1. Introduction
Continuous flow processes have recently emerged as a powerful technology for performing chemical transformations since they ensure some advantages over traditional batch procedures, for example, safer handling of hazardous materials, more efficient heat and mass transfers and precise control of reaction parameters by inline monitoring may be accomplished [
1,
2,
3,
4,
5]. Many research groups have focused attention onto this field favoring a gap closure between academia and industry [
6,
7,
8,
9,
10,
11,
12,
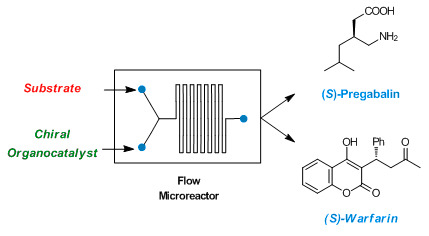
13]. An example of innovative synthetic procedure that could be applied in pharmaceutical industry was reported by Seeberger and coworkers. They recently published the continuous flow synthesis of five Active Pharmaceutical Ingredients (APIs) (Rolipram, Pregabalin, Phenibut, Baclofen, and Gabapentin) in racemic form. The flow system used to produce all these APIs is based on different modular synthetic platforms, operating under a wide range of reaction conditions. By following different linking pathways of the modules, the synthetic platform gives access to several small molecule drugs in multigrams scale without requiring intermediate purification [
14]. However, racemic mixtures of chiral APIs need to be resolved, since usually only one enantiomer is more active or effective. For this reason, the development of an enantioselective synthesis of chiral APIs would be highly desirable.
Asymmetric organocatalysis represents a powerful strategy for performing stereoselective transformations ensuring high levels of enantioselectivity [
15,
16]. The extremely fast growth of metal-free catalytic methodologies and novel organocatalytic stereoselective transformations has opened new avenues in the synthesis of complex chiral molecules, and has offered unique possibilities to develop more sustainable processes [
17]. Since organocatalysis involves simple organic molecules as catalysts, the contamination of final products with heavy metals is intrinsically avoided; thus, the combination of organocatalysis with flow chemistry would represent an effective methodology for the synthesis
in continuo of enantiomerically pure APIs.
The use of chiral organocatalysts under flow conditions is, however, still underdeveloped. In the last few years, the use of organocatalytic chiral reactors has received considerable attention, leading to the development of different supported chiral metal-free catalysts employed in continuous-flow processes [
18,
19,
20,
21]. Also our group contributed to this field and realized packed-bed and monolithic reactors containing solid supported chiral catalysts such as cinchona derivates [
22] and chiral imidazolidinones [
23,
24,
25]. On the other hand, homogeneous organocatalytic reactions performed under continuous flow conditions, for example in microreactors, are much less explored, the very few studies being almost exclusively limited to proline derivatives [
26,
27,
28,
29]. Recently, we have reported the use of a chiral bifunctional catalyst in glass microreactors, exploiting the potentiality of microfluidic technique to successfully accomplish enantioselective Michael type additions reactions [
30]. Commercially available glass microreactors have extensively been used for performing continuous flow organic transformations. Due to the narrow size of the channels (internal diameter less than 1 mm), flow microreactors feature extremely efficient heat and mass transfers and allow running chemical reactions very rapidly, with residence times of few minutes only.
In this work, we wish to demonstrate the possibility of using simple HPLC connections (PEEK or PTFE tubing, inner diameter 0.1–0.5 mm) instead of traditional glass microreactors to perform stereoselective organocatalyzed continuous flow reactions. In this context, the stereoselective synthesis in continuo of (S)-Pregabalin intermediate and (S)-Warfarin through two organocatalytic strategies has been reported.
2. Results and Discussion
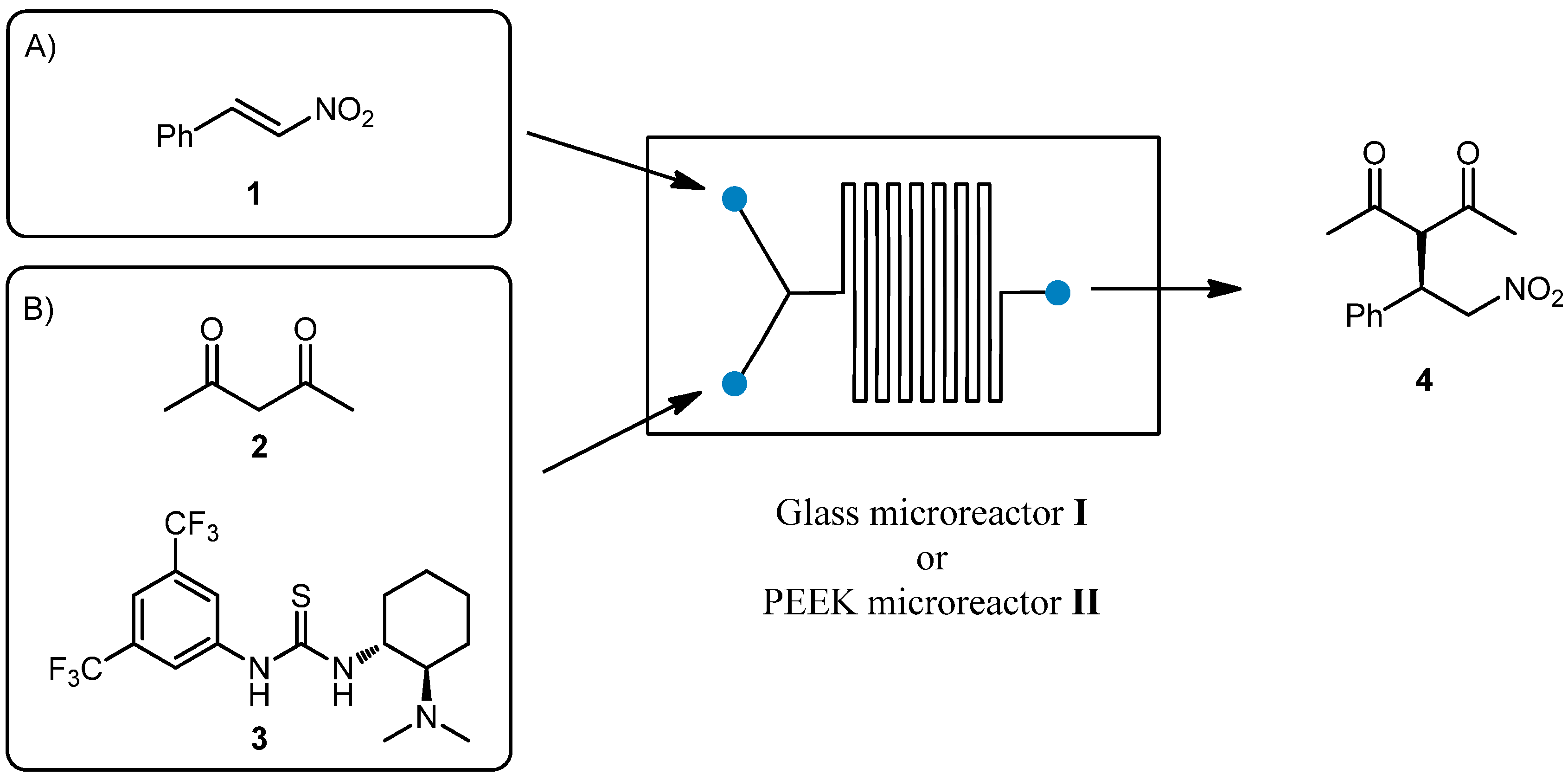
In a preliminary experiment, we compared the use of PEEK microreactor
II with the use of a commercially available glass microreactor
I (
Figure 1), recently used by our group in catalytic Michael reactions. In our previous work [
30], the addition of acetylacetone
2 to nitrostyrene
1 catalyzed by Takemoto thiourea
3 [
31,
32] (
Scheme 1) was performed in a Chemtrix Labtrix
® Start Standard platform system (Glass microreactor
I, Chemtrix SOR 3223, channel width 0.3 mm and channel depth 0.12 mm, total volume 10 μL, 70 °C and 5 min residence time) to give the corresponding product
4 in 91% yield and 80% ee.
Figure 1.
Examples of (a) glass microreactor I and (b) PEEK (Polyetheretherketone) microreactor II.
Figure 1.
Examples of (a) glass microreactor I and (b) PEEK (Polyetheretherketone) microreactor II.
Scheme 1.
The addition of acetylacetone 2 to nitrostyrene 1 catalyzed by Takemoto thiourea 3.
Scheme 1.
The addition of acetylacetone 2 to nitrostyrene 1 catalyzed by Takemoto thiourea 3.
In this work, the glass microreactor was replaced with a commercially available PEEK tubing designed for HPLC applications (PEEK microreactor II, inner diameter 0.127 mm, length 79 cm, total volume 10 μL) that was coiled in a bundle and immersed in a preheated oil bath. A Chemix Fusion 100 syringe pump equipped with two Hamilton gastight 1 mL syringes was used to feed the reagents into the microreactor through a T-junction (Syringe A: nitrostyrene 1; Syringe B: acetylacetone 2 and Takemoto thiourea 3).
Preliminary results are reported in
Table 1.
Table 1.
Comparison between glass microreactor I and PEEK (Polyetheretherketone) microreactor II.
Table 1.
Comparison between glass microreactor I and PEEK (Polyetheretherketone) microreactor II.
| Entry | Reactor | T (°C) | Flow Rate (μL/min) a | Residence Time (min) | Conversion (%) b | ee (%) c |
|---|
| 1 | Glass microreactor I | RT | 2 | 5 | 32 | 47 |
| 2 | Glass microreactor I | 70 | 2 | 5 | 91 | 80 |
| 3 | PEEK microreactor II | RT | 2 | 5 | 76 | 80 |
| 4 | PEEK microreactor II | 65 | 2 | 5 | 99 | 75 |
| 5 | PEEK microreactor II | 65 | 4 | 2.5 | 99 | 77 |
Gratifying, product 4 was obtained in 76% yield and 80% ee even at room temperature when PEEK microreactor II was employed (Entry 3 vs. Entry 2). Better yields were obtained when the temperature was increased to 65 °C, where a complete conversion was observed without loss of enantioselectivity (Entry 4). Even halving the residence time (Entry 5) allowed obtaining comparable yield and ee.
On the basis of these satisfactory results, we decided to explore the use of PEEK microreactor technology to perform organocatalyzed stereoselective synthesis of APIs. In particular, the stereoselective addition of diethylmalonate
6 to aliphatic nitroalkene
5 catalyzed by Takemoto thiourea
3 for the synthesis of (
S)-Pregabalin intermediate
7 is reported in
Section 2.1; the use of chiral primary amine
10 derived from cinchona alkaloids for the continuous flow stereoselective synthesis of (
S)-Warfarin
11 is highlighted in
Section 2.2.
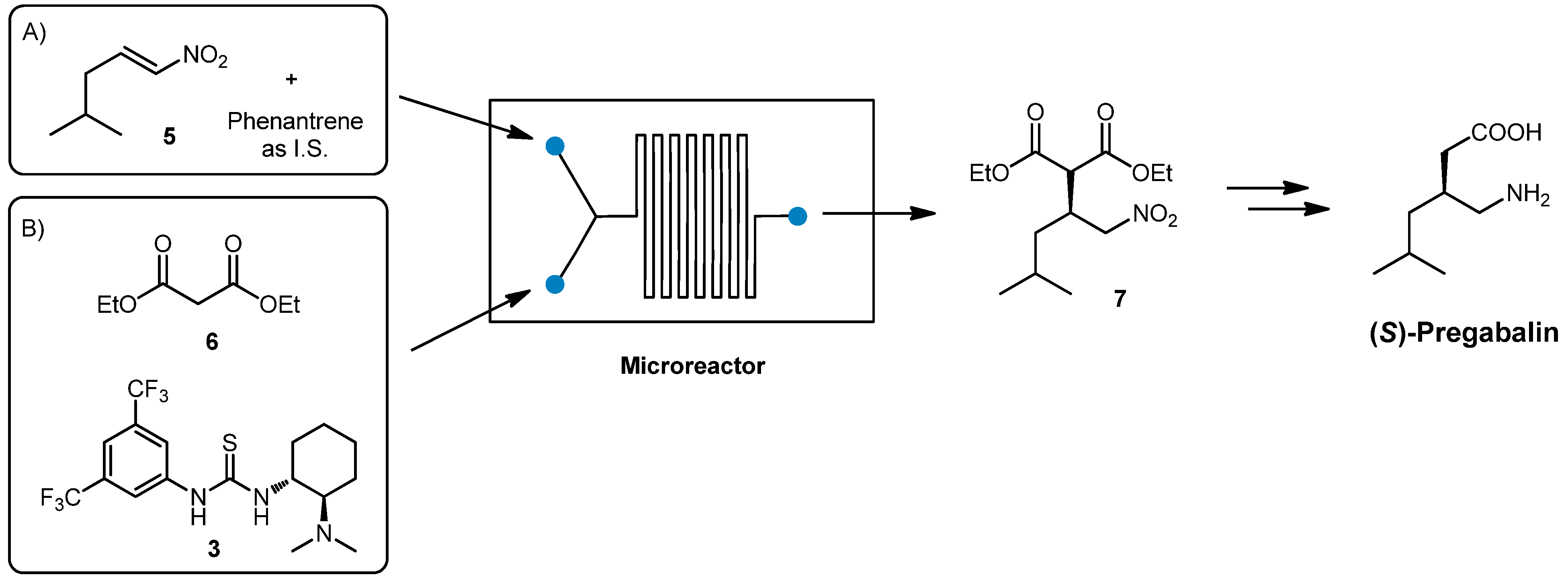
2.1. Chiral Thiourea Organocatalyst: Continuous Flow Synthesis of (S)-Pregabalin Intermediate
(
S)-Pregabalin, marketed under the trade name Lyrica, is a wide-administered anticonvulsant and antiepileptic drug. As previously reported in the literature, the stereoselective nucleophilic addition of diethylmalonate
6 to nitroalkene
5 promoted by chiral thiourea
3 could be considered a suitable strategy for the preparation of chiral intermediate
7 [
33,
34]. Reduction of the nitro moiety followed by hydrolysis and decarboxylation, affords (
S)-Pregabalin (
Scheme 2).
In our initial experiments, we employed PEEK microreactor
II with a total volume of 10 μL in order to demonstrate the compatibility of the organocatalytic methodology with continuous flow processes. A syringe pump equipped with two syringes was used to feed microreactor
II with the reagents through a T-junction. Conversion and enantiomeric excess were calculated through high-performance liquid chromatography (HPLC) analysis using phenantrene as an internal standard and with preliminary calibration experiments (Syringe A: nitroalkene
5 and internal standard; Syringe B: diethylmalonate
6 and Takemoto thiourea
3). Microreactor
II was coiled in a bundle and immersed in a preheated oil bath. After a screening of reagent concentrations and reaction temperatures (
Table 2), it was found that the product was formed in 36% conversion and 79% enantiomeric excess using a residence time of 10 min (Entry 8,
Table 2). It is worth noting that the same reaction performed in a single batch afforded product
7 in 88% yield and 81% ee after 48 h at room temperature (literature data [
31]); in our hands, product
7 was recovered in >98% yield and 77% ee after 48 h at 60 °C. To further compare the flow process to the batch process, we performed the same reaction at 70 °C and stopped it after 10 min reaction (residence time under continuous flow conditions); product
7 was recovered in 8% yield only (75% ee).
Scheme 2.
Synthesis of (S)-Pregabalin in flow.
Scheme 2.
Synthesis of (S)-Pregabalin in flow.
Table 2.
Preliminary results for the synthesis of (
S)-Pregabalin intermediate
7: 10 μL PEEK microreactor
II, flow rate 1 μL/min, residence time 10 min.
![Symmetry 07 01395 i001]()
Table 2.
Preliminary results for the synthesis of (S)-Pregabalin intermediate 7: 10 μL PEEK microreactor II, flow rate 1 μL/min, residence time 10 min. ![Symmetry 07 01395 i001]()
| Entry | 5 [M] a | 6 [M] a | T (°C) | Conversion (%) b | ee (%) c |
|---|
| 1 | 0.5 | 1.0 | 25 | 0 | - |
| 2 | 0.5 | 1.0 | 65 | 35 | 77 |
| 3 | 0.5 | 1.0 | 100 | 28 | 71 |
| 4 | 0.5 | 2.0 | 100 | 32 | 69 |
| 5 | 0.5 | 2.0 | 120 | 22 | 59 |
| 6 | 0.5 | neat | 105 | 19 | 76 |
| 7 | 0.5 | neat | 135 | 19 | 72 |
| 8 | 1.0 | 3.3 | 60 | 36 | 79 |
| 9 | 1.0 | 3.3 | 80 | 34 | 76 |
| 10 | 1.0 | neat | 60 | 25 | 77 |
| 11 | 1.0 | neat | 85 | 24 | 73 |
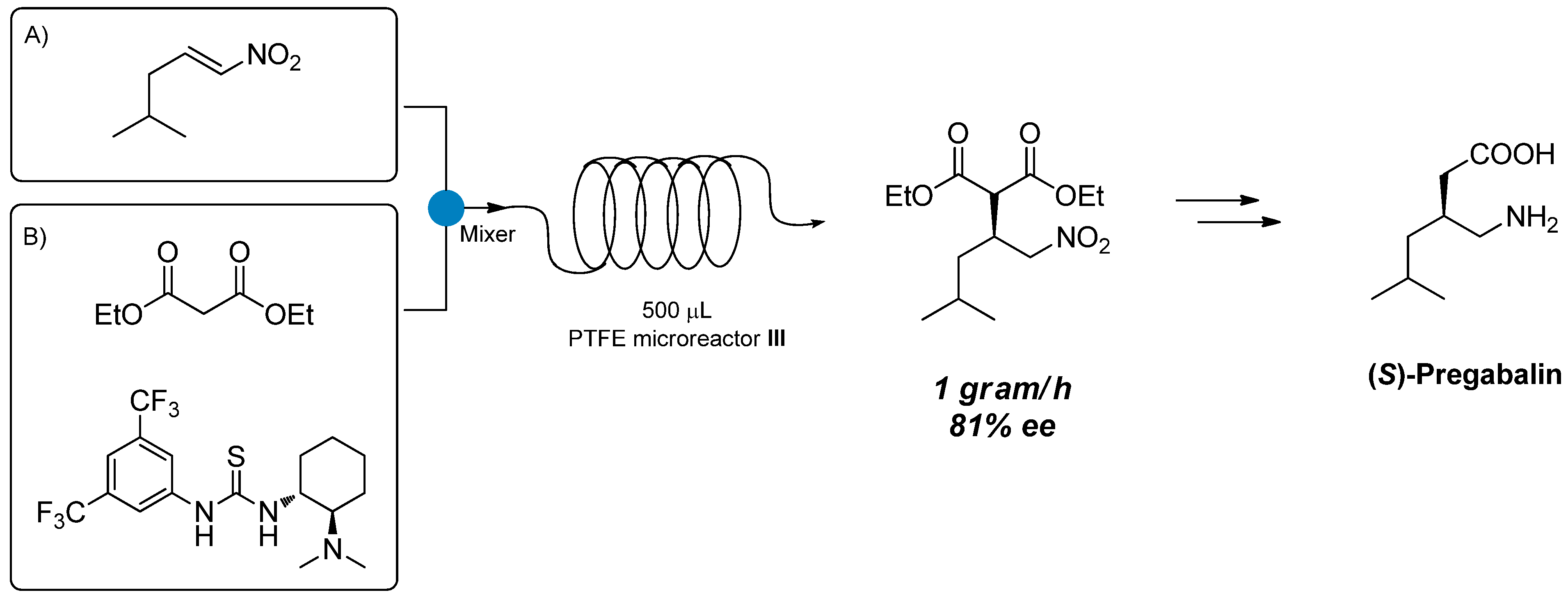
Attempts to scale up the reaction by increasing the size of the microreactor were conducted using PTFE microreactor
III (inner diameter 0.58 mm, length 189 cm, total volume 500 μL) (
Scheme 3). By performing the reaction under the previously optimized reaction conditions, we were able to obtain product
7 in 40% conversion and 79% ee (Entry 3,
Table 3), thus confirming the results obtained using microreactor
II (
Table 3). When the residence time was lowered from 10 to 5 and 2 min (by increasing the flow rate), conversions and ee remained unchanged (Entries 4 and 5); no improvements in terms of conversion were observed when the temperature was increased to 80 °C (Entry 6). We then decided to performed a gram-scale synthesis of (
S)-Pregabalin intermediate
7: under the best reaction conditions (60 °C, 2 min residence time) we were able to produce 1 g of
7 in 81% ee, in one hour only using a very simple flow apparatus (
Scheme 3).
Scheme 3.
Gram-scale synthesis of 7 in 500 μL microreactor III.
Scheme 3.
Gram-scale synthesis of 7 in 500 μL microreactor III.
Table 3.
500 μL PTFE microreactor
III, screening of different flow rates.
![Symmetry 07 01395 i002]()
Table 3.
500 μL PTFE microreactor III, screening of different flow rates. ![Symmetry 07 01395 i002]()
| Entry | Total Flow Rate (μL/min) a | Residence Time (min) | T (°C) | Conversion (%) b | ee (%) c |
|---|
| 1 | 2 | 30 | 60 | 55 | 76 |
| 2 | 4 | 15 | 60 | 56 | 76 |
| 3 | 6 | 10 | 60 | 40 | 79 |
| 4 | 12 | 5 | 60 | 37 | 79 |
| 5 | 30 | 2 | 60 | 37 | 80 |
| 6 | 30 | 2 | 80 | 13 | 77 |
2.2. Chiral Primary Amine Organocatalyst: Continuous Flow Synthesis of (S)-Warfarin
(
S)-Warfarin, known with the brand name of Coumadin or Jantoves, is an oral anticoagulant, a drug that inhibits the clotting of blood. Since it can be prepared through different organocatalytic strategies [
35,
36], we identified the nucleophilic addition of 4-hydroxy-coumarin
8 to benzalacetone
9 catalyzed by cinchona-derived primary amine
10 [
37] as the most straightforward methodology compatible with flow microreactors.
A solvent screening for the homogeneous reaction in batch was first performed in order to select the best reaction medium to provide high ee (see
Table S1 of ESI for further details). It was found that dioxane was the best solvent for the addition of 4-hydroxy-coumarin
8 to benzalacetone
9 promoted by cinchona-derived primary amine
10 (10 mol%) in the presence of trifluoroacetic acid as a co-catalyst, that afforded product
11 in 99% yield and 96% ee after 24 h at 30 °C (Entry 6,
Table S1).
In order to optimize the reaction conditions in flow, we employed PEEK microreactor
II; the flow apparatus was settled as illustrated in
Scheme 4. A syringe pump equipped with two syringes was used to feed microreactor
II with the reagents through a T-junction (Syringe A: coumarin
8; Syringe B: benzalacetone
9, chiral catalyst
10 and trifluoroacetic acid). Microreactor
II was coiled in a bundle and immersed in a preheated oil bath. Conversion was determined by
1H-NMR spectroscopy of the crude mixture while enantiomeric excess through HPLC on chiral stationary phase.
Scheme 4.
Synthesis of (S)-Warfarin in flow.
Scheme 4.
Synthesis of (S)-Warfarin in flow.
Different combinations of flow rates and reaction temperatures were investigated (
Table 4). It should be noted that, due to low solubility profile of coumarin
8, it was necessary to work under high dilution conditions (0.05 M) in order to prevent coumarin precipitation in the syringe. As shown in
Table 4, the conversion increased with increasing temperature, while the ee remained constantly high (>90%). Optimum reaction conditions were found to be 75 °C and 1 μL/min flow rate (corresponding to 10 min residence time), which lead to the formation of the desired product
11 with 61% conversion and 93% ee (Entry 5,
Table 4).
With the aim to scale up the reaction we tried to synthetize compound
11 using the 500 μL PTFE microreactor
III under the best reaction conditions previously found. However, the reaction conditions settled in microreactor
II showed to be not reproducible: by performing the reaction in dioxane at 75 °C with a residence time of 10 min (obtained with 50 μL/min flow rate), the product was isolated in 95% ee but in 29% yield only (Entry 1,
Table 5). An increase of the residence time did not have a beneficial effect in terms of both yield (20% yield) and enantioselectivity (Entries 2 and 3,
Table 5).
Table 4.
Screening of flow rate and temperature for 10 μL PEEK microreactor
II.
![Symmetry 07 01395 i003]()
Table 4.
Screening of flow rate and temperature for 10 μL PEEK microreactor II. ![Symmetry 07 01395 i003]()
| Entry | Total Flow Rate (μL/min) a | Residence Time (min) | T (°C) | Conversion (%) b | ee (%) c |
|---|
| 1 | 2 | 5 | 30 | 15 | 95 |
| 2 | 1 | 10 | 40 | 38 | 95 |
| 3 | 1 | 10 | 50 | 51 | 95 |
| 4 | 1 | 10 | 60 | 55 | 95 |
| 5 | 1 | 10 | 75 | 61 | 93 |
| 6 | 1 | 10 | 90 | 65 | 90 |
Table 5.
Synthesis of
11 in 500 μL PTFE microreactor
III.
![Symmetry 07 01395 i004]()
Table 5.
Synthesis of 11 in 500 μL PTFE microreactor III. ![Symmetry 07 01395 i004]()
| Entry | Total Flow Rate (μL/min) a | Residence Time (min) | Yield (%) b | ee (%) c |
|---|
| 1 | 50 | 10 | 29 | 95 |
| 2 | 17 | 30 | 20 | 86 |
| 3 | 8 | 60 | 20 | 81 |
It is possible that different microfluidic properties of a 0.58 mm inner diameter tubing (PTFE microreactor
III) with respect to the 0.127 mm inner diameter tubing (PEEK microreactor
II) could account for the failure of the scale up process. We then decided to explore the “numbering up” (or “scale out”) technique, which consists of connecting several microreactors of the same dimension in parallel. As depicted in
Scheme 5, by using a stainless-steel splitter (2 inlet and 4 outlet), four PEEK microreactors
II were connected in parallel and immersed in a preheated oil bath. A 0.05 M solution of coumarin
8 and a 0.1 M solution of benzalacetone
9 together with catalyst
10 and trifluoroacetic acid, were fluxed into the splitter with a flow rate of 4 μL/min (10 min residence time in each microreactor); the microreactors were kept at 75 °C and the reactors outcome was collected into a vial. Product
11 was isolated in 36% yield and 91% ee. Attempts to collect the four reaction outcomes separately showed that the four microreactors afforded slightly different yields. At this stage we believe that the mixing in the splitter is not efficient and it may account for the lower yield; further studies and optimization works on the differently components of the device are currently going on. In addition, the necessity of working under dilution conditions, due to the low solubility of coumarin, prevents the possibility to produce a large amount of the desired product in a short reaction time.
Scheme 5.
Scale-out process: 4 × 10 μL PEEK microreactors II.
Scheme 5.
Scale-out process: 4 × 10 μL PEEK microreactors II.
Despite the unsatisfactory results obtained in the
Warfarin scale up process, few issues deserve a comment: by using a small-scale reactor it is possible to easily monitor the reaction course (by
1H-NMR and/or HPLC analysis) thus establishing the best reaction conditions very quickly by adjusting some simple reaction parameters such as temperature and flow rate. After the large-scale reaction, a purification step is required in order to separate the desired product from unreacted starting material and homogeneous catalyst. This purification step (that is also usually required in the batch process) somehow affects the appealing of the continuous-flow process and more efforts are required to circumvent this problem. One possible solution could be represented by the use of supported catalysts, that is a well-established methodology when
meso- or
mini-reactors are used [
18], but much less exploited in the case of microreactors [
38]. The combination of an immobilized catalyst and a complete conversion of the starting material (or an easy separation of the desired product) could allow a very attractive procedure for large-scale production of interesting intermediates.
3. Experimental
3.1. General Methods
Reactions were monitored by analytical thin-layer chromatography (TLC) using silica gel 60 F254 pre-coated glass plates (0.25 mm thickness) and visualized using UV light. Flash chromatography was carried out on silica gel (230–400 mesh). Proton NMR spectra were recorded on spectrometers operating at 300 MHz (Bruker Fourier 300 or AMX 300 (Bruker, Billerica, MA, USA)). Proton chemical shifts are reported in ppm (δ) with the solvent reference relative to tetramethylsilane (TMS) employed as the internal standard (CDCl3 δ = 7.26 ppm).
Enantiomeric excess determinations were performed under below reported conditions with Agilent 1200 or 1100 series HPLC (Agilent, Santa Clara, CA, USA).
Reagents mixtures were fed to continuous flow reactors using Syringe Pump Chemtrix Chemix Fusion 100 (Chemtrix, Geleen, The Netherlands).
3.2. Materials
Dry solvents were purchased and stored under nitrogen over molecular sieves (bottles with crown caps). Commercial grade reagents and solvents were used without further purifications. Quinine (anhydrous, technical grade 98%) and trifluorocetic acid (technical grade 99%) were purchase from Sigma-Aldrich (St. Louis, MO, USA) and used without further purifications.
Trans-nitrostyrene 1 was recrystallized from ethanol, 4-hydroxy-coumarin 8 was recrystallized from ethyl acetate, and benzalacetone 9 was recrystallized from hexane.
Trans-4-methyl-1-nitropent-1-ene [
33] was prepared according to published procedure.
3.3. Microreactors
Microreactors were prepared using tubing for HPLC connections purchased from Sigma-Aldrich.
PEEK microreactor II: PEEK tubing, inner diameter 0.127 mm, length 79 cm, total volume 10 µL.
PTFE microreactor III: PTFE tubing, inner diameter 0.58 mm, length 189 cm, total volume 500 µL.
3.4. Preparation of Catalysts 3 and 10
Chiral Takemoto thiourea
3 [
31] and 9-amino-9-deoxy-
epi-quinine
10 [
39] are known compounds and were prepared according to published procedures.
3.5. General Procedure for Continuous Flow Synthesis of Compound 4 Using PEEK Microreactor II
Syringe A was filled with a mixture of nitrostyrene 1 (2 mmol, 298 mg) dissolved in dry toluene (1 mL). Syringe B was loaded with a mixture of chiral thiourea 3 (0.2 mmol, 83 mg) and acetylacetone 2 (4 mmol, 0.41 mL) dissolved in dry toluene (0.6 mL). Syringes A and B were connected to a syringe pump and the reagents were pumped into microreactor II at the desired flow rate (mL/min) and temperature. Three reactor volumes were discarded before starting sample collection in order to achieve steady-state conditions. Reaction outcome was collected into a vial at room temperature and analyzed through 1H-NMR spectroscopy and HPLC in order to determine conversion and enantiomeric excess.
Compound
4 is known [
32]; it was purified by flash column chromatography on silica gel (eluent: Hexane/AcOEt = 8/2) to afford a colorless oil.
TLC Rf = 0.35 (Hexane/AcOEt = 8/2, stained yellow with KMnO4).
1H-NMR (300 MHz, CDCl3): δ 1.95 (s, 3H), 2.29 (s, 3H), 4.25–4.29 (m, 1H), 4.38 (d, J = 10.7 Hz, 1H), 4.60–4.69 (m, 2H), 7.09–7.21 (m, 2H), 7.21–7.39 (m, 3H).
The enantiomeric excess was determined by HPLC on chiral stationary phase with Daicel Chiralcel OD-H column: eluent Hexane/iPrOH = 98/2, flow rate 0.8 mL/min, λ = 280 nm, τminor = 7.9 min, τmajor = 10.8 min.
3.6. General Procedure for Continuous Flow Synthesis of (S)-Pregabalin Intermediate 7 Using PEEK Microreactor II
Syringe A was filled with a mixture of nitroalkene 5 (2 mmol, 258 mg) and phenantrene (1 mmol, 178 mg) dissolved in dry toluene (1 mL). Syringe B was loaded with a mixture of chiral thiourea 3 (0.2 mmol, 83 mg) and diethylmalonate 6 (6.6 mmol, 1 mL). Syringes A and B were connected to a syringe pump and the reagents were pumped into microreactor II at the desired flow rate (μL/min) at the desired temperature. Three reactor volumes were discarded before starting sample collection in order to achieve steady-state conditions. Reaction outcome was collected into a vial at room temperature and analyzed through 1H-NMR spectroscopy and HPLC in order to determine conversion and enantiomeric excess.
Compound
7 is known [
32]; it was purified by flash column chromatography on silica gel (eluent: Hexane/AcOEt = 8/2) to afford a colorless oil.
TLC Rf = 0.26 (Hexane/AcOEt = 8/2, stained yellow with KMnO4).
1H-NMR (300 MHz, CDCl3): δ 4.70 (dd, J = 13, 5 Hz, 1H), 4.51 (dd, J = 13, 6 Hz, 1H), 4.28–4.20 (m, 4H), 3.62 (d, J = 5 Hz, 1H), 3.00–2.95 (m, 1H), 1.72.166 (m, 1H), 1.38–1.28 (m, 8H), 0.96–0.92 (m, 6H).
The enantiomeric excess was determined by HPLC on chiral stationary phase with Daicel Chiralcel OD-H column: eluent Hexane/iPrOH = 98/2, flow rate 0.8 mL/min, λ = 280 nm, τminor = 7.9 min, τmajor=10.8 min.
3.7. General Procedure for Continuous Flow Synthesis of (S)-Warfarin 11 Using PEEK Microreactor II
Syringe A was filled with a mixture of 4-hydroxy-coumarin 8 (0.2 mmol, 32 mg) dissolved in dry dioxane (1 mL). Syringe B was loaded with a mixture of chiral primary amine 10 (0.02 mmol, 6 mg), trifluoroacetic acid (0.03 mmol, 3 μL) and benzalacetone 9 (0.4 mmol, 58 mg) dissolved in dry dioxane (1 mL). Syringes A and B were connected to a syringe pump and the reagents were pumped into microreactor II at the desired flow rate (μL/min) and temperature. Three reactor volumes were discarded before starting sample collection in order to achieve steady-state conditions. Reaction outcome was collected into a vial at room temperature and analyzed through 1H-NMR spectroscopy and HPLC in order to determine conversion and enantiomeric excess.
Compound
11 is known [
37]; it was purified by flash column chromatography on silica gel (eluent: Hexane/AcOEt = 7/3) to afford a white-off solid.
TLC Rf = 0.28 (Hexane/AcOEt = 7/3, stained yellow with KMnO4).
Compound 22 was found to exist in rapid equilibrium with a pseudo-diastereomeric hemiketal form in solution. However, the equilibrium is very rapid and therefore no pseudo-diastereomers were observed during HPLC analysis using the mixture of hexane/2-propanol containing 0.1% TFA as the eluent.
1H-NMR (300 MHz, CDCl3): δ 7.98 (dd, 1H), 7.93 (dd, 2.38H), 7.85 (dd, 3.15H), 7.58–7.50 (m, 7.83H), 7.22–7.4 (m, 54.13H), 4.73 (dd, 1.24H), 4.32 (dd, J = 3.6, 2.87H), 4.12–4.18 (m, 5.53H), 3.9 (dd, 1.55H), 3.35 (dd, 1.8H), 3.2 (bs, 2.76H), 2.6–2.4 (m, 10.6H), 2.09–2.00 (dd, 5H), 1.76 (s, 12H), 1.71 (s, 10H).
The enantiomeric excess was determined by HPLC on chiral stationary phase with Daicel Chiralpack AD column: eluent Hexane/iPrOH = 8/2 + 0.1% TFA, flow rate 0.8 mL/min, λ = 280 nm, τminor = 7.8 min, τmajor = 19.5 min.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

