Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General Characteristics of IDPs
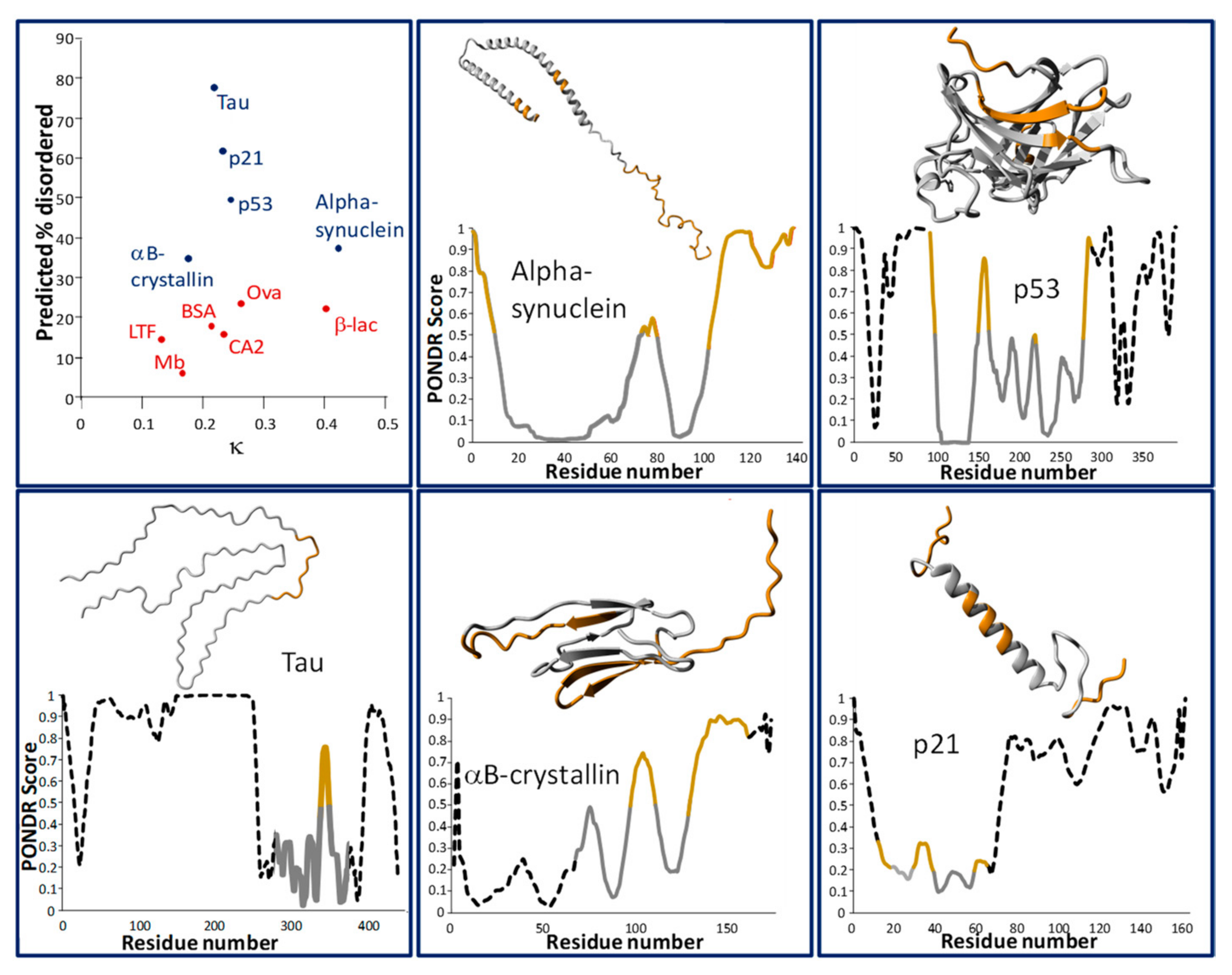
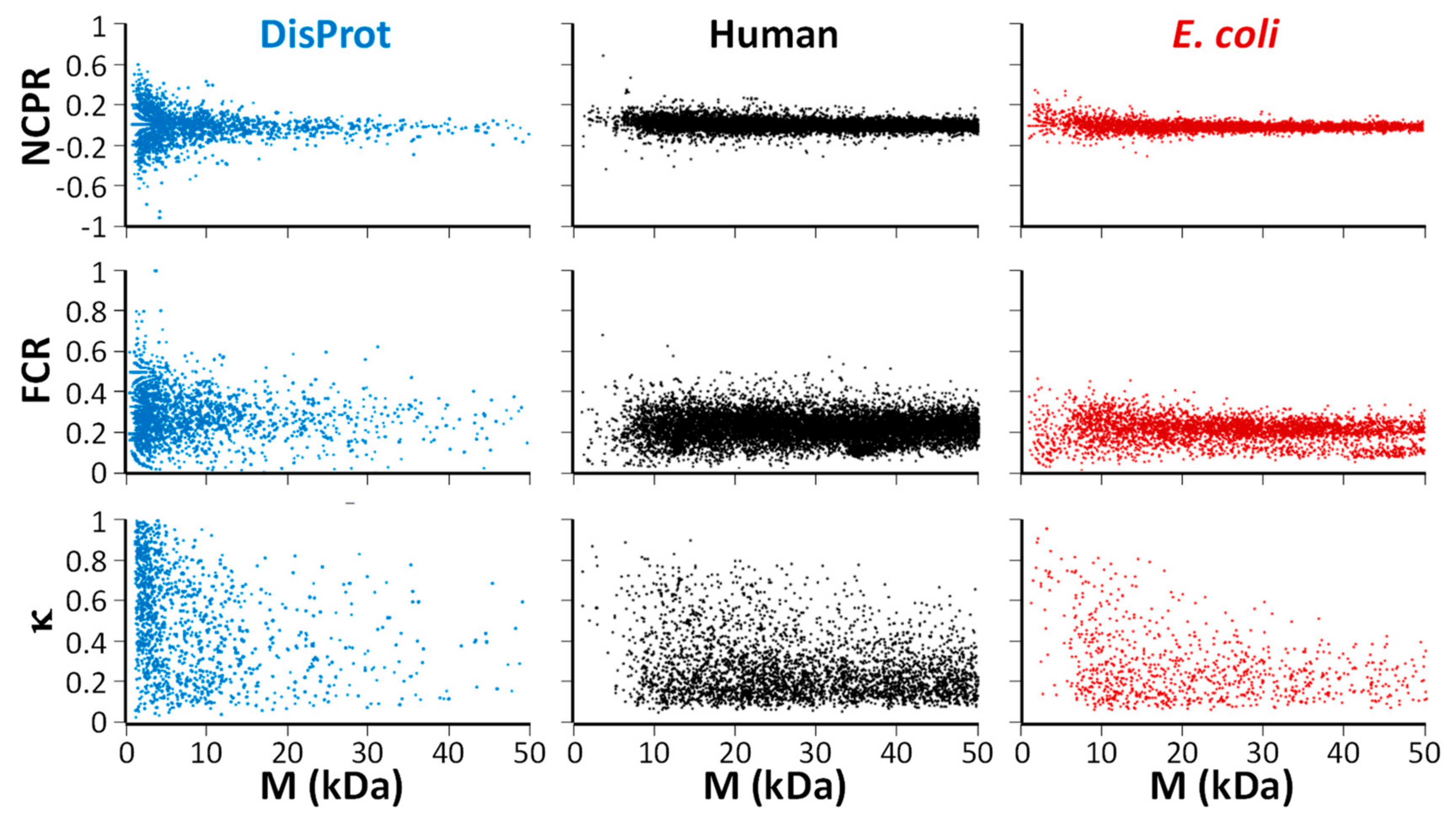
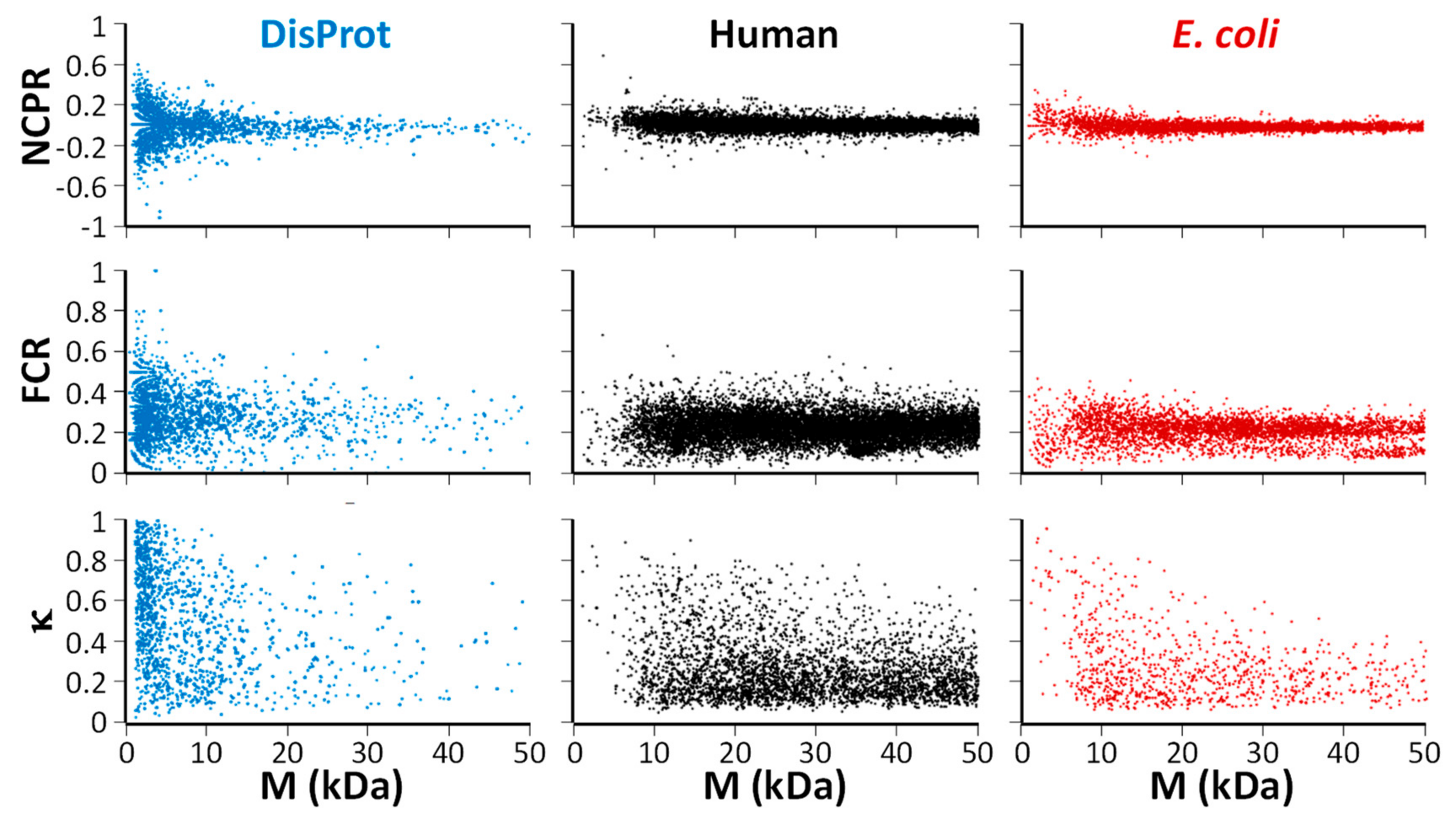
2.1. Physicochemical (Sequence) Characteristics
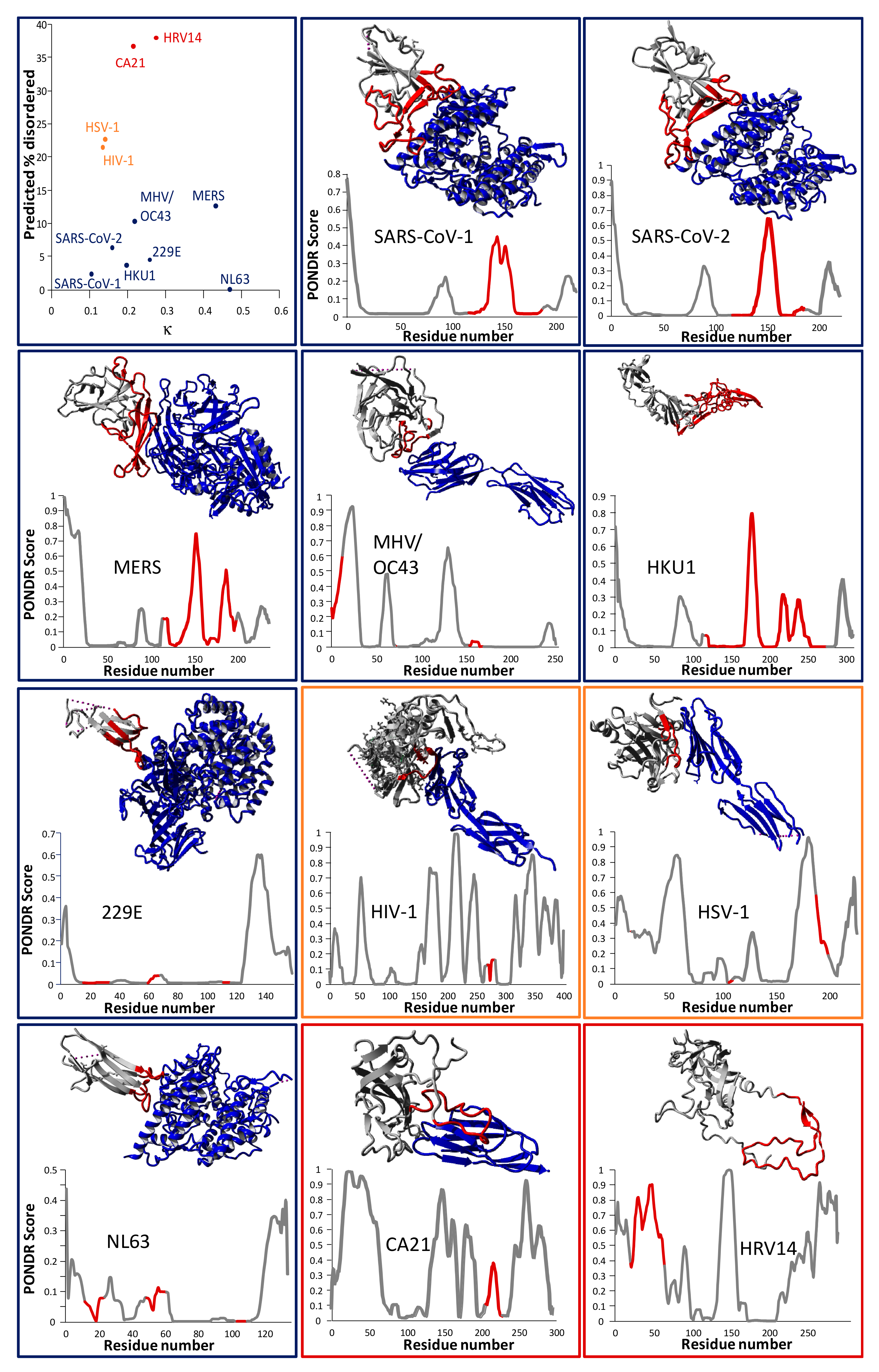
2.2. Evolutionary Characteristics
3. Analysis of IDPs
3.1. Computational Methods for Sequence-Based Prediction of Disorder
3.2. Experimental Methods for Structural Characterization of IDPs
4. Conclusions and Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Stuchfield, D.; Barran, P. Unique insights to intrinsically disordered proteins provided by ion mobility mass spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Receveur-Brechot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins Struct. Funct. Bioinform. 2006, 62, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.D.R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struc. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Pauwels, K.; Lebrun, P.; Tompa, P. To be disordered or not to be disordered: Is that still a question for proteins in the cell? Cell. Mol. Life Sci. 2017, 74, 3185–3204. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. Lausanne 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP(121–231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP(29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef] [Green Version]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Maity, B.K.; Das, A.K.; Dey, S.; Moorthi, U.K.; Kaur, A.; Dey, A.; Surendran, D.; Pandit, R.; Kallianpur, M.; Chandra, B.; et al. Ordered and Disordered Segments of Amyloid-β Drive Sequential Steps of the Toxic Pathway. ACS Chem. Neurosci. 2019, 10, 2498–2509. [Google Scholar] [CrossRef]
- Viles, J.H. Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev. 2012, 256, 2271–2284. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; la Penna, G. Metal Ions and Intrinsically Disordered Proteins and Peptides: From Cu/Zn Amyloid-β to General Principles. Acc. Chem. Res. 2014, 47, 2252–2259. [Google Scholar] [CrossRef]
- Wongkongkathep, P.; Han, J.Y.; Choi, T.S.; Yin, S.; Kim, H.I.; Loo, J.A. Native Top-Down Mass Spectrometry and Ion Mobility MS for Characterizing the Cobalt and Manganese Metal Binding of α-Synuclein Protein. J. Am. Soc. Mass Spectrom. 2018, 29, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Lermyte, F.; Everett, J.; Brooks, J.; Bellingeri, F.; Billimoria, K.; Sadler, P.J.; O’Connor, P.B.; Telling, N.D.; Collingwood, J.F. Emerging Approaches to Investigate the Influence of Transition Metals in the Proteinopathies. Cells 2019, 8, 1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lermyte, F.; Everett, J.; Lam, Y.P.Y.; Wootton, C.A.; Brooks, J.; Barrow, M.P.; Telling, N.D.; Sadler, P.J.; O’Connor, P.B.; Collingwood, J.F. Metal Ion Binding to the Amyloid β Monomer Studied by Native Top-Down FTICR Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Sal, G.D. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Ballestar, E.; Wolffe, A.P. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur. J. Biochem. 2001, 268, 1–6. [Google Scholar] [CrossRef]
- Hite, K.C.; Kalashnikova, A.A.; Hansen, J.C. Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Sci. 2012, 21, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, U.F.; Lim, J.; Zhang, Q.; Wasik, M.A.; Yang, D.; Swaminathan, K. Transcriptional repressor domain of MBD1 is intrinsically disordered and interacts with its binding partners in a selective manner. Sci. Rep. 2014, 4, 4896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, M.A.; Webb, H.D.; Sinanan, L.M.; Scarsdale, J.N.; Walavalkar, N.M.; Ginder, G.D.; Williams, D.C. An intrinsically disordered region of methyl-CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucleic Acids Res. 2015, 43, 3100–3113. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Na, I.; Kim, J.S.; Son, S.H.; Choi, S.; Lee, S.E.; Kim, J.H.; Jang, K.; Alterovitz, G.; Chen, Y.; et al. Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 2019, 5, eaav9810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Kovacs, D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. 2010, 88, 167–174. [Google Scholar] [CrossRef]
- Machida, K.; Kono-Okada, A.; Hongo, K.; Mizobata, T.; Kawata, Y. Hydrophilic Residues 526KNDAAD531 in the FlexibleC-terminal Region of the Chaperonin GroEL Are Criticalfor Substrate Protein Folding within the Central Cavity*. J. Biol. Chem. 2008, 283, 6886–6896. [Google Scholar] [CrossRef] [Green Version]
- Kazakov, A.S.; Markov, D.I.; Gusev, N.B.; Levitsky, D.I. Thermally induced structural changes of intrinsically disordered small heat shock protein Hsp22. Biophys. Chem. 2009, 145, 79–85. [Google Scholar] [CrossRef]
- Sudnitsyna, M.V.; Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. The role of intrinsically disordered regions in the structure and functioning of small heat shock proteins. Curr. Protein Pept. Sci. 2012, 13, 76–85. [Google Scholar] [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins Struct. Funct. Genet. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Christopher, M.D. Mapping Long-Range Interactions in α-Synuclein using Spin-Label NMR and Ensemble Molecular Dynamics Simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Mapping Long-Range Interactions in α-Synuclein using Spin-Label NMR and Ensemble Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstation. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. ReviewBeyond aggregation: Pathological phse transitions in neurodegenerative disease. Science 2020, 370, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.W.; Holehouse, A.S.; Grace, C.R.; Hughes, A.; Pappu, R.V.; Mittag, T. Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. J. Am. Chem. Soc. 2016, 138, 15323–15335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soranno, A.; Koenig, I.; Borgia, M.B.; Hofmann, H.; Zosel, F.; Nettels, D.; Schuler, B. Single-molecule spectroscopy reveals polymer effects of disordered proteins in crowded environments. Proc. Natl. Acad. Sci. USA 2014, 111, 4874–4879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zosel, F.; Soranno, A.; Buholzer, K.J.; Nettels, D.; Schuler, B. Depletion interactions modulate the binding between disordered proteins in crowded environments. Proc. Natl. Acad. Sci. USA 2020, 117, 13480–13489. [Google Scholar] [CrossRef]
- Mukherjee, S.K.; Gautam, S.; Biswas, S.; Kundu, J.; Chowdhury, P.K. Do Macromolecular Crowding Agents Exert Only an Excluded Volume Effect? A Protein Solvation Study. J. Phys. Chem. B 2015, 119, 14145–14156. [Google Scholar] [CrossRef]
- Goh, G.K.M.; Dunker, A.K.; Uversky, V.N. Protein intrinsic disorder toolbox for comparative analysis of viral proteins. BMC Genom. 2008, 9, S4. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.K.M.; Dunker, A.K.; Uversky, V.N. Understanding Viral Transmission Behavior via Protein Intrinsic Disorder Prediction: Coronaviruses. J. Pathog. 2012, 2012, 738590. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.M.; Dunker, A.K.; Uversky, V.N. Prediction of Intrinsic Disorder in MERS-CoV/HCoV-EMC Supports a High Oral-Fecal Transmission. PLoS. Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.M.; Dunker, A.K.; Uversky, V.N. Shell disorder, immune evasion and transmission behaviors among human and animal retroviruses. Mol. Biosyst. 2015, 11, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Shell disorder analysis predicts greater resilience of the SARS-CoV-2 (COVID-19) outside the body and in body fluids. Microb. Pathog. 2020, 144, 104177. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.K.M.; Dunker, A.K.; Foster, J.A.; Uversky, V.N. Rigidity of the Outer Shell Predicted by a Protein Intrinsic Disorder Model Sheds Light on the COVID-19 (Wuhan-2019-nCoV) Infectivity. Biomolecules 2020, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Prather, K.A.; Marr, L.C.; Schooley, R.T.; McDiarmid, M.A.; Wilson, M.E.; Milton, D.K. Airborne transmission of SARS-CoV-2. Science 2020, 6514. [Google Scholar] [CrossRef]
- Goldman, E. Exaggerated risk of transmission of COVID-19 by fomites. Lancet Infect. Dis. 2020, 20, 892–893. [Google Scholar] [CrossRef]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Giovine, P.D.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor Nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Bator, C.M.; Bowman, V.D.; Rieder, E.; He, Y.; Hebert, B.; Bella, J.; Baker, T.S.; Wimmer, E.; Kuhn, R.J.; et al. Interaction of coxsackievirus A21 with its cellular receptor, ICAM-1Citation formats. J. Virol. 2001, 75, 2444–2451. [Google Scholar] [CrossRef] [Green Version]
- Grunert, H.P.; Wolf, K.U.; Langner, K.D.; Sawitzky, D.; Habermehl, K.O.; Zeichhardt, H. Internalization of human rhinovirus 14 into HeLa and ICAM-1-transfected BHK cells. Med. Microbiol. Immunol. 1997, 186, 1–9. [Google Scholar] [CrossRef]
- Jean, J.R.; Jacomy, H.; Desforges, M.; Vabret, A.; Freymuth, F.; Talbot, P.J. Human respiratory coronavirus OC43: Genetic stability and neuroinvasion. J. Virol. 2004, 78, 8824–8834. [Google Scholar] [CrossRef] [Green Version]
- Mahase, E. Coronavirus: Covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. BMJ 2020, 368, m641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Ranst, M.V. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission even. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripperger, T.J.; Uhrlaub, J.L.; Watanabe, M.; Wong, R.; Castaneda, Y.; Pizzato, H.A.; Thompson, M.R.; Bradshaw, C.; Weinkauf, C.C.; Bime, C.; et al. Orthogonal SARS-CoV-2 Serological Assays Enable Surveillance of Low-Prevalence Communities and Reveal Durable Humoral Immunity. Immunity 2020, 53, 725–733. [Google Scholar]
- Wajnberg, A.; Amanat, F.; Firpo, A.; Altman, D.R.; Bailey, M.J.; Mansour, M.; McMahon, M.; Meade, P.; Mendu, D.R.; Muellers, K.; et al. Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science 2020. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Lane, H.C. Four Decades of HIV/AIDS—Much Accomplished, Much to Do. N. Engl. J. Med. 2020, 383, 1–4. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 2019, 20, 330–346. [Google Scholar] [CrossRef]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Dosztanyi, Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018, 27, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.W.; Uversky, V.N.; Dunker, A.K. Protein intrinsic disorder and influenza virulence: The 1918 H1N1 and H5N1 viruses. Protein Peptide Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. BBA Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.S.; Weinzierl, R.O.J. Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System. Life 2020, 10, 109. [Google Scholar] [CrossRef]
- Navarro-Paya, C.; Sanz-Hernandez, M.; de Simone, A. In Silico Study of the Mechanism of Binding of the N-Terminal Region of α Synuclein to Synaptic-Like Membranes. Life 2020, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Sala, D.; Cosentino, U.; Ranaudo, A.; Greco, C.; Moro, G. Dynamical Behavior and Conformational Selection Mechanism of the Intrinsically Disordered Sic1 Kinase-Inhibitor Domain. Life 2020, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, S.; Gapsys, V.; Gajda, M.J.; Zweckstetter, M.; de Groot, B.L.; Grubmuller, H. Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput. 2015, 11, 5513–5524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Gapsys, V.; Kim, H.Y.; Bessonov, S.; Hsiao, H.H.; Mohlmann, S.; Klaukien, V.; Ficner, R.; Becker, S.; Urlaub, H.; et al. Phosphorylation drives a dynamic switch in serine/arginine-rich proteins. Structure 2013, 21, 2162–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An arsenal of methods for the experimental characterization of intrinsically disordered proteins-How to choose and combine them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef]
- Xing, Y.; Takemaru, K.; Liu, J.; Berndt, J.D.; Zheng, J.J.; Moon, R.T.; Xu, W. Crystal structure of a full-length beta-catenin. Structure 2008, 16, 478–487. [Google Scholar] [CrossRef] [Green Version]
- de Genst, E.J.; Guilliams, T.; Wellens, J.; O’Day, E.M.; Waudby, C.A.; Meehan, S.; Dumoulin, M.; Hsu, S.T.; Cremades, N.; Verschueren, K.H.; et al. Structure and properties of a complex of α-synuclein and a single-domain camelid antibody. J. Mol. Biol. 2010, 402, 326–343. [Google Scholar] [CrossRef]
- Abskharon, R.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-Terminal β-Sheet Conversion in the Crystal Structure of the Human Prion Protein Bound to a Nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Nannenga, B.L.; de la Cruz, M.J.; Liu, J.; Sawtelle, S.; Calero, G.; Reyes, F.E.; Hattne, J.; Gonen, T. The collection of MicroED data for macromolecular crystallography. Nat. Protoc. 2016, 11, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawaya, M.R.; Rodriguez, J.; Cascio, D.; Collazo, M.J.; Shi, D.; Reyes, F.E.; Hattne, J.; Gonen, T.; Eisenberg, D.S. Ab initio structure determination from prion nanocrystals at atomic resolution by MicroED. Proc. Natl. Acad. Sci. USA 2016, 113, 11232–11236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz, M.J.; Hattne, J.; Shi, D.; Seidler, P.; Rodriguez, J.; Reyes, F.E.; Sawaya, M.R.; Cascio, D.; Weiss, S.C.; Kim, S.K.; et al. Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat. Methods 2017, 14, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konrat, R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felli, I.C.; Pierattelli, R. Novel methods based on 13C detection to study intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 115–125. [Google Scholar] [CrossRef]
- Chhabra, S.; Fischer, P.; Takeuchi, K.; Dubey, A.; Ziarek, J.J.; Boeszoermenyi, A.; Mathieu, D.; Bermel, W.; Davey, N.E.; Wagner, G.; et al. 15N detection harnesses the slow relaxation property of nitrogen: Delivering enhanced resolution for intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E1710–E1719. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; de Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterlund, N.; Moons, R.; Ilag, L.L.; Sobott, F.; Graslund, A. Native Ion Mobility-Mass Spectrometry Reveals the Formation of β-Barrel Shaped Amyloid-β Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc. 2019, 141, 10440–10450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theillet, F.X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical Properties of Cells and Their Effects on Intrinsically Disordered Proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef] [PubMed]
- Leney, A.C.; Heck, A.J. Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, L.; Ahadi, E.; Rodriguez, A.D.; Vahidi, S. Unraveling the Mechanism of Electrospray Ionization. Anal. Chem. 2013, 85, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Kuprowski, M.C.; Konermann, L. Signal response of coexisting protein conformers in electrospray mass spectrometry. Anal. Chem. 2007, 79, 2499–2506. [Google Scholar] [CrossRef]
- Frimpong, A.K.; Abzalimov, R.R.; Uversky, V.N.; Kaltashov, I.A. Characterization of intrinsically disordered proteins with electrospray ionization mass spectrometry: Conformational heterogeneity of alpha-synuclein. Proteins 2010, 78, 714–722. [Google Scholar] [CrossRef]
- Testa, L.; Brocca, S.; Grandori, R. Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins. Anal. Chem. 2011, 83, 6459–6463. [Google Scholar] [CrossRef]
- Testa, L.; Brocca, S.; Santambrogio, C.; D’Urzo, A.; Habchi, J.; Longhi, S.; Uversky, V.N.; Grandori, R. Extracting structural information from charge-state distributions of intrinsically disordered proteins by non-denaturing electrospray-ionization mass spectrometry. Intrinsically Disord. Proteins 2013, 1, e25068. [Google Scholar] [CrossRef] [Green Version]
- Beveridge, R.; Covill, S.; Pacholarz, K.J.; Kalapothakis, J.M.; MacPhee, C.E.; Barran, P.E. A mass-spectrometry-based framework to define the extent of disorder in proteins. Anal. Chem. 2014, 86, 10979–10991. [Google Scholar] [CrossRef]
- Beveridge, R.; Phillips, A.S.; Denbigh, L.; Saleem, H.M.; MacPhee, C.E.; Barran, P.E. Relating gas phase to solution conformations: Lessons from disordered proteins. Proteomics 2015, 15, 2872–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santambrogio, C.; Natalello, A.; Brocca, S.; Ponzini, E.; Grandori, R. Conformational Characterization and Classification of Intrinsically Disordered Proteins by Native Mass Spectrometry and Charge-State Distribution Analysis. Proteomics 2019, 19, e1800060. [Google Scholar] [CrossRef] [PubMed]
- Borysik, A.J.; Kovacs, D.; Guharoy, M.; Tompa, P. Ensemble Methods Enable a New Definition for the Solution to Gas-Phase Transfer of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2015, 137, 13807–13817. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, R.; Migas, L.G.; Das, R.K.; Pappu, R.V.; Kriwacki, R.W.; Barran, P.E. Ion Mobility Mass Spectrometry Uncovers the Impact of the Patterning of Oppositely Charged Residues on the Conformational Distributions of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2019, 141, 4908–4918. [Google Scholar] [CrossRef] [Green Version]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.C.; Wexler, B.B.; Rogers, D.J.; Hite, K.C.; Panchenko, T.; Ajith, S.; Black, B.E. DNA binding restricts the intrinsic conformational flexibility of methyl CpG binding protein 2 (MeCP2). J Biol. Chem. 2011, 286, 18938–18948. [Google Scholar] [CrossRef] [Green Version]
- Chanthamontri, C.; Liu, J.; McLuckey, S.A. Charge State Dependent Fragmentation of Gaseous α-Synuclein Cations via Ion Trap and Beam-Type Collisional Activation. Int. J. Mass Spectrom. 2009, 283, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.S.; Gomes, A.F.; Kalapothakis, J.M.; Gillam, J.E.; Gasparavicius, J.; Gozzo, F.C.; Kunath, T.; MacPhee, C.; Barran, P.E. Early stages of insulin fibrillogenesis examined with ion mobility mass spectrometry and molecular modelling. Analyst 2015, 140, 3070–3081. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Lantz, C.; Brown, K.A.; Ge, Y.; Tolic, L.P.; Loo, J.A.; Lermyte, F. Higher-order structural characterisation of native proteins and complexes by top-down mass spectrometry. Chem. Sci. 2020. [Google Scholar] [CrossRef]
- Miraglia, F.; Valvano, V.; Rota, L.; di Primio, C.; Quercioli, V.; Betti, L.; Giannaccini, G.; Cattaneo, A.; Colla, E. Alpha-Synuclein FRET Biosensors Reveal Early Alpha-Synuclein Aggregation in the Endoplasmic Reticulum. Life 2020, 10, 147. [Google Scholar] [CrossRef]
- Visconti, L.; Malagrino, F.; Pagano, L.; Toto, A. Understanding the Mechanism of Recognition of Gab2 by the N-SH2 Domain of SHP2. Life 2020, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, L.J.; Eaton, W.A.; Hofrichter, J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA 2000, 97, 7220–7225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, S.; Saha, S.; Ahmad, B.; Lapidus, L.J. Effects of Mutations on the Reconfiguration Rate of α-Synuclein. J. Phys. Chem. B 2015, 119, 15443–15450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, B.; Chen, Y.; Lapidus, L.J. Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl. Acad. Sci. USA 2012, 109, 2336–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins and the Janus Challenge. Proteomics 2018, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kumar, A.; Yadav, R.; Uversky, V.N.; Giri, R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep.UK 2018, 8, 5822. [Google Scholar] [CrossRef] [Green Version]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lermyte, F. Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview. Life 2020, 10, 320. https://doi.org/10.3390/life10120320
Lermyte F. Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview. Life. 2020; 10(12):320. https://doi.org/10.3390/life10120320
Chicago/Turabian StyleLermyte, Frederik. 2020. "Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview" Life 10, no. 12: 320. https://doi.org/10.3390/life10120320
APA StyleLermyte, F. (2020). Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview. Life, 10(12), 320. https://doi.org/10.3390/life10120320