


Effect of Solid Ratio and Particle Size on Dissolution of Heat-Activated Lizardite at Elevated Pressures and Moderate Temperatures

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
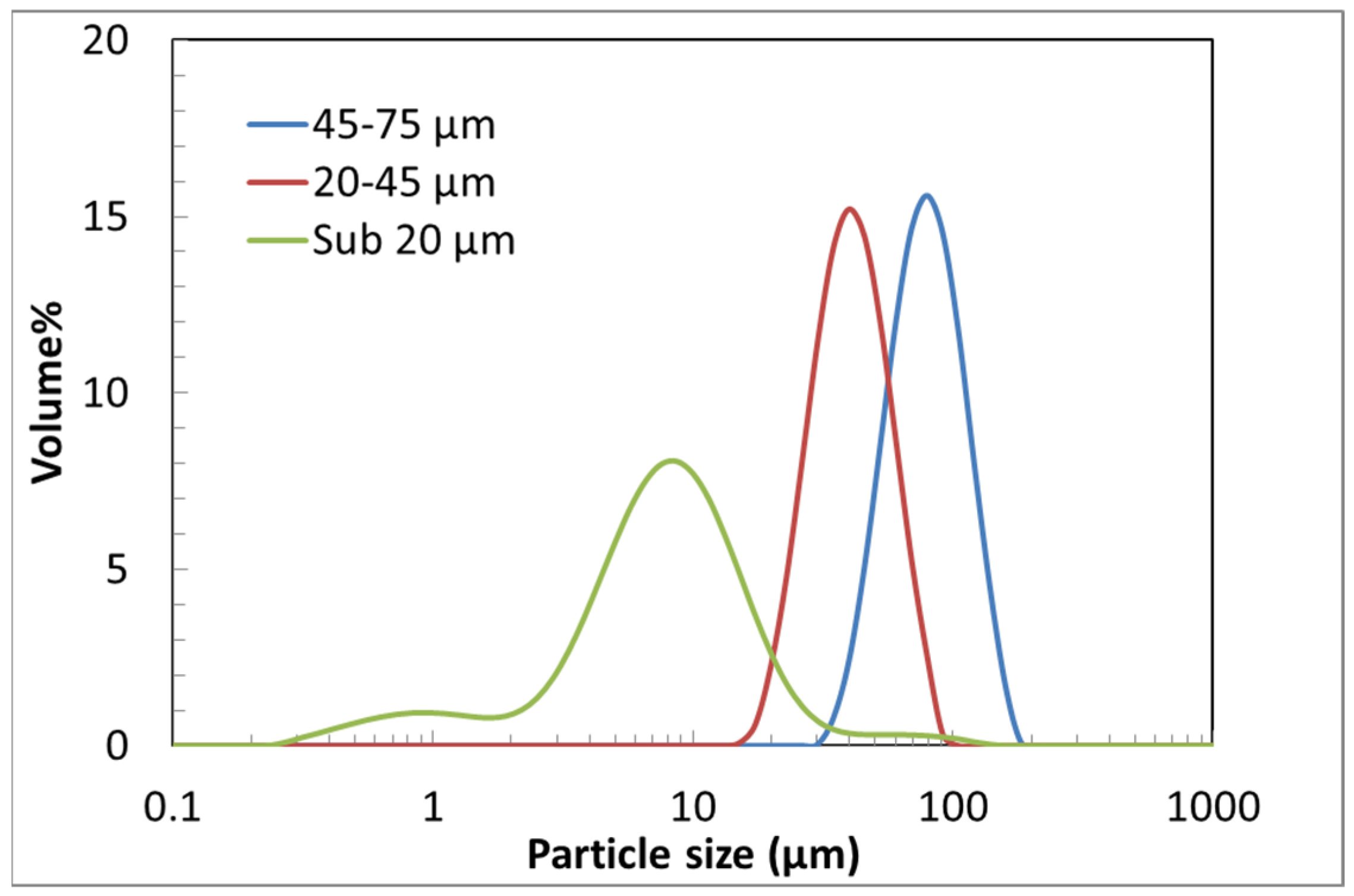
3.1. Particle Size Analysis
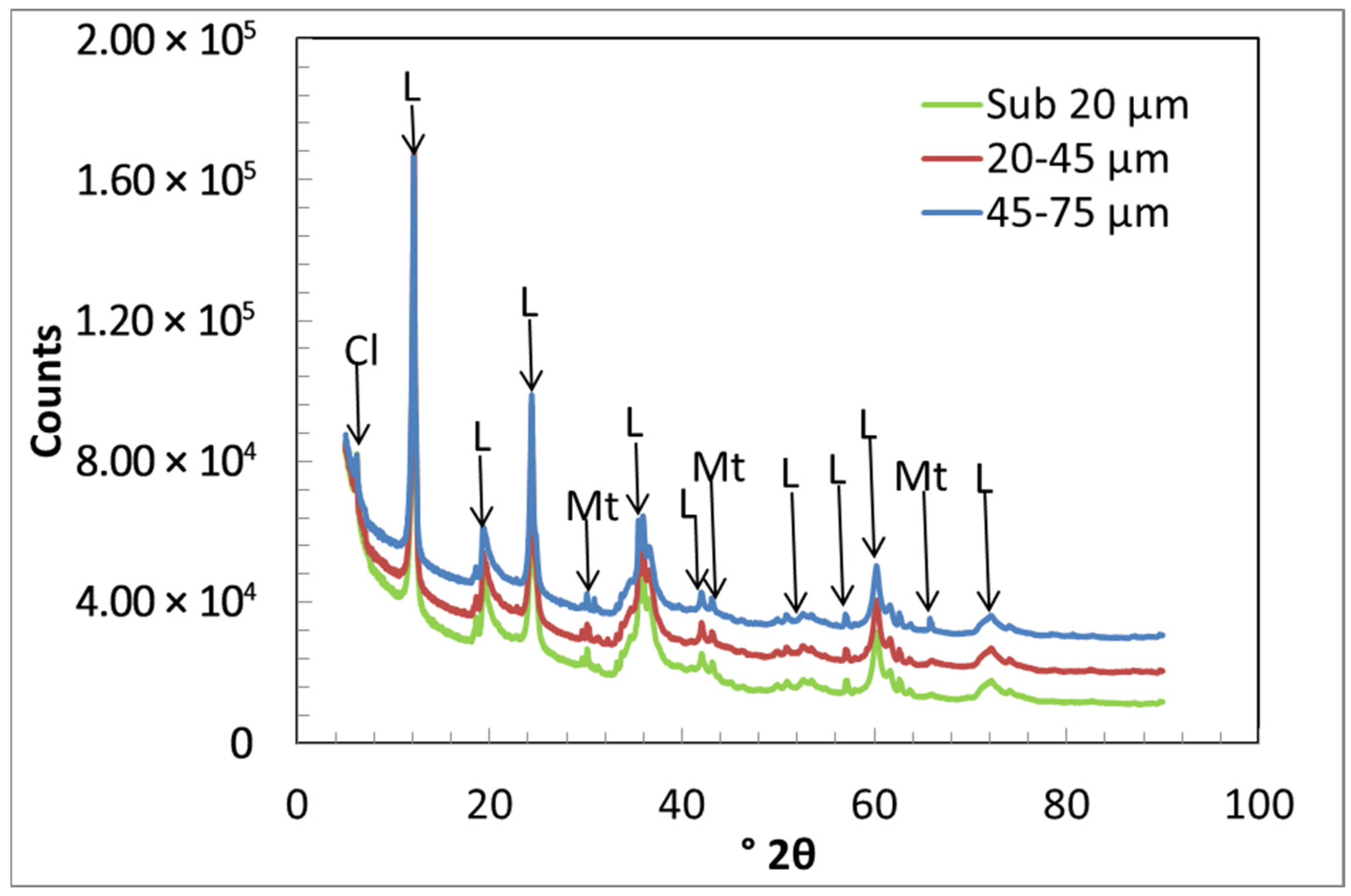
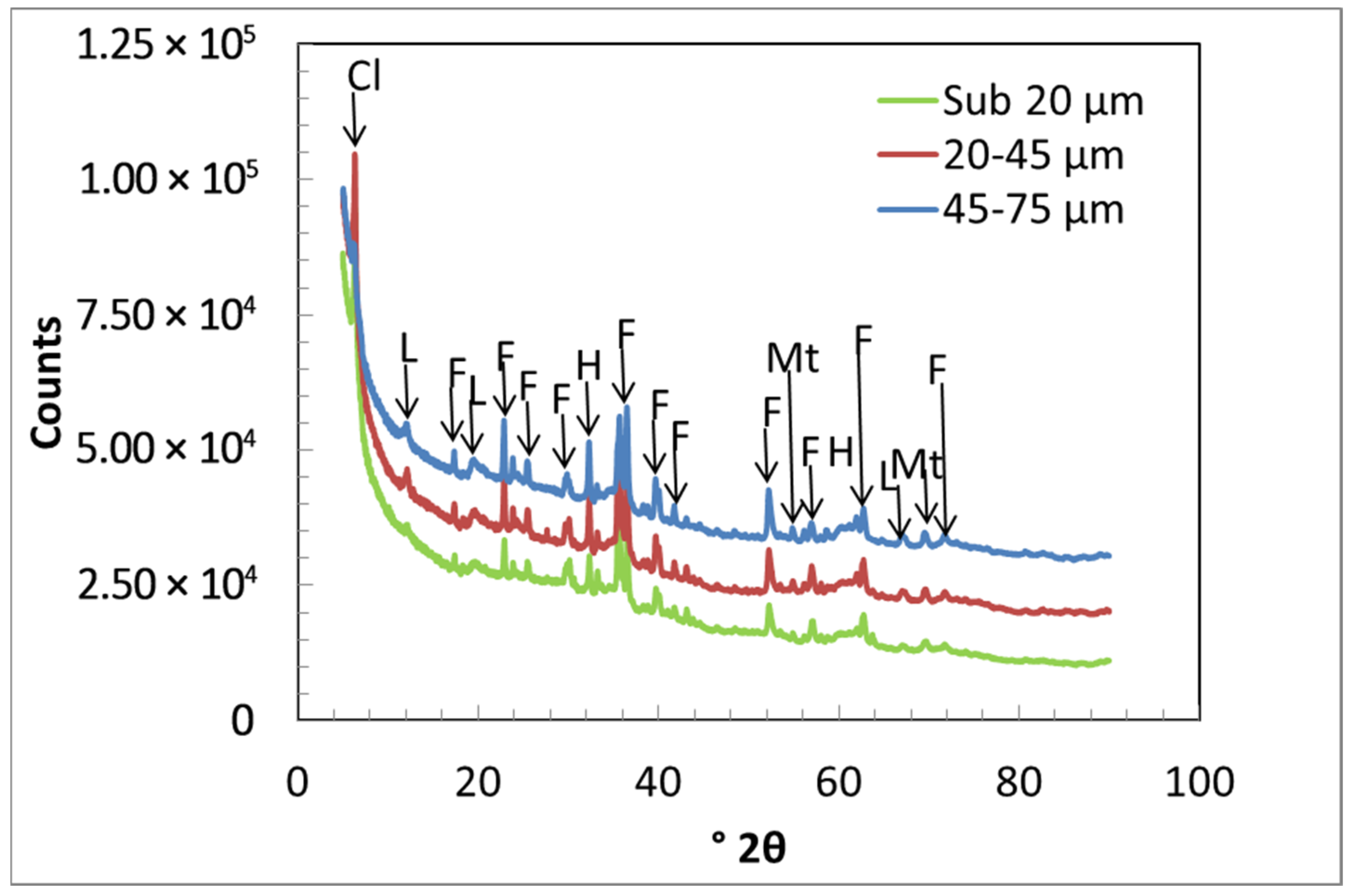
3.2. X-ray Analysis
3.3. Elemental Analysis of Supernatant Solutions with ICP-OES
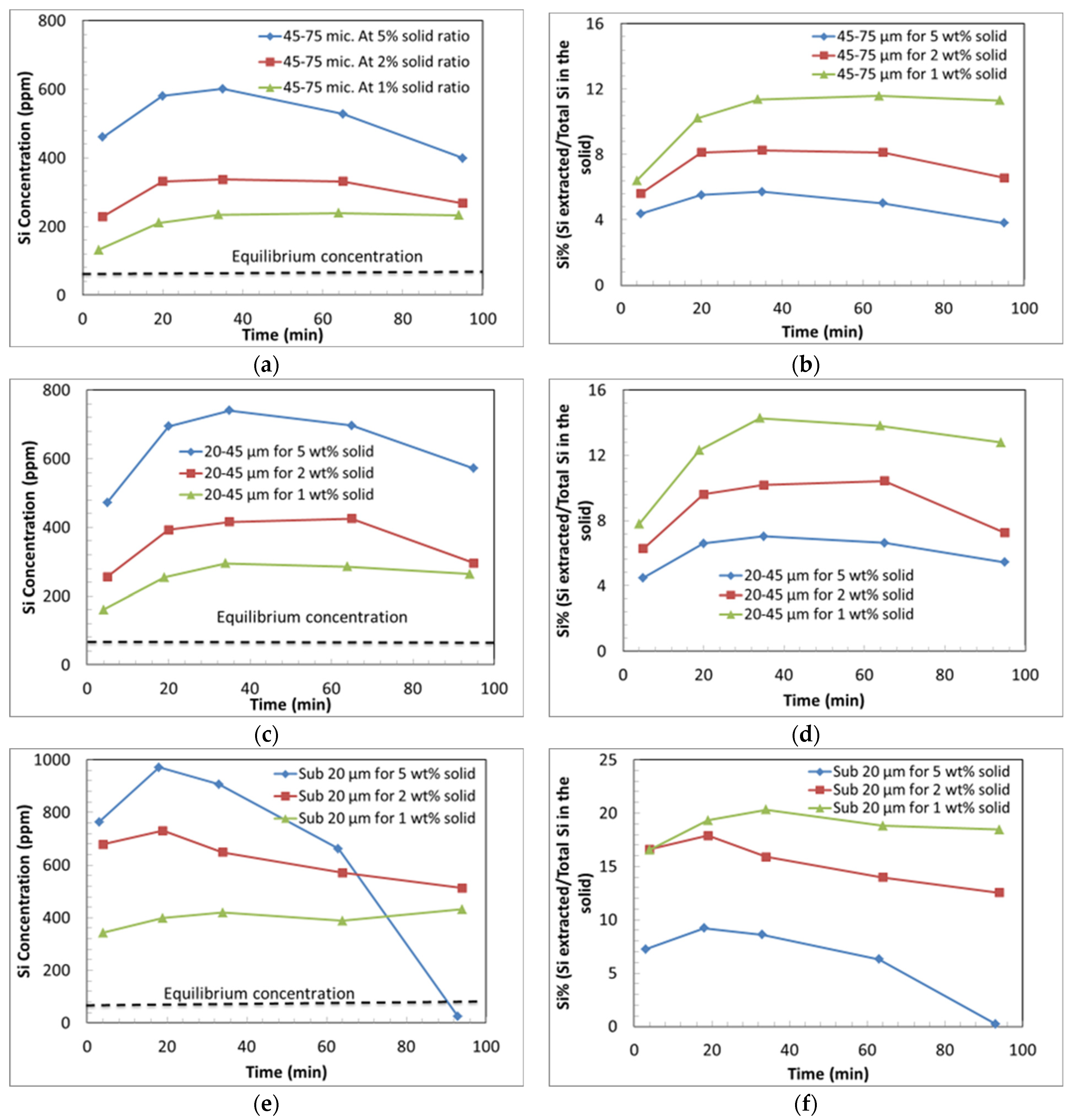
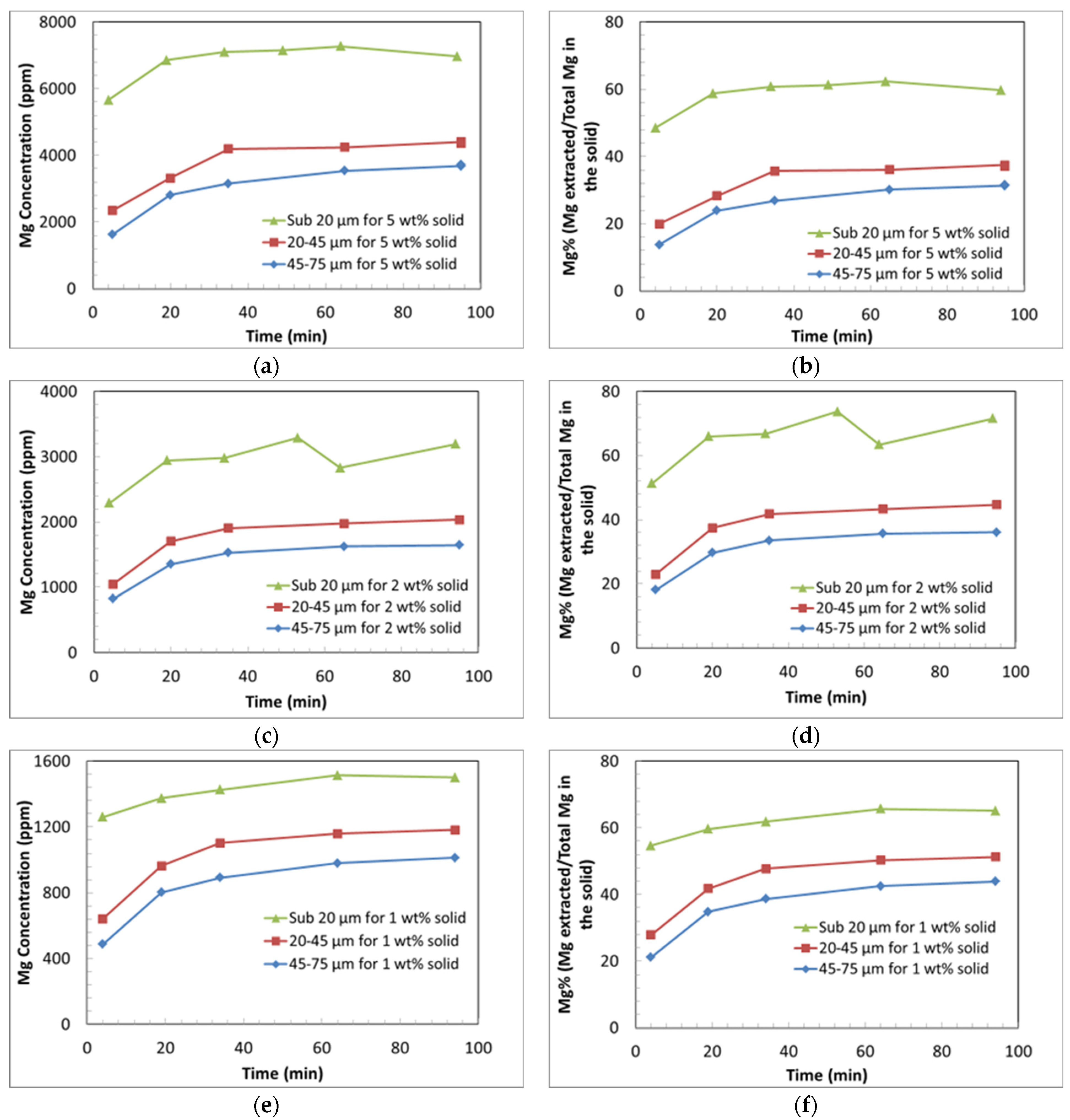
3.3.1. Effect of Liquid-to-Solid Ratio
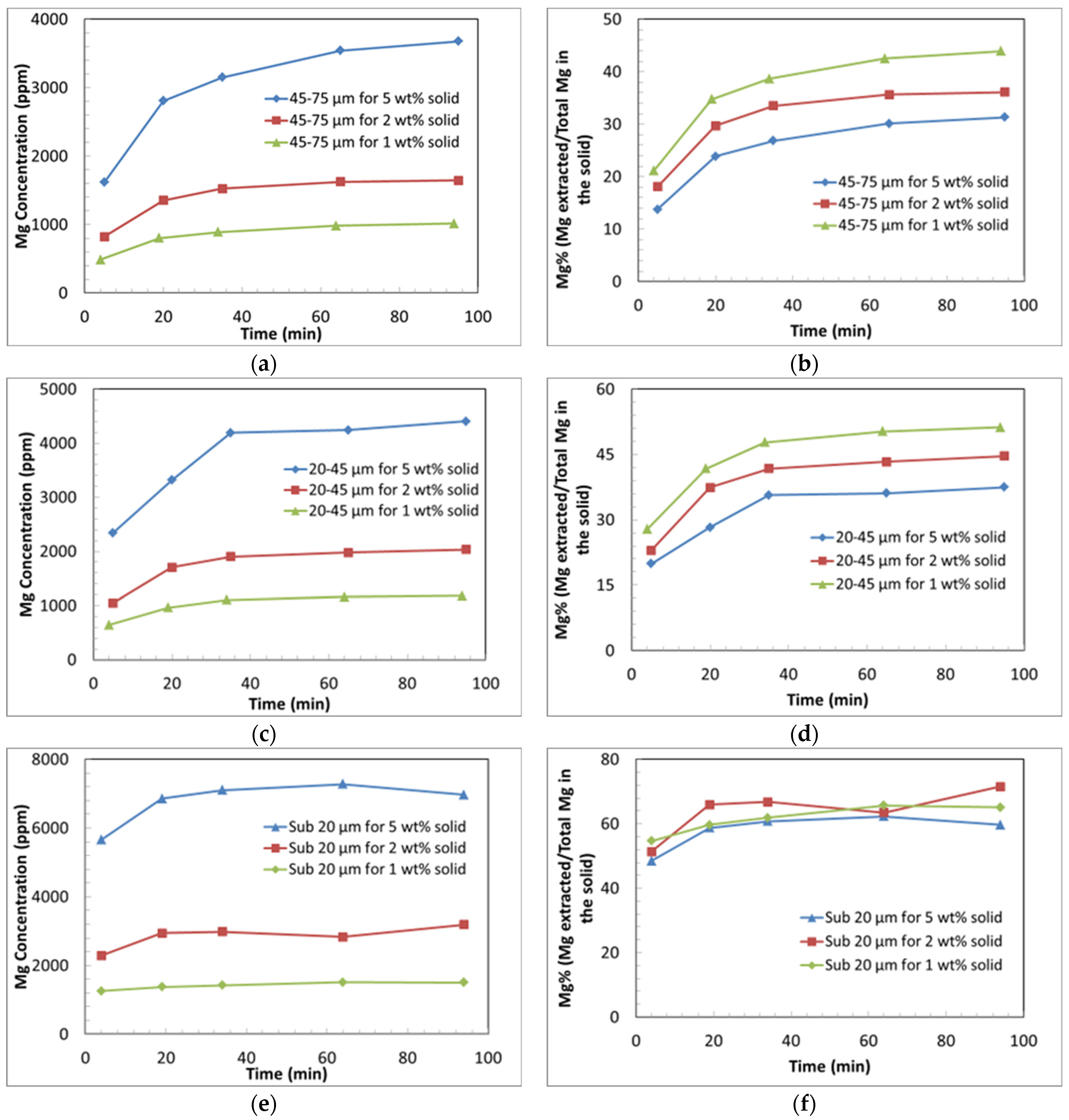
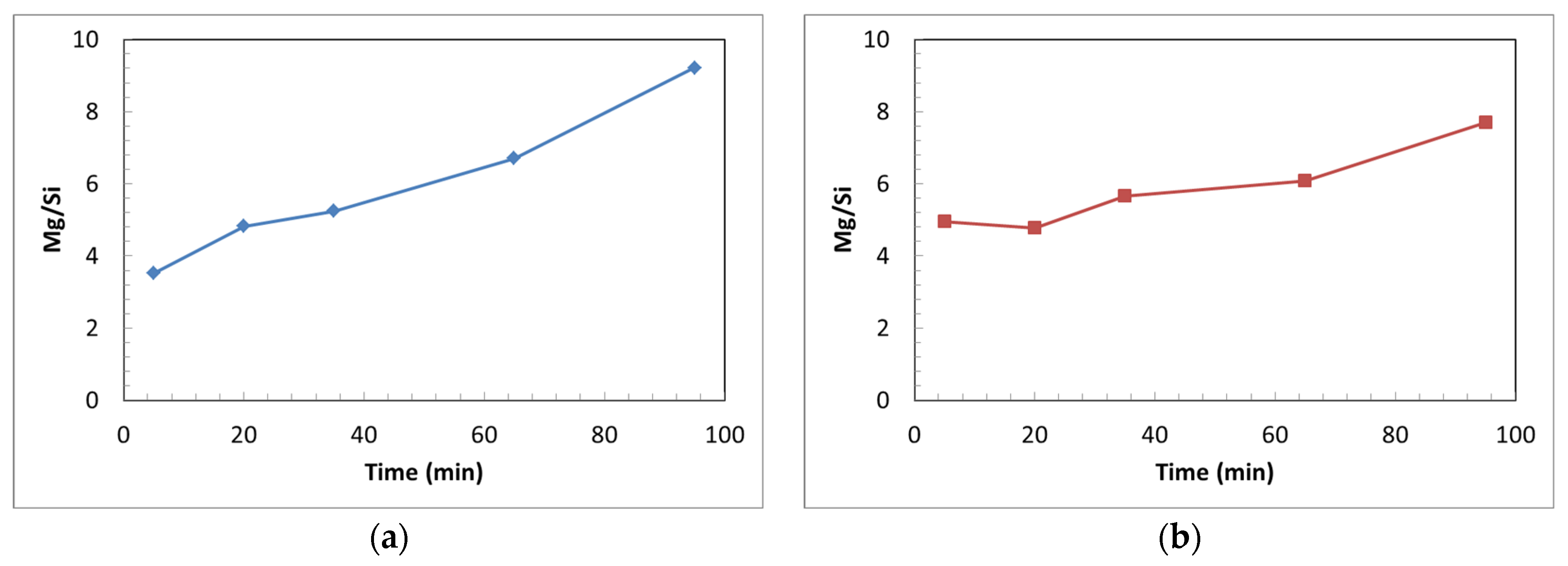
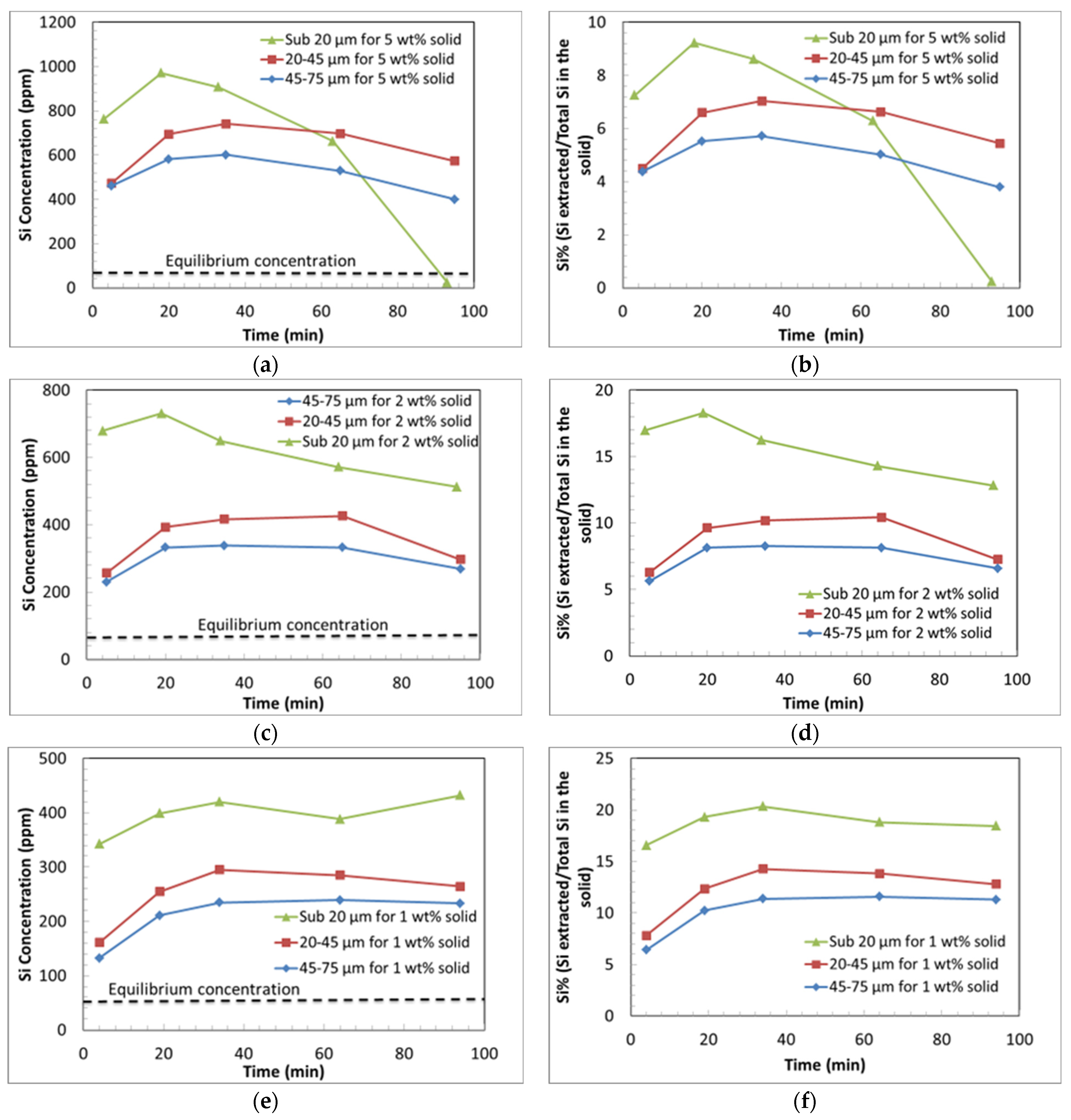
3.3.2. Effect of Particle Size on Si and Mg Extraction
- (a)
- The dissolution behaviour of Mg comprises two main stages: an initial rapid extraction, followed by a dramatic decrease in the dissolution rate.
- (b)
- The Mg concentration increases monotonically with time. This indicates that magnesium carbonate precipitation is greatly hindered compared to ARC conditions.
- (c)
- The extent of Mg extraction for coarse particles decreases with increasing solids loading. This is partly due to the effect of pH on extraction kinetics, as the increased solids loading results in a greater rise in pH over the course of the experiment.
- (d)
- The decrease in the extraction rate and extent with the increasing solids content is not fully explained by the effect of pH alone. Another possible contributing factor is the higher rate of deposition of Si, which may form a passivating layer, hindering further Mg extraction.
- (e)
- The high surface area of the fine particles (sub-20 µm) in the experiments leads to a high extent of Mg extraction regardless of the solids loading. A maximum extent of Mg extraction of approximately 65% was achieved using fine particles (sub-20 µm).
- (f)
- The low solids loading experiments produced a greater extent of Si extraction compared to the experiments with a higher solids loading.
- (g)
- The dissolution trends for Si also show two distinct steps; an initially rapid rate of extraction of Si, followed by a declining concentration of Si, indicative of the precipitation of Si from the solution.
- (h)
- It is hypothesised that amorphous silica precipitates at a greater rate in the higher solids loading experiments.
- (i)
- Silica gel was observed in all the collected samples at the time of collection and amorphous silica precipitation is predicted by the modelling. It is hypothesised that the silica gel transformed during the reaction into amorphous silica, which contributes to the formation of a Si passivating layer.
- (j)
- The extent of Mg extraction per unit weight of mineral was enhanced by decreasing the solids loading (due to lowered pH and the reduction in the extent of Si re-precipitation), which facilitates the additional leaching of Mg compared with higher solids loading experiments.
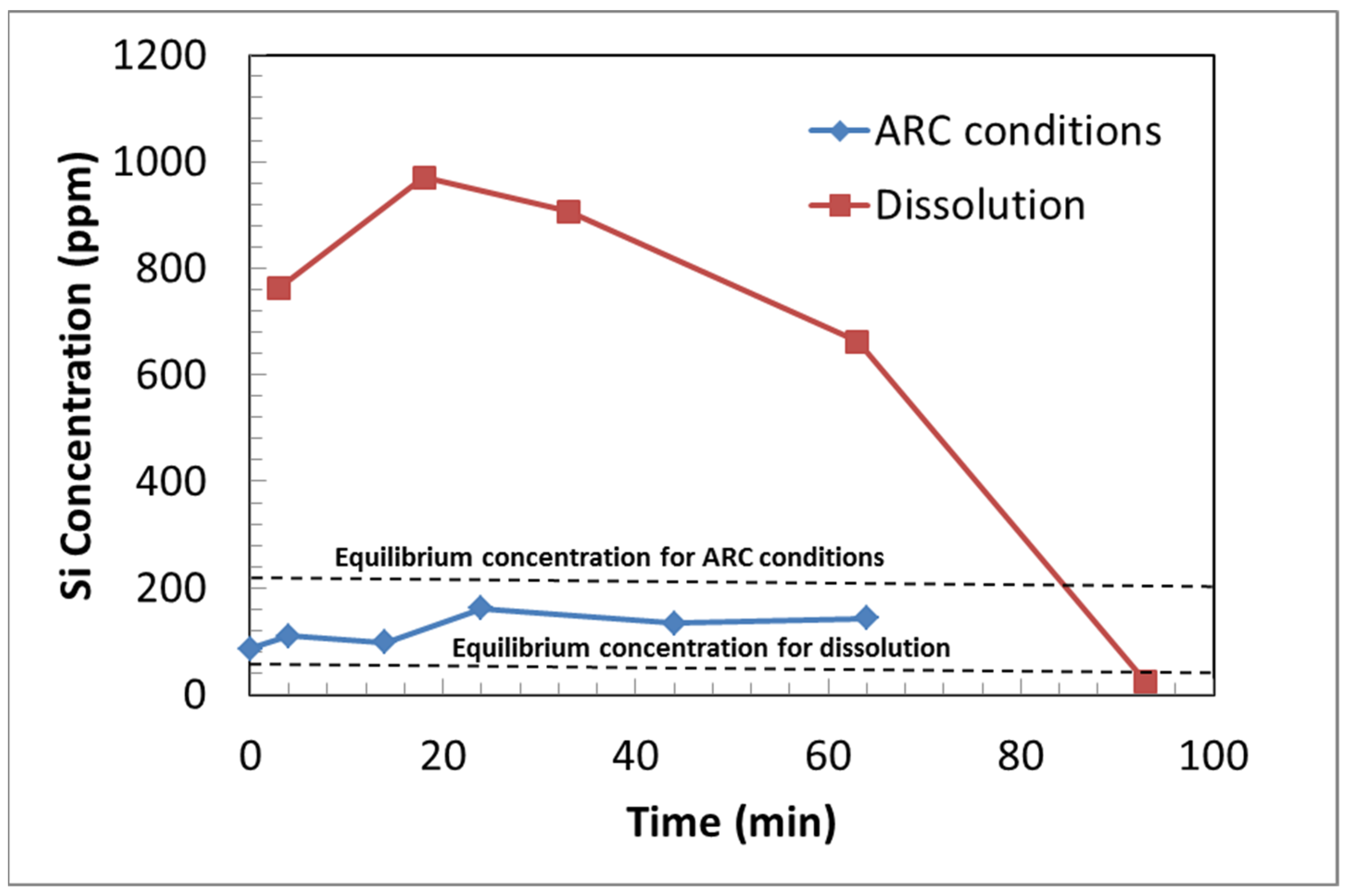
- (k)
- The Si concentrations are considerably higher in the low-temperature (40 °C) dissolution experiments compared to those under ARC conditions (150 °C, 130 bar). The results indicate the potential formation of Mg silicate phases under ARC conditions, which might explain the discrepancy in reaction extents in the two systems.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alexander, G.; Maroto-Valer, M.M.; Gafarova-Aksoy, P. Evaluation of reaction variables in the dissolution of serpentine for mineral carbonation. Fuel 2007, 86, 273–281. [Google Scholar] [CrossRef]
- Guyot, F.; Daval, D.; Dupraz, S.; Martinez, I.; Ménez, B.; Sissmann, O. CO2 geological storage: The environmental mineralogy perspective. Comptes Rendus Geosci. 2011, 343, 246–259. [Google Scholar] [CrossRef]
- Stockmann, G.J.; Wolff-Boenisch, D.; Gislason, S.R.; Oelkers, E.H. Do carbonate precipitates affect dissolution kinetics? 1: Basaltic glass. Chem. Geol. 2011, 284, 306–316. [Google Scholar] [CrossRef]
- Bales, R.C.; Morgan, J.J. Dissolution kinetics of chrysotile at pH 7 to 10. Geochim. Cosmochim. Acta 1985, 49, 2281–2288. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Gislason, S.R.; Matter, J. Mineral Carbonation of CO2. GeoScienceWorld Elem. 2008, 4, 333–337. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Comans, R.N.J. Carbon Dioxide Sequestration by Mineral Carbonation. Literature Review; Energy research Centre of The Netherlands ECN: Petten, The Netherlands, 2003; pp. 1–52. [Google Scholar]
- Luce, R.W.; Bartlett, R.W.; Parks, G.A. Dissolution kinetics of magnesium silicates. Geochim. Cosmochim. Acta 1972, 36, 35–50. [Google Scholar] [CrossRef]
- Teir, S.; Revitzer, H.; Eloneva, S.; Fogelholm, C.-J.; Zevenhoven, R. Dissolution of natural serpentinite in mineral and organic acids. Int. J. Miner. Process. 2007, 83, 36–46. [Google Scholar] [CrossRef]
- Gao, W.; Wen, J.; Li, Z. Dissolution Kinetics of Magnesium from Calcined Serpentine in NH4Cl Solution. Ind. Eng. Chem. Res. 2014, 53, 7947–7955. [Google Scholar] [CrossRef]
- Prigiobbea, V.; Hänchen, M.; Werner, M.; Baciocchi, R.; Mazzotti, M. Mineral carbonation process for CO2 sequestration. Energy Procedia 2009, 1, 4885–4890. [Google Scholar] [CrossRef]
- Hänchena, M.; Prigiobbe, V.; Storti, G.; Seward, T.M.; Mazzotti, M. Dissolution kinetics of fosteritic olivine at 90–150 °C including effects of the presence of CO2. Geochim. Cosmochim. Acta 2006, 70, 4403–4416. [Google Scholar] [CrossRef]
- Rosso, J.J.; Rimstidt, J.D. A high resolution study of forsterite dissolution rates. Geochim. Cosmochim. Acta 2000, 64, 797–811. [Google Scholar] [CrossRef]
- Oelkers, E.H. An experimental study of forsterite dissolution rates as a function of temperature and aqueous Mg and Si concentrations. Chem. Geol. 2001, 175, 485–494. [Google Scholar] [CrossRef]
- Awad, A.; Groos, A.F.K.V.; Guggenheim, S. Forsteritic olivine: Effect of crystallographic direction on dissolution kinetics. Geochim. Cosmochim. Acta 2000, 64, 1765–1772. [Google Scholar] [CrossRef]
- Daval, D.; Hellmann, R.; Martinez, I.; Gangloff, S.; Guyot, F. Lizardite serpentine dissolution kinetics as a function of pH and temperature, including effects of elevated pCO2. Chem. Geol. 2013, 351, 245–256. [Google Scholar] [CrossRef]
- Lin, F.-C.; Clemency, C.V. The dissolution kinetics of brucite, antigorite, talc, and phlogopite at room temperature and pressure. Am. Mineral. 1981, 66, 801–806. [Google Scholar]
- Giammar, D.E.; Bruant, R.G., Jr.; Peters, C.A. Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem. Geol. 2005, 217, 257–276. [Google Scholar] [CrossRef]
- Chen, Y.; Brantley, S.L. Dissolution of forsteritic olivine at 65 °C and 2 < pH < 5. Chem. Geol. 2000, 165, 267–281. [Google Scholar]
- Guthrie, G.D.; Carey, J.W.; Bergfeld, D.; Byler, D.; Chipera, S.; Ziock, H.-J.; Lackner, K. Geochemical aspects of the carbonation of magnesium silicates in an aqueous medium. In Proceedings of the Conference: Workshop on Carbon Sequestration Science, Morgantown, WV, USA, 22–24 May 2001; pp. 1–14. [Google Scholar]
- Pasquier, L.-C.; Mercier, G.; Blais, J.-F.; Cecchi, E.; Kentish, S. Reaction Mechanism for the Aqueous-Phase Mineral Carbonation of Heat-Activated Serpentine at Low Temperatures and Pressures in Flue Gas Conditions. Environ. Sci. Technol. 2014, 48, 5163–5170. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Hariharana, S.; Mazzotti, M. Flue gas CO2 mineralization using thermally activated serpentine: From single- to double-step carbonation. Phys. Chem. Chem. Phys. 2014, 16, 24978–24993. [Google Scholar] [CrossRef]
- Julcour, C.; Bourgeois, F.; Bonfils, B.; Benhamed, I.; Guyot, F.; Bodénan, F.; Petiot, C.; Gaucher, É.C. Development of an attrition-leaching hybrid process for direct aqueous mineral carbonation. Chem. Eng. J. 2015, 262, 716–726. [Google Scholar] [CrossRef]
- Daval, D.; Sissmann, O.; Menguy, N.; Saldi, G.D.; Guyot, F.; Martinez, I.; Corvisier, J.; Garcia, B.; Machouk, I.; Knauss, K.; et al. Influence of amorphous silica layer formation on the dissolution rate of olivine at 90 °C and elevated pCO2. Chem. Geol. 2011, 284, 193–209. [Google Scholar] [CrossRef]
- Vieira, K.R.M.; Arce, G.L.; Luna, C.M.; Facio, V.O.; Carvalho, A.J.; Neto, T.G.S.; Ávila, I. Understanding the acid dissolution of Serpentinites (Tailings and waste rock) for use in indirect mineral carbonation. S. Afr. J. Chem. Eng. 2022, 40, 154–164. [Google Scholar] [CrossRef]
- Galina, N.R.; Arce, G.L.A.F.; Maroto-Valer, M.; Ávila, I. Experimental Study on Mineral Dissolution and Carbonation Efficiency Applied to pH-Swing Mineral Carbonation for Improved CO2 Sequestration. Energies 2023, 16, 2449. [Google Scholar] [CrossRef]
- Li, J.; Hitch, M. Mechanical activation of ultramafic mine waste rock in dry condition for enhanced mineral carbonation. Miner. Eng. 2016, 95, 1–4. [Google Scholar] [CrossRef]
- Li, J.; Hitch, M. Ultra-fine grinding and mechanical activation of mine waste rock using a planetary mill for mineral carbonation. Int. J. Miner. Process. 2017, 158, 18–26. [Google Scholar] [CrossRef]
- Ke, X.; Baki, V.A.; Skevi, L. Mechanochemical activation for improving the direct mineral carbonation efficiency and capacity of a timber biomass ash. J. CO2 Util. 2023, 68, 102367. [Google Scholar] [CrossRef]
- Dufourny, A.; Julcour, C.; Esvan, J.; Cassayre, L.; Laniesse, P.; Bourgeois, F. Observation of the depassivation effect of attrition on magnesium silicates’ direct aqueous carbonation products. Front. Clim. 2022, 4, 946735. [Google Scholar] [CrossRef]
- PHREEQC. Thermodynamic Software; Appelo, P.A., Ed.; United States Geological Survey (USGS): Washington, DC, USA, 1999. [Google Scholar]
- Abu Fara, A. Experimental and Modelling Studies on Direct Aqueous Carbonation of Thermally Activated Lizardite. Ph.D. Thesis, The University of Newcastle, Newcastle, Australia, 2019. [Google Scholar]
- Farhang, F.; Oliver, T.K.; Rayson, M.; Brent, G.; Stockenhuber, M.; Kennedy, E. Experimental study on the precipitation of magnesite from thermally activated serpentine for CO2 sequestration. Chem. Eng. J. 2016, 303, 439–449. [Google Scholar] [CrossRef]
- Farhang, F.; Rayson, M.; Brent, G.; Hodgins, T.; Stockenhuber, M.; Kennedy, E. Insights into the dissolution kinetics of thermally activated serpentine for CO2 sequestration. Chem. Eng. J. 2017, 330, 1174–1186. [Google Scholar] [CrossRef]
- Abu Fara, A.; Rayson, M.R.; Brent, G.F.; Oliver, T.K.; Stockenhuber, M.; Kennedy, E.M. Formation of magnesite and hydromagnesite from direct aqueous carbonation of thermally activated lizardite. Environ. Prog. Sustain. Energy 2019, 38, e13244. [Google Scholar] [CrossRef]
- Mann, J. Serpentine Activation for CO2 Sequestration. Ph.D. Thesis, The University of Sydney, Sydney, Australia, 2014. [Google Scholar]
- Dri, M.; Sanna, A.; Maroto-Valer, M.M. Mineral carbonation from metal wastes: Effect of solid to liquid ratio on the efficiency and characterization of carbonated products. Appl. Energy 2014, 113, 515–523. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Comans, R.N.J.; Witkamp, G.-J. Cost evaluation of CO2 sequestration by aqueous mineral carbonation. Energy Convers. Manag. 2007, 48, 1923–1935. [Google Scholar] [CrossRef]
- Huijgen, W.J.J.; Witkamp, G.-J.; Comans, R.N.J. Mechanisms of aqueous wollastonite carbonation as a possible CO2 sequestration process. Chem. Eng. Sci. 2006, 61, 4242–4251. [Google Scholar] [CrossRef]
- Hariharan, S.; Werner, M.; Hänchen, M.; Mazzotti, M. Dissolution of dehydroxylated lizardite at flue gas conditions: II. Kinetic modeling. Chem. Eng. J. 2014, 241, 314–326. [Google Scholar] [CrossRef]
- Hariharan, S.; Repmann-Werner, M.; Mazzotti, M. Dissolution of dehydroxylated lizardite at flue gas conditions: III. Near-equilibrium kinetics. Chem. Eng. J. 2016, 298, 44–54. [Google Scholar] [CrossRef]
- Wang, X.; Maroto-Valer, M. Integration of CO2 capture and storage based on pH-swing mineral carbonation using recyclable ammonium salts. Energy Procedia 2011, 4, 4930–4936. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sieve Bin (of Raw Lizardite), µm | d10 (µm) | d50 (µm) | d90 (µm) |
|---|---|---|---|
| 45–75 | 47.2 | 73.2 | 112 |
| 20–45 | 27.5 | 43.0 | 66.8 |
| Sub-20 | 2.1 | 8.2 | 19.2 |
| Crystalline Phases % | Amorphous Phase % | ||||||
|---|---|---|---|---|---|---|---|
| Particle Size (µm) | Clinochlore | Lizardite | Forsterite | Magnetite | Hematite | Total Crystalline % | |
| 45–75 | 3.68 | 7.19 | 14.1 | 0.26 | 0.60 | 25.8 | 74.2 |
| 20–45 | 8.13 | 5.84 | 12.8 | 0.33 | 0.59 | 27.7 | 72.3 |
| <20 | 6.81 | 0.94 | 8.68 | 0.24 | 0.58 | 17.3 | 82.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu Fara, A.; Rayson, M.R.; Brent, G.F.; Oliver, T.K.; Stockenhuber, M.; Kennedy, E.M. Effect of Solid Ratio and Particle Size on Dissolution of Heat-Activated Lizardite at Elevated Pressures and Moderate Temperatures. Minerals 2024, 14, 831. https://doi.org/10.3390/min14080831
Abu Fara A, Rayson MR, Brent GF, Oliver TK, Stockenhuber M, Kennedy EM. Effect of Solid Ratio and Particle Size on Dissolution of Heat-Activated Lizardite at Elevated Pressures and Moderate Temperatures. Minerals. 2024; 14(8):831. https://doi.org/10.3390/min14080831
Chicago/Turabian StyleAbu Fara, Ammar, Mark R. Rayson, Geoff F. Brent, Timothy K. Oliver, Michael Stockenhuber, and Eric M. Kennedy. 2024. "Effect of Solid Ratio and Particle Size on Dissolution of Heat-Activated Lizardite at Elevated Pressures and Moderate Temperatures" Minerals 14, no. 8: 831. https://doi.org/10.3390/min14080831