,4-trifluoro-3-phenylbutanoic acid was recrystallized from toluene. The reaction mixture was analyzed by GC-FLD, after irradiation, then the reaction flask was moved and the temperature of the reaction mixture was 45, °C.Mp 45 °C; [α]D20 = +42 (C = 0.5, CHCl3)(S); ee 92%. 1H NMR (400 MHz, ppm, J, Hz, CDCl3) δ 2.9–3.1 m (2H, CH2); 3.85 m (1H, CHCH3), 7.3 m (4H, C6H4). HPLC 15.85 min (R); 13.5 min (S); Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—173WE001-DC191—7; Mobile Phase: Hexane/IPA/MeOH, 90:5:5; Flow Rate: 0.6 mL/min which corresponds to the compound 5f previously described in the literature.
(R)-4,4,4-trifluoro-3-phenylbutanoic acid ((R)-3f). Mp 45 °C; [α]D20 = −43 (C = 0.5, CHCl3)(S); ee 47%. 1H NMR (400 MHz, ppm, J, Hz, CDCl3) δ 2.9–3.1 m (2H, CH2); 3.85 m (1H, CHCH3), 7.3 m (4H, C6H4). HPLC 15.851 min (R); 13.5 min (S); Column: Chiralcel OJ-H (250 × 4.6 mm, 5 mkm)—173WE001-DC191—7; Mobile Phase: Hexane/IPA/MeOH, 90:5:5; Flow Rate: 0.6 mL/min. which corresponds to the compound 3f previously described in the literature.
2.2.1. Docking
As a model structure, the crystal structure with the record number 1HQD in the Protein Data Bank was chosen [https://www.rcsb.org/structure/1HQD] (accessed on 22 August 2001). The structure had a good resolution of −2.30 Å and already contained a covalently bound ligand, which best suited our purposes.
For the best imitation of the transition state, the covalent docking method was chosen. Calculations were conducted using the Schrödinger software package [Schrödinger Release 2024-1: CovDock, Schrödinger, LLC, New York, NY, USA 2024]. Ester forms of acids were considered as ligands. The compounds were protonated and energy-minimized using the LigPrep module under the force field of OPLS4, and the remaining parameters were set as default. The protein did not undergo mutation treatment during covalent virtual screening, and the default parameters of the Protein Preparation Wizard module were used to preprocess the protein structure. The structures were processed to remove water (since the compounds we are studying are larger in volume and have a different chemical structure) and calcium ions, as they were not involved in the reaction and could interfere with accurate calculations; also, hydrogen atoms were optimized, and energy was minimized directly.
We used the CovDock module to specify the receptor-active amino acid residues (Ser87) and reaction type (Nucleophhilic Addition to a double bond), specified the interfaced pocket based on the original ligand position of the crystal structure, and performeded covalent virtual screening in Pose Prediction (Thorough) mode.
2.2.2. Molecular Dynamics Simulation
Calculations were conducted using the Schrödinger software package [Schrödinger Release 2024-1: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, USA, 2024. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, USA 2024]. The complexes obtained after molecular docking were used for the MD simulation. Each system was placed into the center of a periodic orthorhombic box, which was then filled with TIP3P water molecules. A minimum 1 nm distance was maintained between the nearest atom of the complex and the edge of the simulation box, so that the complex could fully immerse with water and rotate freely. Then, to neutralize the system and mimic the cellular environment (pH = 7), Na+ and Cl− ions were added to bring the ionic concentration to 150 mM. Here, the solvent molecules are replaced with monoatomic ions, randomly. Next, the obtained complex was energy-minimized, which also relieved any steric clashes. For each complex, a molecular dynamics simulation in water was carried out for 20 ns using an NPT ensemble, OPLS4 force field at a temperature of 300 K. To accurately reproduce the transitional state complex, the carbonyl oxygen of the ligand and His286 were additionally charged.
3. Results
3.1. Stereochemical Studies of Arylcarboxylic Acids
These recommendations have changed opportunities and strategies for marketing and patenting established drugs. They force us to consider stereochemistry when searching for new drugs. Enantiomers require the use of “specialized chiral techniques for their correct identification, characterization, resolution and measurement”. Tools for identification and quantification may include optical rotation measurements, chiral chromatography, optical rotational dispersion, circular dichroism, and NMR with chiral shift reagents. Under these conditions, chiral technologies were formed and developed, primarily the “chiral switching” method, which expands the patent protection of a drug, since a patent for a racemate is non-patentable, and a patent for an eutomer can extend the validity of a patent for a drug. Based on new guidelines in the 1990s, most pharmaceutical companies and research institutions began to focus on single enantiomers early on when they identified a potential chiral drug. The authors demonstrated that within 10 years of the 1992 FDA guidelines, there was a clear shift toward the development of single enantiomeric drugs. As a result, global sales of single-enantiomeric drugs have increased by 13% per year since 2000.
3.2. Determination of the Absolute Configuration by Enzymatic Deracemization
The configuration of arylcarboxylic acids was determined using a combination of enzymatic kinetic resolution and chiral HPLC. Enzymatic kinetic resolution by transesterification of racemic secondary alcohols in the presence of Amano PS proceeds. This empirical model is an extension of Prelog’s rule, which is based on the assumption that enantioselectivity is proportional to the size difference between large (L) and medium (M) substituents in the substrate, as well as the structure of the lipase. The physical essence of Kazlauskas’s rule is determined by the structure of the active center of lipases, which has two pockets—one larger, the other smaller. Based on the structure of the active center of the enzyme, the orientation of the carboxylic acid ester occurs, and esterification/hydrolysis of the corresponding ester occurs. According to Kazlauskas’s rule, biocatalytic resolution of arylcarboxylic acids should be (S)-selective, leading to the formation of (S)-arylcarboxylic acids and unreacted esters of (R)-arylcarboxylic acids (Figure 2) [10,11,12]. Empirically, as was previously shown with a very large number of examples, transesterification of arylcarboxylic acids in the presence of lipases such as Amano PS and a number of other lipases always leads to the formation of (R)-esters and (S)-arylcarboxylic acids. Enzymatic-kinetic resolution is a fairly reliable method for determining the absolute configuration of secondary alcohols and arylcarboxylic acids, tested on a large number of examples and described in many articles and monographs [10,11].
4. Discussion
4.1. Determination of Optical Purity and Absolute Configuration of Enantiomers by Chiral HPLC
Chiral HPLC and enzymatic analysis can determine the absolute configuration of arylcarboxylic acids. Biocatalytic separation and chiral HPLC methods have many similarities. First of all, they are able to predict how the deracemization of arylcarboxylic acids will occur. When using a chiral column, Chiralcel OJ-H (S)-alcohols are more strongly retained by the chiral stationary phase, so the alcohol with the (R)-configuration elutes first (Figure 5). This is due to the fact that the (S)-acid forms a strong hydrogen bond with the chiral sugar contained in the stationary phase [10,11]. In the case of enzymatic esterification of secondary alcohol catalyzed by lipases, the (R)-acid preferentially enters into the esterification reaction and forms (R)-ester, while the (S)-acid remains unreacted, as a result of which the reaction mixture is easily separated using column chromatography (Figure 6). The resolution of carboxylic acid esters into enantiomers was carried out by enzymatic hydrolysis under kinetic control conditions in the presence of Burkholderia cepacia lipase. The reaction was brought to 50% conversion; then, the reaction was stopped by filtering the biocatalyst.
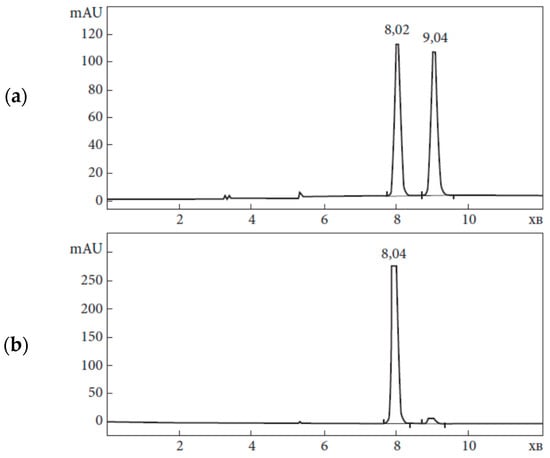
Figure 5.
An example of the analysis of 3-(4-fluorophenyl)-2-methylpropanoic acid by chiral HPLC: (a)—racemate; (b)—(R)-2a (chiral column Chiralcel OJ-H (250 × 4.6 mm, with Tris(4-methylbenzoate) cellulose selector deposited on 5 μm silica gel).
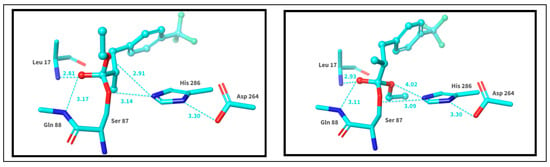
Figure 6.
The catalytic center and the covalently bound transition state complexes. The (S)-enantiomer is presented on the left, while the (R)-enantiomer is showcased on the right. To simplify visual noise, all protons were removed.
The resulting mixture of unreacted ester and acid was separated by column chromatography. For the kinetic transesterification of fluorinated arylcarboxylic acid, the best solvent was tert-butyl methyl ether (MTBE) and the best biocatalyst was Burkholderia cepacia at 35 °C. In this case, completion of the reaction at 50% conversion resulted in the hydrolysis of only the (R)-enantiomer of the racemic mixture. As a result, unreacted (S)-ester and (R)-carboxylic acid were obtained in high chemical yield and high enantiomeric excess in abundance (ee). Subsequent hydrolysis of the (R)-ester gave the (R)-ester configuration. Both (S)- and (R)-enantiomers of arylcarboxylic acids were thus obtained.
Therefore, the development of convenient express methods for determining the absolute configuration of arylcarboxylic acid is undoubtedly an interesting and useful practical task. The chiral HPLC, enzymatic analysis, and X-ray diffraction analysis provide a convergent determination of the absolute configuration of fluorinated arylcarboxylic acid. Biocatalytic resolution and chiral HPLC methods have many similarities. First of all, they are able to predict how deracemization of the secondary alcohol will occur.
4.2. Molecular Modeling of the of Enzymatic Resolution Process
Molecular modeling helps explain differences in enantioselectivity in the hydrolysis of racemic arylcarboxylic acid esters. Modeling of tetrahedral intermediates of racemic arylcarboxylic acid esters revealed that the two enantiomers bind differently to the Burkholderia cepacia lipase-binding site. A conformational search reveals several different binding modes for both enantiomers. In the lowest energy-binding mode (all the hydrogen bonds necessary for the reaction to proceed are formed, causing the fast-reacting enantiomer to place its large substituent in a hydrophobic, so-called HA pocket, similar to that found in the crystal structure of Bulrkholderia cepacia lipase. The Bulkholderia cepacia lipase-binding site was complex, with a secondary alcohol-like inhibitor. On the other hand, the slow-reacting enantiomer inserts its large substituent into a partially hydrophilic, so-called HH pocket.
The catalytic site 1HQD consists of residues Ser87, His286, and Asp264; Leu17 and Gln88 are also involved in catalysis. To create Michaelis complexes, ester ligands were covalently linked to the protein structure and assessed by covalent docking. After computer assessments, an analysis of the docking structures was carried out. Although the difference between the estimates is insignificant, it can be concluded that the (S)-enantiomer exhibits a greater tendency to form a transition–intermediate complex. As we can see, HIS286, in the case of the (S)-enantiomer, interacts better with the oxygen of the resulting complex, stabilizing it.
Depending on the size and shape of the substrate, the large substituent (L) of the fast-reacting enantiomer occupies a large hydrophobic pocket, the amino acid residues of which are Phe119, Val123, Ala247, Leu248, Thr251, and Val266. Its medium-sized substituent (M) fits into the HH pocket formed by the residues Tyr23, Tyr29, Leu287, Ile290, and Leu293. This binding is consistent with the results of kinetic measurements, i.e., even compounds with bulky L-substituents are catalyzed by the Bulkholderia cepacia lipase-binding site. Such a large substituent displaces Leu287 from the original position, reduces the rotational entropy of the His286 side chain, and interacts with the terminal CH3 group of the M substituent. The potential energy of the (S,R)-12-acyl-lipase-binding site of Bulkholderia cepacia exceeds the average energy calculated for the remaining complexes without a bulky substituent in the ortho-position. Apparently, the bulky substituent in the ortho-position destabilizes the complex.
Conformational analysis revealed the bound substrate with T2 and T3 predominantly in synclinal (±(60 ± 30°)) and T1 and T4 in antiperiplanar (±(180 ± 30°)) conformations. The orientations of the side chains of amino acid residues in the binding pocket in most cases deviate slightly from their position in the free enzyme. The largest deviation was observed for the imidazole ring His286. In some complexes, it is oriented approximately ±30° from the position determined in the crystal structures of the free enzyme (67° in LIP2). These data are consistent with the high similarity of the crystal structures of the native enzyme and its complexes.
The difference in the potential energies of the diastereomeric complexes (acyl- Bulkholderia cepacia lipase-binding site—alcohol) correctly predicted the fast-reacting enantiomer in all cases (Figure 6).
4.3. Molecular Dynamics Simulation
An analysis of the MD trajectories of the complexes was conducted. In the (S)-enantiomer, stable hydrogen bonds with Gln88 (100% of the time), Leu17 (100% of the time), and His286 (95% of the time) were maintained throughout the 20 ns simulation. Additionally, a Pi–cation interaction between the ligand’s aromatic ring and His286 was present (17% of the time). In contrast, in the case of the (R)-enantiomer, stable hydrogen bonds were retained with Gln88 (99% of the time) and Leu17 (99% of the time), while His286 reoriented away from the ligand after 0.48 ns, leading to conformational changes in the substrate, resulting in an unproductive complex. From this, it can be inferred that the S conformation is more favorable for the formation of the transition–state complex (Figure 7).
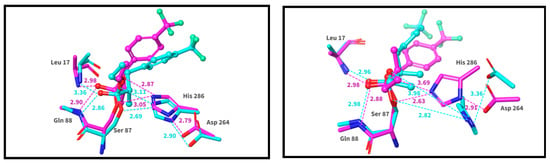
Figure 7.
The difference between the first and the last step of the ligand conformation in the MD simulation of the transition complex. The start of the simulation is shown in cyan; the end of the simulation is shown in magenta colors. To simplify visual noise, all protons were removed, even both polar hydrogens from histidine.
5. Conclusions
A number of racemic 3-arylalkanoic acids were successfully separated into enantiomers with an optical purity of >94% by lipase-catalyzed hydrolysis of the corresponding ethyl esters of arylcarboxylic acids. High values of the enantiopurity of 3-arylalkanoic acids catalyzed by hydrolase were achieved thanks to the optimization of the reaction conditions. In addition, it was determined that the substituents in the phenyl ring, acids, have an effect on the achievable enantioselectivity, which indicates that these hydrolases can more easily tolerate steric influence in the aryl group than in the 3-alkyl group.
In the work, we were able to reproduce the formation of the Michaelis complex for Burkholderia cepacia lipase. The results of covalent docking showed a slight gain in energy and interactions for the (S)-conformation. MD showed an even greater difference between the two stereoisomers. The change in the minimum distance in the trajectory from 3.93 to 4.09 is insignificant, and one can speak of the stability of this fundamentally important interaction. For the (R) conformation, towards the end of the trajectory, this interaction is completely lost, which confirms the key role of this amino acid residue, which is part of the catalytic triad, not only in the catalysis of the processes occurring but also in the discrimination of one of the stereoisomers. The mechanism of this process will be studied further.
Author Contributions
A.O.K., O.F., Y.G. and O.I.K. wrote this paper: experimental design, writing, funding acquisition, project administration, laboratory investigations, and spectroscopic analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
Author Yuliia Gurova was employed by the company Enamine Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- U.S. Food and Drug Administration. Development of New Stereoisomeric Drugs. 1 May 1992. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 8 August 2024).
- European Medicines Agency Investigation of Chiral Active Substances. 1 October 1993. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/investigation-chiral-active-substances_en.pdf (accessed on 8 August 2024).
- Calcaterra, A.D.; Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Caner, H.; Groner, E.; Levy, L. Trends in the development of chiral drugs. Drug Discov. Today 2004, 9, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 7, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Chiral organophosphorus pharmaceuticals: Properties and Application. Symmetry 2023, 15, 1550. [Google Scholar] [CrossRef]
- Kim, C.; Lee, J.; Cho, J.; Oh, Y.; Choi, J.K.; Choi, E.; Park, J.; Kim, M.-J. Kinetic and Dynamic Kinetic Resolution of Secondary Alcohols with Ionic-Surfactant-Coated Burkholderia cepacia Lipase: Substrate Scope and Enantioselectivity. J. Org. Chem. 2013, 78, 2571–2578 doiorg/101021/jo3027627. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.L. Enzymes in the synthesis of chiral drugs. Enzyme Microb. Technol. 1993, 15, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Deasy, R.E.; Brossat, M.; Moody, T.S.; Maguire, A.R. Lipase catalysed kinetic resolutions of 3-aryl alkanoic acids. Tetrahedron Asymmetry 2011, 22, 47–61. [Google Scholar] [CrossRef][Green Version]
- Irurre, J., Jr.; Casas, J.; Messeguer, A. Resistance of the 2,2,2-trifluoroethoxy aryl moiety to the cytochrome P-450 metabolism in rat liver microsomes. Bioorg. Med. Chem. Lett. 1993, 3, 179–182. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, L.; Wang, C.-J.; Zhang, X. Rh-Catalyzed highly enantioselective synthesis of 3-arylbutanoic acids. Angew. Chem. Int. Ed. 2007, 46, 2623–2626. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.R.; Kazlauskas, R.J. Hydrolases in Organic Synthesis, Regio- and Stereoselective Biotransformations, 2nd ed.; Wiley: Hoboken, NJ, USA, 2006; Print ISBN:9783527310296, Online ISBN:9783527607549. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).