CRISPR-Cas9: A Powerful Tool to Efficiently Engineer Saccharomyces cerevisiae



Abstract
:1. Introduction
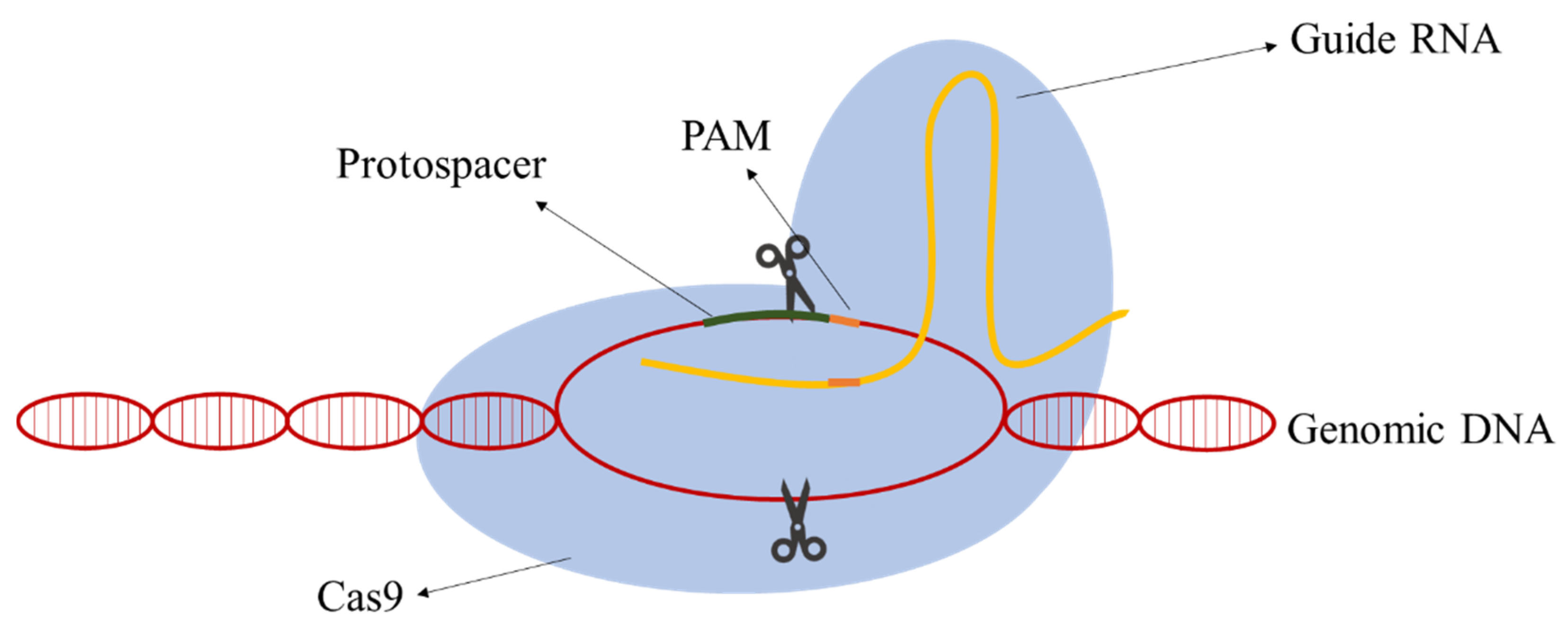
2. The CRISPR-Cas9 System Components
2.1. Cas9 Protein
2.2. Guide RNA
2.3. Donor DNA
3. CRISPR-Cas9 Applications in Saccharomyces cerevisiae
3.1. Knockin and Knockout
3.2. Cell Factory Development
3.3. Innovative CRISPR Toolkits in Saccharomyces cerevisiae
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.; Larsson, C.; van Maris, A.; Pronk, J. Metabolic engineering of yeast for production of fuels and chemicals. Curr. Opin. Biotechnol. 2013, 24, 398–404. [Google Scholar] [CrossRef]
- Bernardi, B.; Wendland, J. Homologous Recombination: A GRAS Yeast Genome Editing Tool. Fermentation 2020, 6, 57. [Google Scholar] [CrossRef]
- Kuijpers, N.G.A.; Solis-Escalante, D.; Bosman, L.; van den Broek, M.; Pronk, J.T.; Daran, J.M.; Daran-Lapujade, P. A versatile, efficient strategy for assembly of multi-fragment expression vectors in Saccharomyces cerevisiae using 60 bp synthetic recombination sequences. Microb. Cell Fact. 2013, 12, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijpers, N.G.A.; Chroumpi, S.; Vos, T.; Solis-Escalante, D.; Bosman, L.; Pronk, J.T.; Daran, J.M.; Daran-Lapujade, P. One-step assembly and targeted integration of multigene constructs assisted by the I-SceI meganuclease in Saccharomyces cerevisiae. FEMS Yeast Res. 2013, 13, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, J.L.; Rodrigues, L.R. Synthetic Biology: Perspectives in Industrial Biotechnology. In Foundations of Biotechnology and Bioengineering, Volume 1—Current Developments in Biotechnology & Bioengineering; Elsevier: Amsterdam, The Netherlands, 2017; pp. 239–269. [Google Scholar]
- Rodrigues, J.L.; Ferreira, D.; Rodrigues, L.R. Synthetic biology strategies towards the development of new bioinspired technologies for medical applications. In Bioinspired Materials for Medical Applications; Elsevier: Amsterdam, The Netherlands, 2017; pp. 451–497. [Google Scholar]
- Roberts, R.J. How restriction enzymes became the workhorses of molecular biology. Proc. Natl. Acad. Sci. USA 2005, 102, 5905–5908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Luo, M.L.; Leenay, R.T.; Beisel, C.L. Current and future prospects for CRISPR-based tools in bacteria. Biotechnol. Bioeng. 2016, 113, 930–943. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, J.; Norville, J.; Mali, P.; Rios, X.; Aach, J.; Church, G. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Z.; Xiao, H.; Liang, J.; Zhang, L.; Xiong, X.; Sun, N.; Si, T.; Zhao, H. Homology-Integrated CRISPR-Cas (HI-CRISPR) System for One-Step Multigene Disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 2015, 4, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Mans, R.; van Rossum, H.M.; Wijsman, M.; Backx, A.; Kuijpers, N.G.A.; van den Broek, M.; Daran-Lapujade, P.; Pronk, J.T.; van Maris, A.J.A.; Daran, J.-M.G. CRISPR/Cas9: A molecular Swiss army knife for simultaneous introduction of multiple genetic modifications in Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Generoso, W.C.; Gottardi, M.; Oreb, M.; Boles, E. Simplified CRISPR-Cas genome editing for Saccharomyces cerevisiae. J. Microbiol. Methods 2016, 127, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Stovicek, V.; Borodina, I.; Förster, J. General rights CRISPR-Cas system enables fast and simple genome editing of industrial Saccharomyces cerevisiae strains. Metab. Eng. Commun. 2015, 2, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Ryan, O.W.; Skerker, J.M.; Maurer, M.J.; Li, X.; Tsai, J.C.; Poddar, S.; Lee, M.E.; DeLoache, W.; Dueber, J.E.; Arkin, A.P.; et al. Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 2014, 3, 1–15. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Integr. Plant Biol. 2014, 56, 343–349. [Google Scholar] [CrossRef]
- Jakočinas, T.; Bonde, I.; Herrgård, M.; Harrison, S.J.; Kristensen, M.; Pedersen, L.E.; Jensen, M.K.; Keasling, J.D. Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab. Eng. 2015, 28, 213–222. [Google Scholar] [CrossRef]
- Quan, J.; Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011, 6, 242–251. [Google Scholar] [CrossRef]
- Reider Apel, A.; Wehrs, M.; Sachs, D.; Li, R.A.; Tong, G.J.; Garber, M.; Nnadi, O.; Zhuang, W.; Hillson, N.J.; Keasling, J.D.; et al. A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 496–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.E.; DeLoache, W.C.; Cervantes, B.; Dueber, J.E. A Highly Characterized Yeast Toolkit for Modular, Multipart Assembly. ACS Synth. Biol. 2015, 4, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Ronda, C.; Maury, J.; Jakočiunas, T.; Baallal Jacobsen, S.A.; Germann, S.M.; Harrison, S.J.; Borodina, I.; Keasling, J.D.; Jensen, M.K.; Nielsen, A.T. CrEdit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae. Microb. Cell Fact. 2015, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Pedersen, L.E.; Weber, T.; Lee, S.Y. CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synth. Syst. Biotechnol. 2016, 1, 118–121. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wei, Z.; Dominguez, A.; Li, Y.; Wang, X.; Qi, L.S. CRISPR-ERA: A comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics 2015, 31, 3676–3678. [Google Scholar] [CrossRef]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef]
- Sadhu, M.J.; Bloom, J.S.; Day, L.; Kruglyak, L. CRISPR-directed mitotic recombination enables genetic mapping without crosses. Science 2016, 352, 1113–1116. [Google Scholar] [CrossRef] [Green Version]
- Gorter De Vries, A.R.; Couwenberg, L.G.F.; Van Den Broek, M.; De La Torre Cortés, P.; Ter Horst, J.; Pronk, J.T.; Daran, J.M.G. Allele-specific genome editing using CRISPR-Cas9 is associated with loss of heterozygosity in diploid yeast. Nucleic Acids Res. 2019, 47, 1362–1372. [Google Scholar] [CrossRef]
- Martin, F.; Sánchez-Hernández, S.; Gutiérrez-Guerrero, A.; Pinedo-Gomez, J.; Benabdellah, K. Biased and unbiased methods for the detection of off-target cleavage by CRISPR/Cas9: An overview. Int. J. Mol. Sci. 2016, 17, 1507. [Google Scholar] [CrossRef]
- Waldrip, Z.J.; Jenjaroenpun, P.; DeYoung, O.; Nookaew, I.; Taverna, S.D.; Raney, K.D.; Tackett, A.J. Genome-wide Cas9 binding specificity in Saccharomyces cerevisiae. PeerJ 2020, 8, e9442. [Google Scholar] [CrossRef]
- Tsai, C.S.; Kong, I.I.; Lesmana, A.; Million, G.; Zhang, G.C.; Kim, S.R.; Jin, Y.S. Rapid and marker-free refactoring of xylose-fermenting yeast strains with Cas9/CRISPR. Biotechnol. Bioeng. 2015, 112, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Stovicek, V.; Holkenbrink, C.; Borodina, I. CRISPR/Cas system for yeast genome engineering: Advances and applications. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Garst, A.D.; Bassalo, M.C.; Pines, G.; Lynch, S.A.; Halweg-Edwards, A.L.; Liu, R.; Liang, L.; Wang, Z.; Zeitoun, R.; Alexander, W.G.; et al. Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 2017, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Qiu, M.; Chan, E.; Zhu, L.; Luo, Y. Minimum Length of Sequence Homology Required forin VivoCloning by Homologous Recombination in Yeast. Plasmid 1997, 38, 91–96. [Google Scholar] [CrossRef]
- Da Silva, N.A.; Srikrishnan, S. Introduction and expression of genes for metabolic engineering applications in Saccharomyces cerevisiae. FEMS Yeast Res. 2012, 12, 197–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-N.; Masison, D.; Eisenberg, E.; Greene, L.E. Application of the FLP/FRT system for conditional gene deletion in yeast Saccharomyces cerevisiae. Yeast 2011, 28, 673–681. [Google Scholar] [CrossRef]
- Yamanishi, M.; Matsuyama, T. A modified cre-lox genetic switch to dynamically control metabolic flow in Saccharomyces cerevisiae. ACS Synth. Biol. 2012, 1, 172–180. [Google Scholar] [CrossRef]
- Solis-Escalante, D.; Kuijpers, N.G.A.; van der Linden, F.H.; Pronk, J.T.; Daran, J.-M.; Daran-Lapujade, P. Efficient simultaneous excision of multiple selectable marker cassettes using I-SceI-induced double-strand DNA breaks in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.J.; Kim, S.C. Minimization of the Escherichia coli genome using the Tn5-targeted Cre/loxP excision system. Methods Mol. Biol. 2007, 416, 261–277. [Google Scholar] [CrossRef]
- Komatsu, M.; Uchiyama, T.; Omura, S.; Cane, D.E.; Ikeda, H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Yuan, X.; Liang, L.; Fang, J.; Chen, Y.; He, W.; Xue, T. Using CRISPR/Cas9 for multiplex genome engineering to optimize the ethanol metabolic pathway in Saccharomyces cerevisiae. Biochem. Eng. J. 2019, 145, 120–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Wang, Z.; Zhang, Y.; Shi, S.; Nielsen, J. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Ramos, D.; Gorter De Vries, A.R.; Grijseels, S.S.; Van Berkum, M.C.; Swinnen, S.; Van Den Broek, M.; Nevoigt, E.; Daran, J.M.G.; Pronk, J.T.; Van Maris, A.J.A. A new laboratory evolution approach to select for constitutive acetic acid tolerance in Saccharomyces cerevisiae and identification of causal mutations. Biotechnol. Biofuels 2016, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainha, J.; Gomes, D.; Rodrigues, L.R.; Rodrigues, J.L. Synthetic Biology Approaches to Engineer Saccharomyces cerevisiae towards the Industrial Production of Valuable Polyphenolic Compounds. Life 2020, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Meng, H.; Ma, B.; Tao, X.; Liu, M.; Wang, F.Q.; Wei, D.Z. Immediate, multiplexed and sequential genome engineering facilitated by CRISPR/Cas9 in Saccharomyces cerevisiae. J. Ind. Microbiol. Biotechnol. 2020, 47, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Liang, Y.; Zhang, M.; Ang, E.; Zhao, H. A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae. Metab. Eng. 2016, 33, 19–27. [Google Scholar] [CrossRef]
- Smith, J.D.; Suresh, S.; Schlecht, U.; Wu, M.; Wagih, O.; Peltz, G.; Davis, R.W.; Steinmetz, L.M.; Parts, L.; St. Onge, R.P. Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 2016, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, A.A.; Walter, J.M.; Schubert, M.G.; Kung, S.H.; Hawkins, K.; Platt, D.M.; Hernday, A.D.; Mahatdejkul-Meadows, T.; Szeto, W.; Chandran, S.S.; et al. Efficient Multiplexed Integration of Synergistic Alleles and Metabolic Pathways in Yeasts via CRISPR-Cas. Cell Syst. 2015, 1, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yu, T.; Li, X.; Chen, Y.; Campbell, K.; Nielsen, J.; Chen, Y. Rewiring carbon metabolism in yeast for high level production of aromatic chemicals. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhu, C.; Zhu, M.; Xiong, J.; Ma, H.; Zhuo, M.; Li, S. High production of valencene in Saccharomyces cerevisiae through metabolic engineering. Microb. Cell Fact. 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Kim, H.; Kim, I.J.; Yun, E.J.; Kwak, S.; Jin, Y.-S.; Kim, K.H. Metabolic engineering of Saccharomyces cerevisiae by using the CRISPR-Cas9 system for enhanced fatty acid production. Process Biochem. 2018. [Google Scholar] [CrossRef]
- Huang, S.; Geng, A. High-copy genome integration of 2,3-butanediol biosynthesis pathway in Saccharomyces cerevisiae via in vivo DNA assembly and replicative CRISPR-Cas9 mediated delta integration. J. Biotechnol. 2020, 310, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Xie, W.; Li, X.; Cai, G.; Lu, J.; Xie, G. Metabolic engineering of Saccharomyces cerevisiae using the CRISPR/Cas9 system to minimize ethyl carbamate accumulation during Chinese rice wine fermentation. Appl. Microbiol. Biotechnol. 2020, 104, 4435–4444. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.A.; DeNicola, A.B.; Billingsley, J.M.; Creso, J.G.; Subrahmanyam, V.; Tang, Y. Engineered mitochondrial production of monoterpenes in Saccharomyces cerevisiae. Metab. Eng. 2019, 55, 76–84. [Google Scholar] [CrossRef]
- Hu, Z.; Lin, L.; Li, H.; Li, P.; Weng, Y.; Zhang, C.; Yu, A.; Xiao, D. Engineering Saccharomyces cerevisiae for production of the valuable monoterpene d-limonene during Chinese Baijiu fermentation. J. Ind. Microbiol. Biotechnol. 2020, 47, 511–523. [Google Scholar] [CrossRef]
- Hassing, E.J.; de Groot, P.A.; Marquenie, V.R.; Pronk, J.T.; Daran, J.M.G. Connecting central carbon and aromatic amino acid metabolisms to improve de novo 2-phenylethanol production in Saccharomyces cerevisiae. Metab. Eng. 2019, 56, 165–180. [Google Scholar] [CrossRef]
- Deaner, M.; Alper, H.S. Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab. Eng. 2017, 40, 14–22. [Google Scholar] [CrossRef]
- Dong, C.; Jiang, L.; Xu, S.; Huang, L.; Cai, J.; Lian, J.; Xu, Z. A Single Cas9-VPR Nuclease for Simultaneous Gene Activation, Repression, and Editing in Saccharomyces cerevisiae. ACS Synth. Biol. 2020, 9, 2252–2257. [Google Scholar] [CrossRef]
- Halperin, S.O.; Tou, C.J.; Wong, E.B.; Modavi, C.; Schaffer, D.V.; Dueber, J.E. diversification of all nucleotides in a tunable window. Nature 2018, 560, 248–252. [Google Scholar] [CrossRef]
- Tou, C.J.; Schaffer, D.V.; Dueber, J.E. Targeted diversification in the Saccharomyces cerevisiae genome with CRISPR-guided DNA polymerase I. ACS Synth. Biol. 2020, 9, 1911–1916. [Google Scholar] [CrossRef]
- Mitsui, R.; Yamada, R.; Ogino, H. Improved Stress Tolerance of Saccharomyces cerevisiae by CRISPR-Cas-Mediated Genome Evolution. Appl. Biochem. Biotechnol. 2019, 189, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzadfard, F.; Perli, S.D.; Lu, T.K. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2013, 2, 604–613. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Cámara, E.; Lenitz, I.; Nygård, Y. OPEN A CRISPR activation and interference toolkit for industrial Saccharomyces cerevisiae strain KE6–12. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.D.; Ferreira, R.; Jakočiūnas, T.; Arsovska, D.; Zhang, J.; Ding, L.; Smith, J.D.; David, F.; Nielsen, J.; Jensen, M.K.; et al. Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb. Cell Fact. 2017, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Chang, P.; Yu, H.; Ren, H.; Hong, D.; Li, Z.; Wang, Y.; Song, H.; Huo, Y.; Li, C. Productive Amyrin Synthases for Efficient α-Amyrin Synthesis in Engineered Saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 2391–2402. [Google Scholar] [CrossRef]
- Kiani, S.; Chavez, A.; Tuttle, M.; Hall, R.N.; Chari, R.; Ter-ovanesyan, D.; Qian, J.; Pruitt, B.W.; Beal, J.; Vora, S.; et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 2015, 12, 1051–1054. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knockout and active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; Hamedirad, M.; Hu, S.; Zhao, H. Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Objective | Cas Protein Expression System | gRNA Expression | Phenotype Desired | Transformed Elements | Number of Editions | Ref. |
|---|---|---|---|---|---|---|
| Genome editing | CEN plasmid | pSNR52 | Canavanine resistance | CRISPR-Cas9 plasmid | 1 | [14] |
| Genome editing | 2µ plasmid | pSNR52 plus CRISPR native array | Hydrocortisone production | CRISPR-Cas9 plasmid + Donor DNA | 3 | [15] |
| Genome editing | 2µ plasmid | pSNR52 with 5’ HDV | Cellobiose utilization | CRISPR-Cas9 plasmid + Donor DNA | 3 | [19] |
| Genome editing | Integrated | pSNR52 | Muconic acid production | gRNA plasmid + Donor DNA | 3 | [49] |
| Genome editing | 2µ plasmid | pSNR52 tRNA–gRNA array | Increase free fatty acid production | CRISPR-Cas9 plasmid | 8 | [44] |
| Genome editing | CEN plasmid | pSNR52 | Mevalonate production | CRISPR-Cas9 plasmid + Donor oligos | 5 | [21] |
| Genome editing | 2µ plasmid | pSNR52 | Leucine and Isoleucine auxotrophy | CRISPR-Cas9 plasmid + Donor oligos | 2 | [17] |
| Genome insertion | 2µ plasmid | pSNR52 | BDO production; Xylose utilization | CRISPR-Cas9 plasmid + Donor DNA | Pathway insertion | [48] |
| Genome insertion | 2µ plasmid | ptRNA | Taxadiene production | CRISPR-Cas9 plasmid + Donor DNA | 4 | [23] |
| Genome insertion | CEN plasmid | pSNR52 | β-carotene production | Cas9 plasmid + gRNA plasmid + Donor DNA | 3 | [25] |
| Genome editing and insertion | Cas9 integration | pSNR52 | p-coumaric acid production | gRNA plasmid | 10 | [50] |
| Genome editing | 2µ plasmid | pSNR52 plus CRISPR native array | Increase ethanol production | CRISPR-Cas9 plasmid + Donor DNA plasmid | 3 | [43] |
| Genome editing and insertion | 2µ plasmid | pSNR52 | Enhance synthesis of farnesyl diphosphate | Cas9 plasmid + gRNA plasmids + Donor DNA | 5 | [51] |
| Genome editing and insertion | 2µ plasmid | pSNR52 | Enhance fatty acid production | Cas9 plasmid + gRNA plasmids + Donor DNA | 4 | [52] |
| Genome editing and insertion | 2µ plasmid | pSNR52 | Butanediol production | Cas9 plasmid + gRNA plasmids + Donor DNA | 5 | [53] |
| Genome insertion | 2µ plasmid | pSNR52 | Minimize ethyl carbamate accumulation | CRISPR-Cas9 plasmid + | 1 | [54] |
| Genome editing and integration | Integrated | pSNR52 | Production of monoterpene precursor, geraniol | gRNA plasmid + Donor DNA | 8 | [55] |
| Genome editing and integration | CEN plasmid | pSNR52 | Limonene production | Cas9 plasmid + gRNA plasmid + Donor DNA | 9 | [56] |
| Genome editing and integration | Integrated | pSNR52 | 2-phenylethanol production | gRNA plasmid + Donor DNA | 8 | [57] |
| Gene activation and repression | ARS/CEN plasmid (dCas9 fused with VPR domain) | pSNR52 | Optimize isoprenoids and triacylglycerols biosynthesis | dCas9 plasmid + gRNA plasmid | 4 | [58] |
| Gene activation and repression | ARS/CEN plasmid (Cas9 fused with VPR domain) | pSNR52 | Optimize α-santalene biosynthesis | Cas9 plasmid + truncated gRNA plasmid | 3 | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rainha, J.; Rodrigues, J.L.; Rodrigues, L.R. CRISPR-Cas9: A Powerful Tool to Efficiently Engineer Saccharomyces cerevisiae. Life 2021, 11, 13. https://doi.org/10.3390/life11010013
Rainha J, Rodrigues JL, Rodrigues LR. CRISPR-Cas9: A Powerful Tool to Efficiently Engineer Saccharomyces cerevisiae. Life. 2021; 11(1):13. https://doi.org/10.3390/life11010013
Chicago/Turabian StyleRainha, João, Joana L. Rodrigues, and Lígia R. Rodrigues. 2021. "CRISPR-Cas9: A Powerful Tool to Efficiently Engineer Saccharomyces cerevisiae" Life 11, no. 1: 13. https://doi.org/10.3390/life11010013