
A Novel RNA Synthesis Inhibitor, STK160830, Has Negligible DNA-Intercalating Activity for Triggering A p53 Response, and Can Inhibit p53-Dependent Apoptosis

 ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Apoptosis Assay
2.3. Immunoblotting Analysis
2.4. Quantitative PCR (qPCR) Analysis
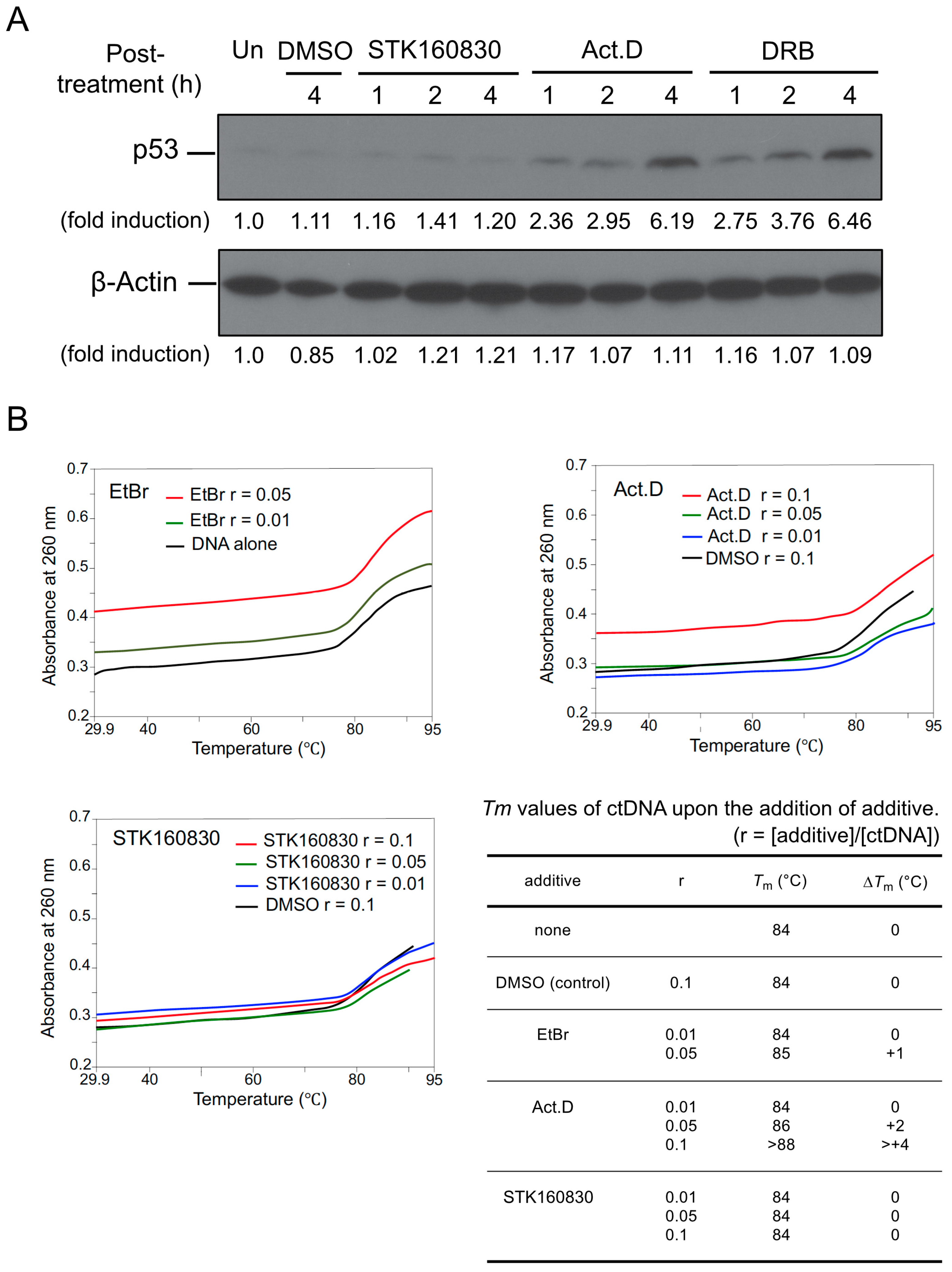
2.5. DNA Melting-Curve Analysis
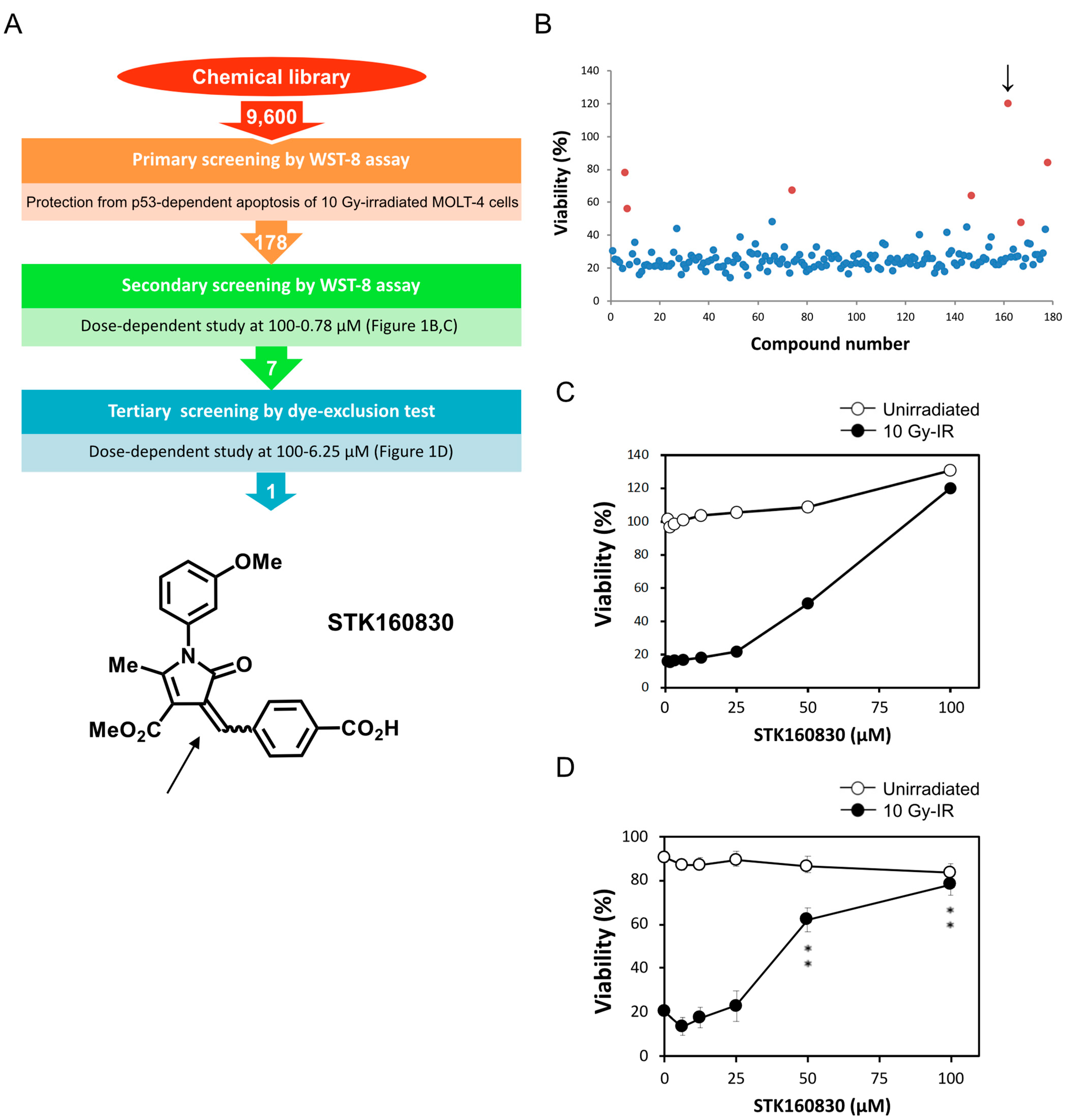
3. Results
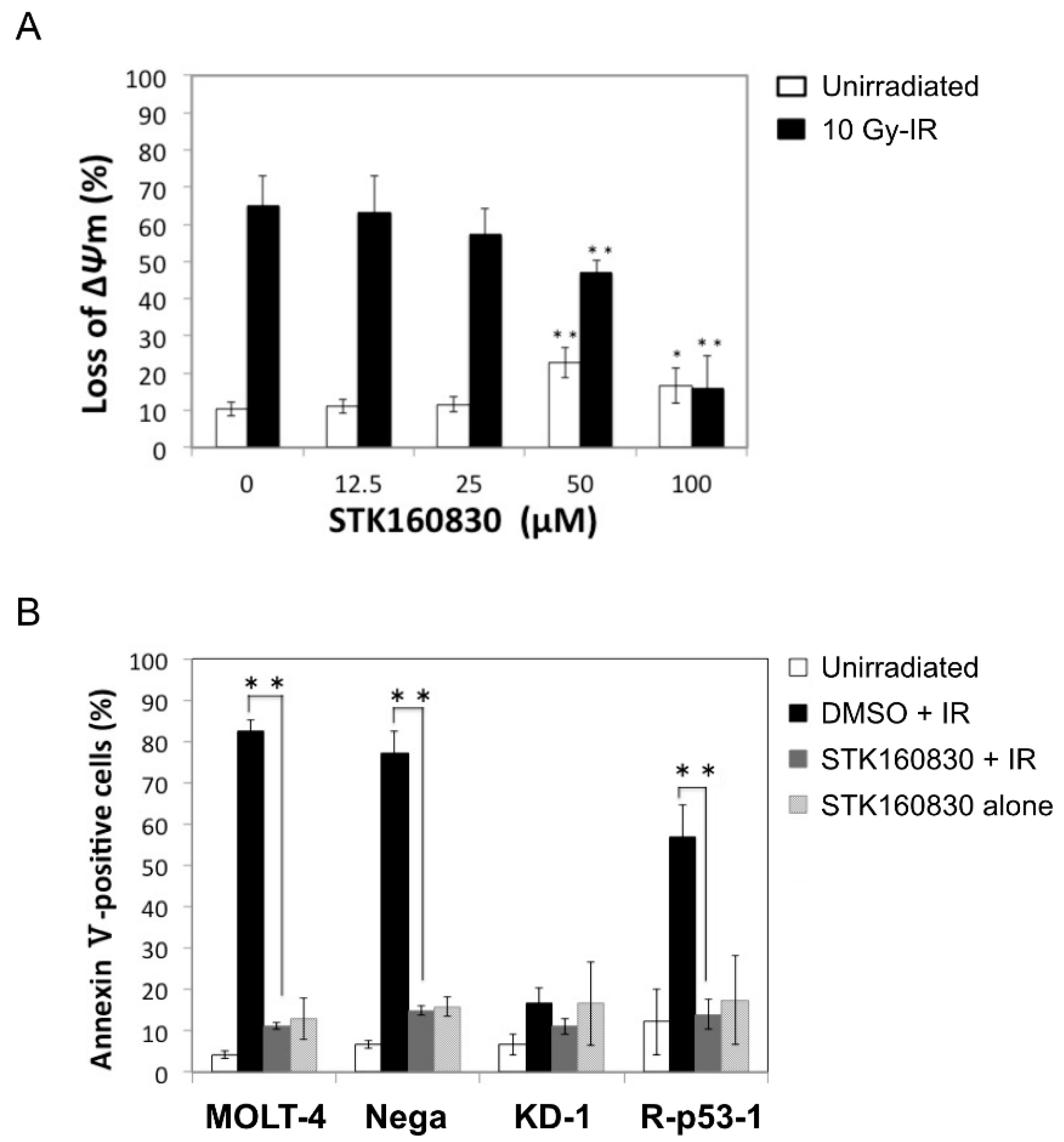
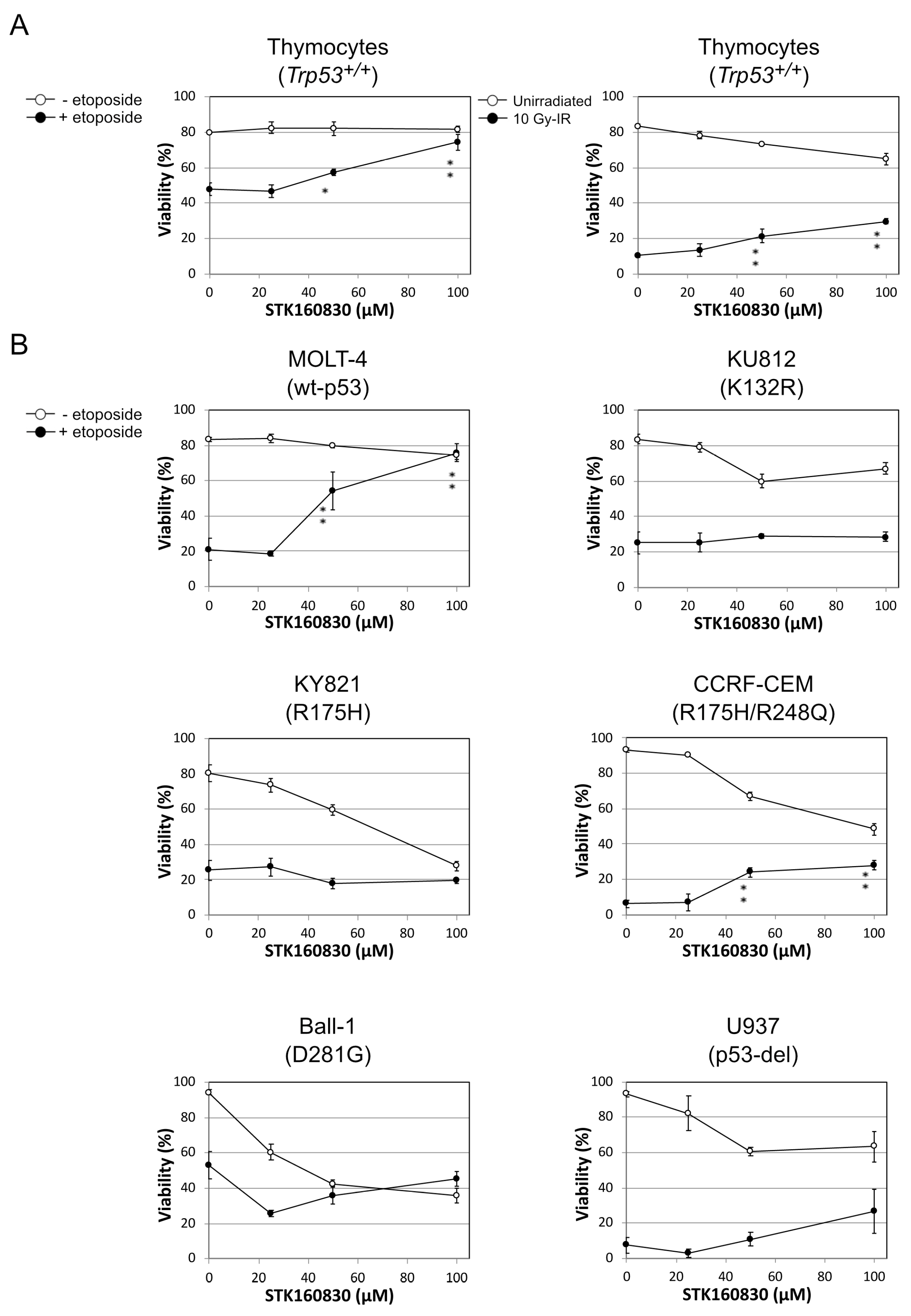
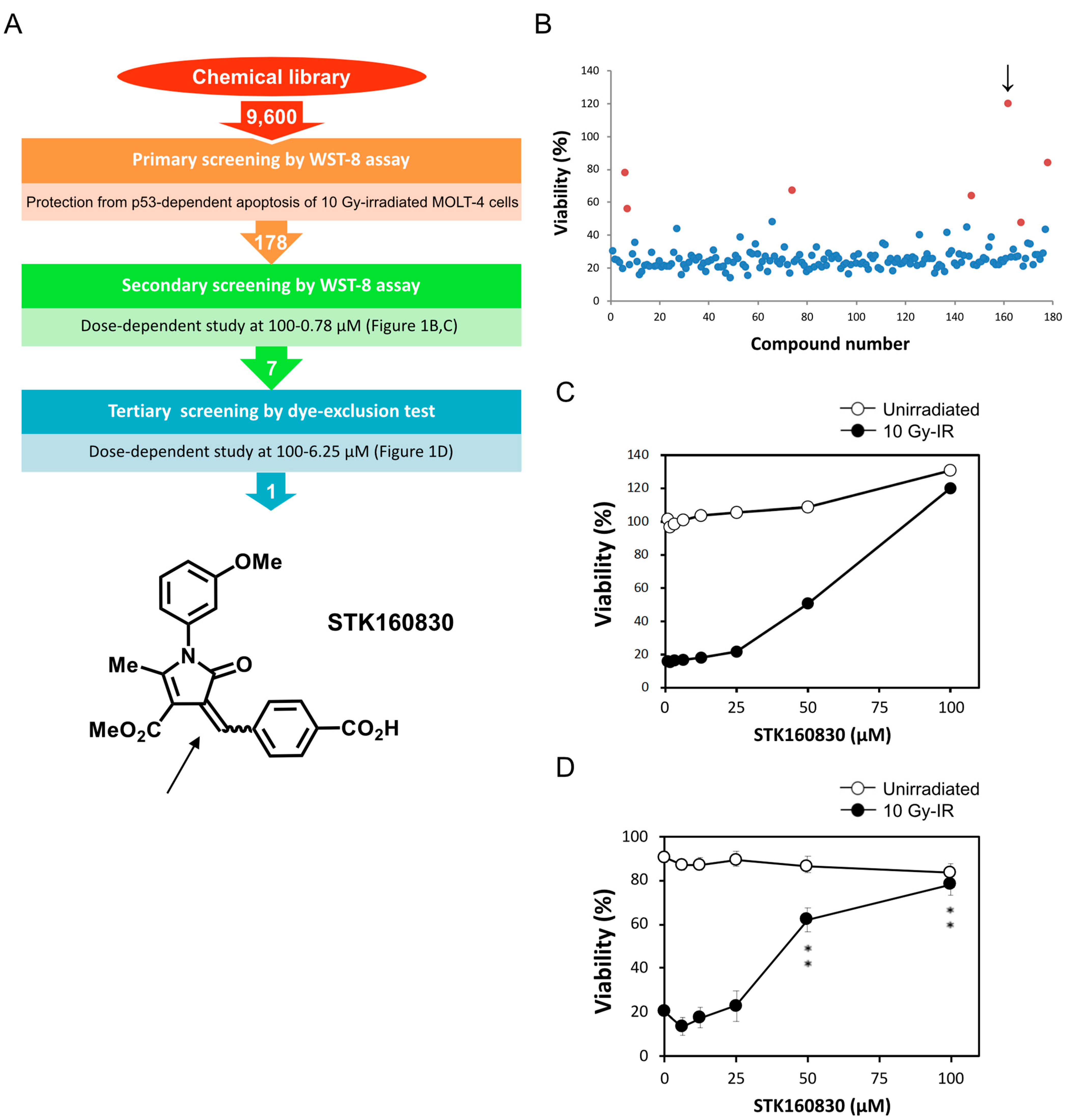
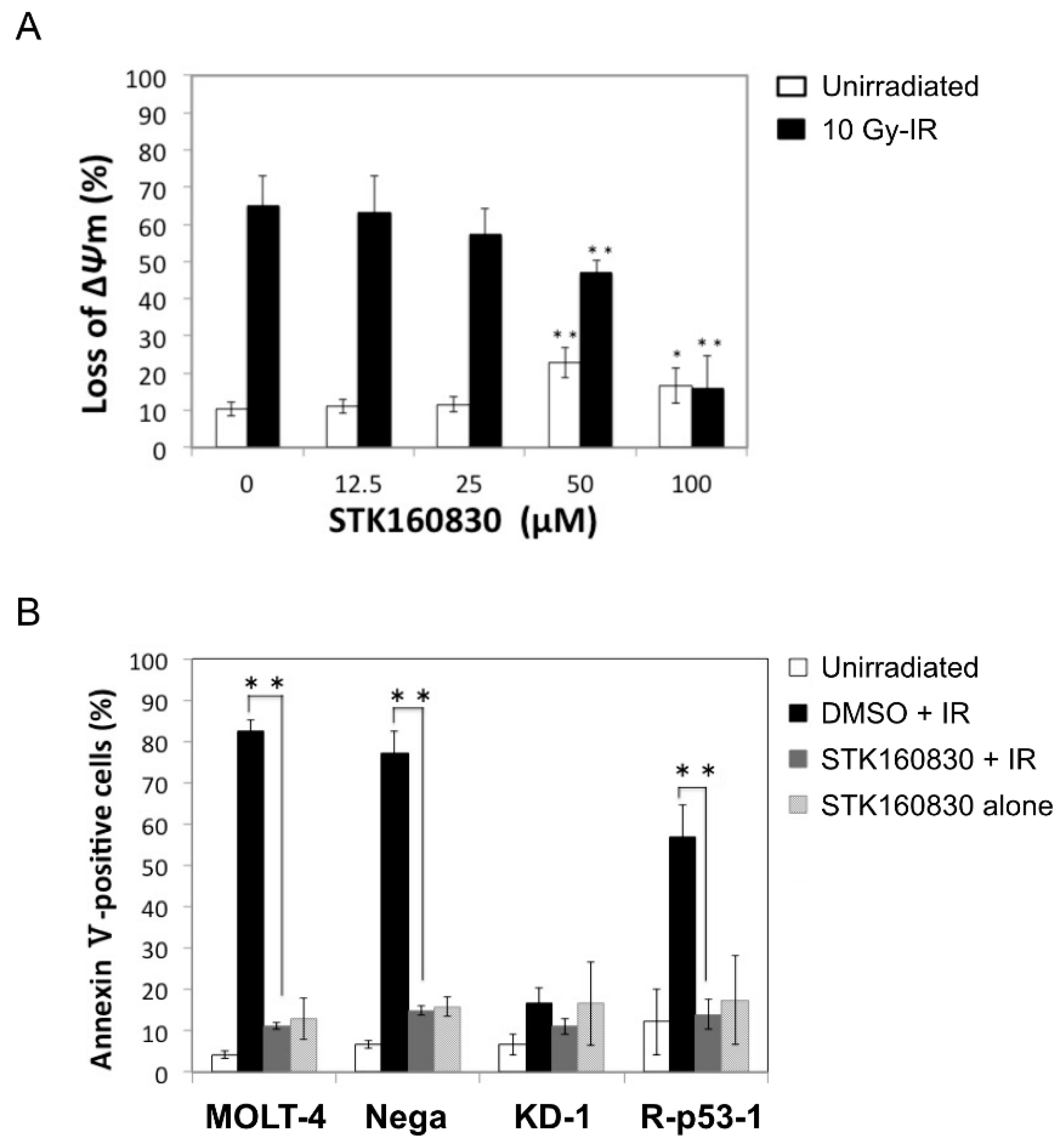
3.1. STK160830 Suppresses DNA Damage-Induced Apoptosis in A p53-Dependent Manner
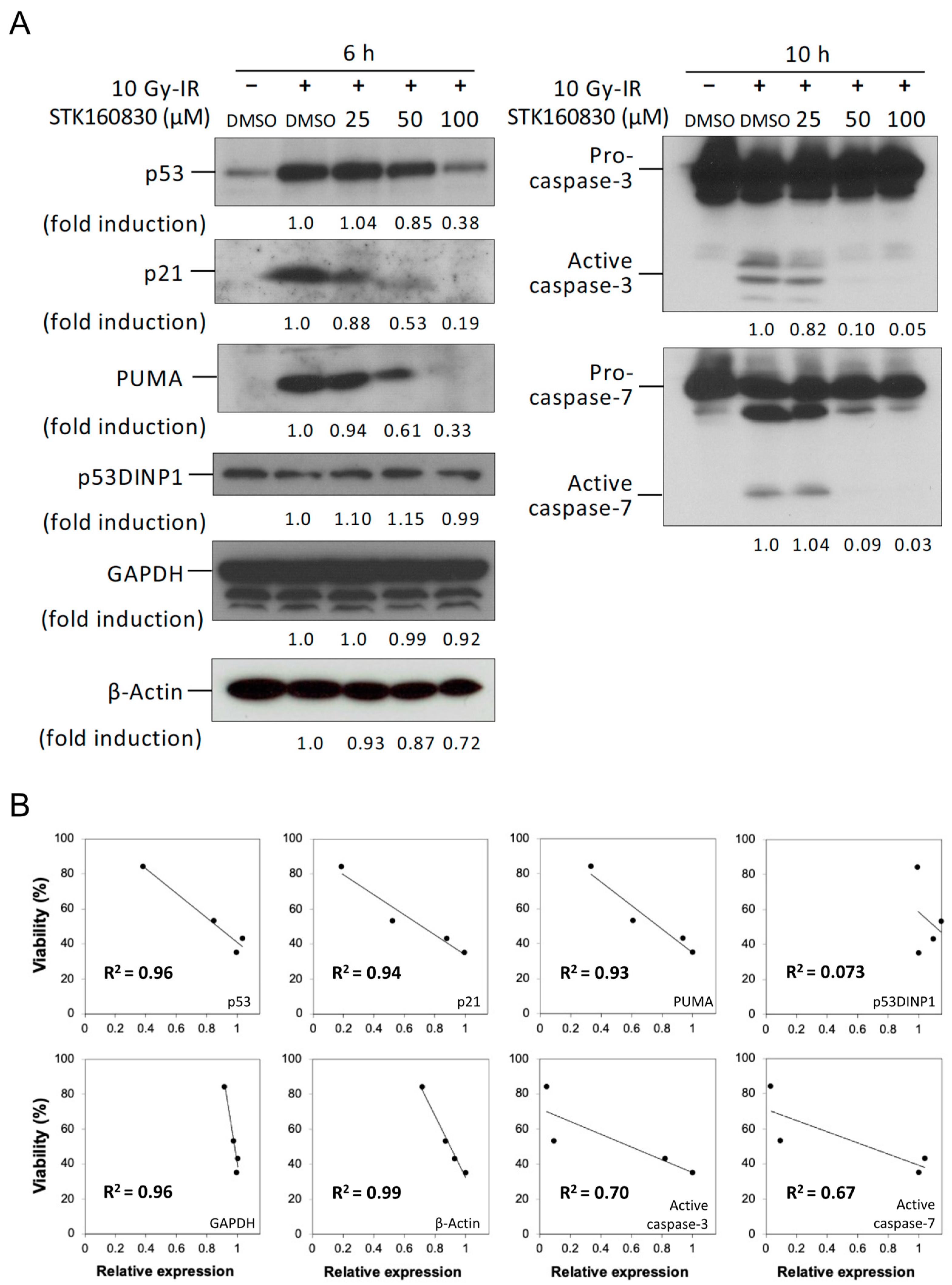
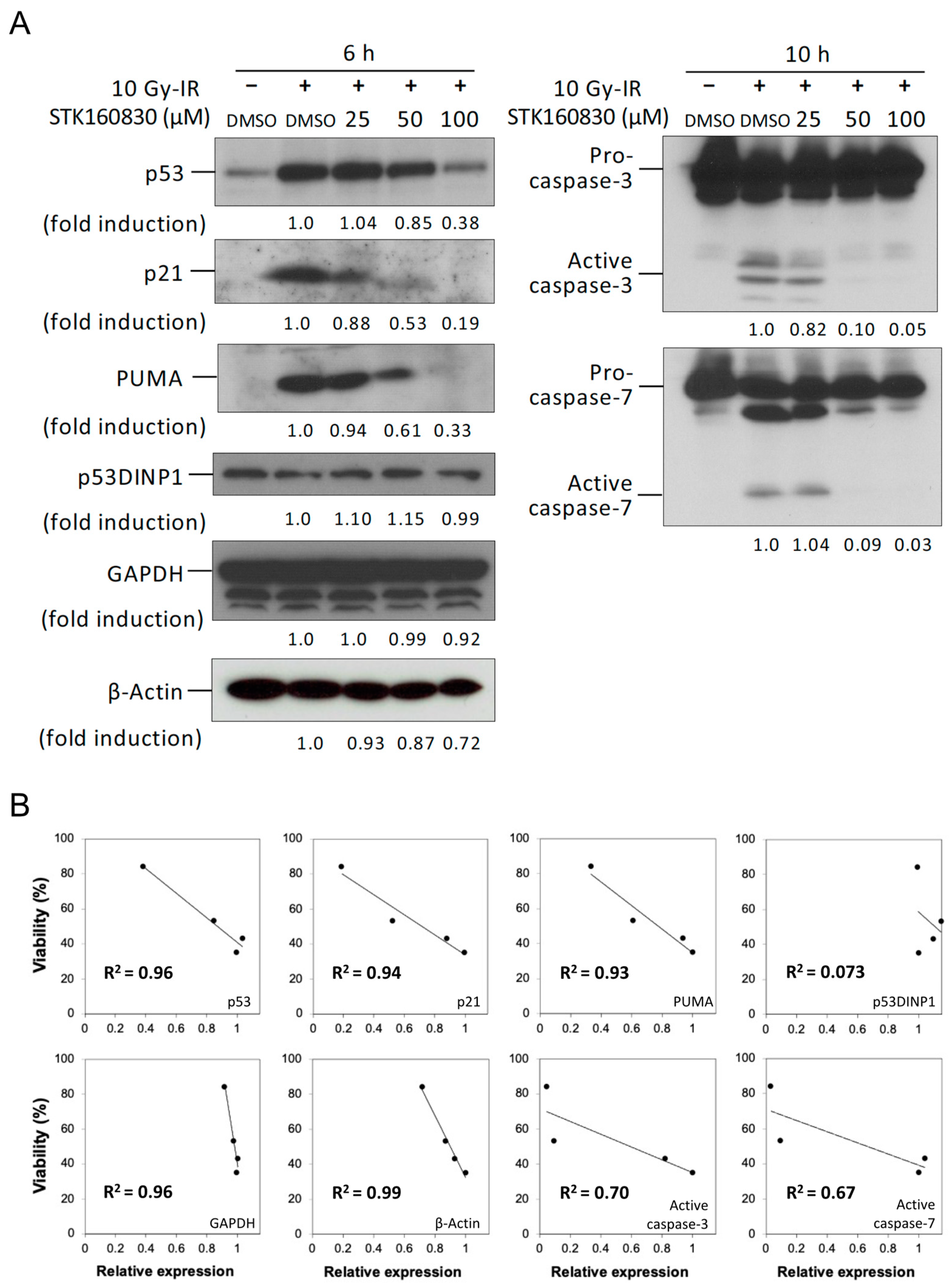
3.2. Correlation between Changes in Intracellular Protein Expression and Viability by STK160830
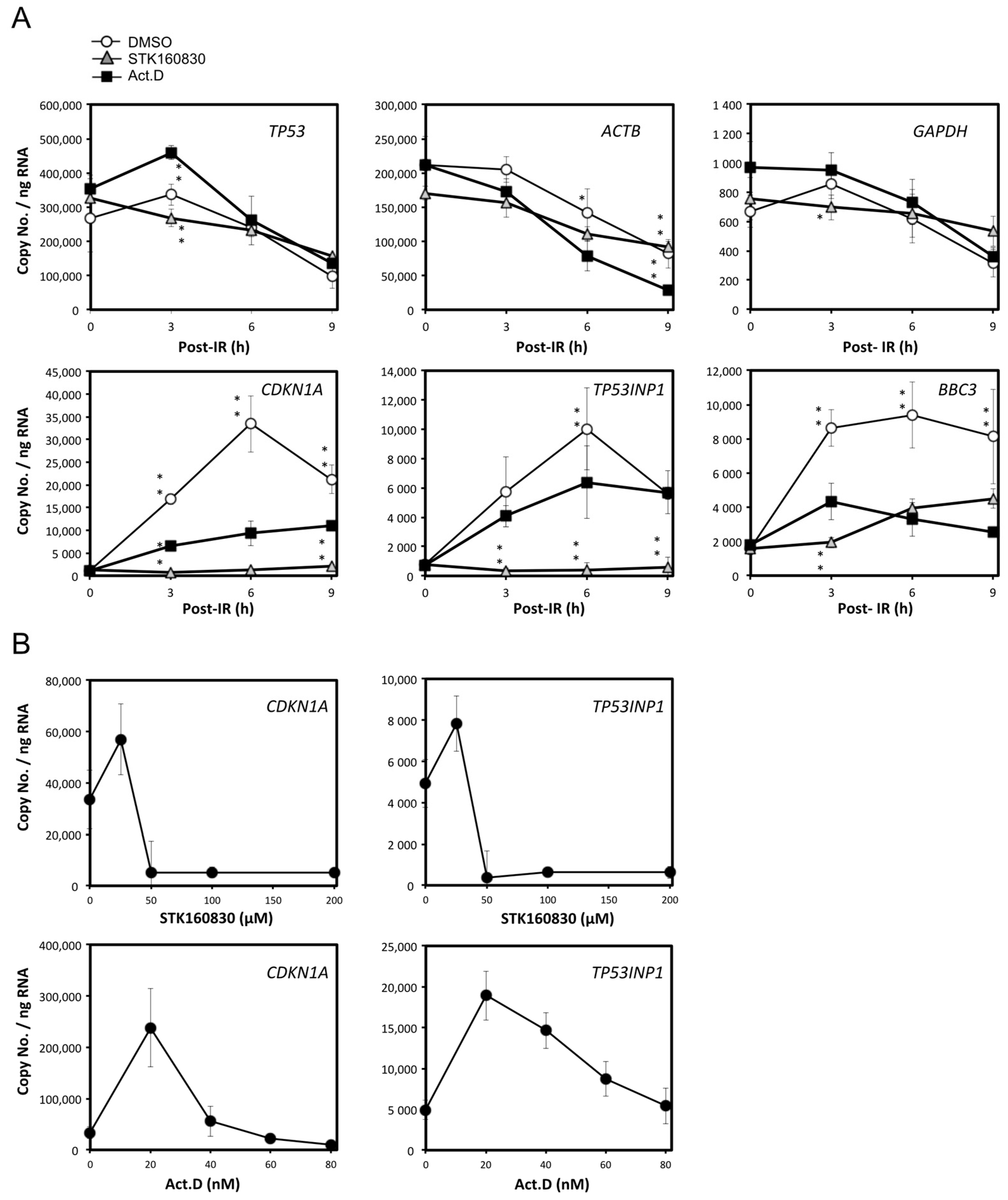
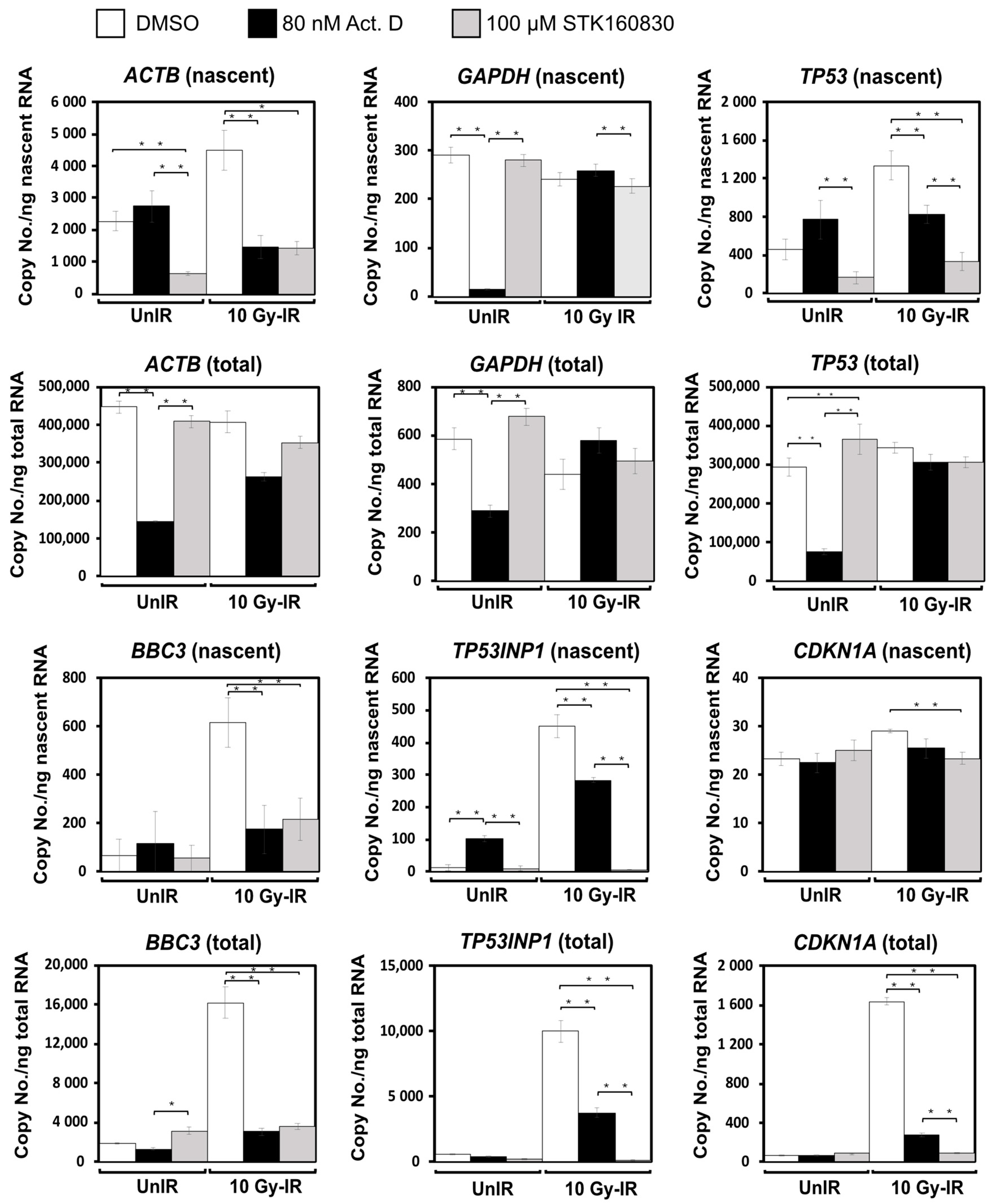
3.3. STK160830 Is A Novel mRNA Synthesis Inhibitor with A Different Spectrum of Action from Act.D
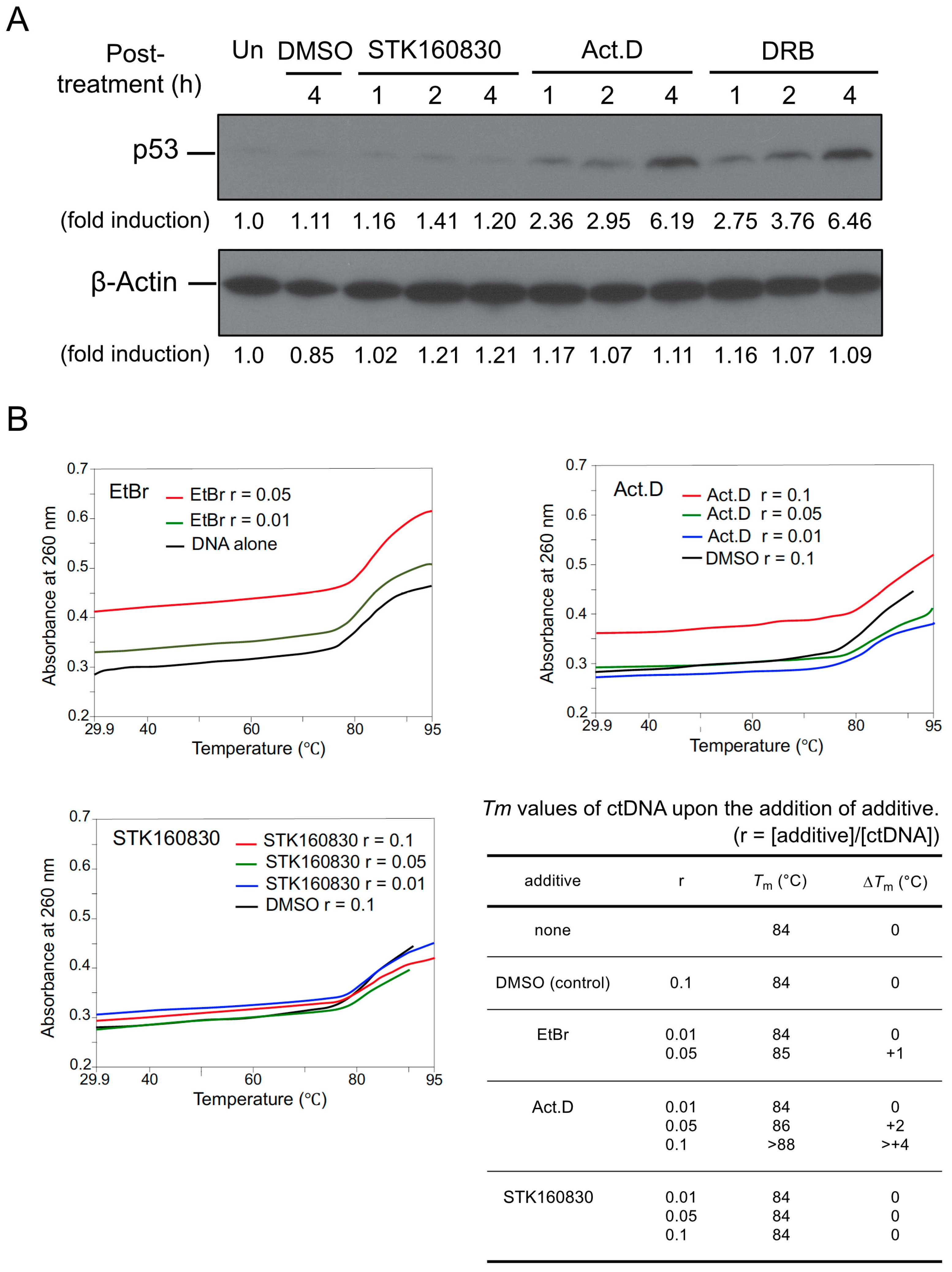
3.4. STK160830 Has Negligible DNA-Intercalating Activity for Triggering A p53 Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chernova, O.B.; Chernov, M.V.; Agarwal, M.L.; Taylor, W.R.; Stark, G.R. The role of p53 in regulating genomic stability when DNA and RNA synthesis are inhibited. Trends Biochem. Sci. 1995, 20, 431–434. [Google Scholar] [CrossRef]
- Ljungman, M.; Zhang, F.; Chen, F.; Rainbow, A.J.; McKay, B.C. Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 1999, 18, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Arima, Y.; Nitta, M.; Kuninaka, S.; Zhang, D.; Fujiwara, T.; Taya, Y.; Nakao, M.; Saya, H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J. Biol. Chem. 2005, 280, 19166–19176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, W.E.; Bradley, M.O. DNA double-stranded breaks in mammalian cells after exposure to intercalating agents. Biochim. Biophys. Acta 1981, 654, 129–134. [Google Scholar] [CrossRef]
- Chen, A.Y.; Liu, L.F. DNA topoisomerases: Essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Baguley, B.C. Topoisomerase II enzymes and mutagenicity. Environ. Mol. Mutagen. 1994, 24, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Chodosh, L.A.; Fire, A.; Samuels, M.; Sharp, P.A. 5,6-Dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J. Biol. Chem. 1989, 264, 2250–2257. [Google Scholar] [CrossRef]
- Prelich, G. RNA polymerase II carboxy-terminal domain kinases: Emerging clues to their function. Eukaryot. Cell 2002, 1, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.J.; Belotserkovskaya, R.; Reinberg, D. Elongation by RNA polymerase II: The short and long of it. Genes Dev. 2004, 18, 2437–2468. [Google Scholar] [CrossRef] [Green Version]
- Sellins, K.S.; Cohen, J.J. Gene induction by gamma-irradiation leads to DNA fragmentation in lymphocytes. J. Immunol. 1987, 139, 3199–3206. [Google Scholar]
- Yamada, T.; Ohyama, H. Radiation-induced interphase death of rat thymocytes is internally programmed (apoptosis). Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1988, 53, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993, 362, 849–852. [Google Scholar] [CrossRef]
- Petrova, G.V.; Donchenko, G.V. Effect of alpha-tocopherol and its derivatives on actinomycin D induced apoptosis of rat thymocytes. Ukr. Biokhim. Zh. (1999) 2005, 77, 72–77. [Google Scholar]
- Kasai, T.; Ohguchi, K.; Nakashima, S.; Ito, Y.; Naganawa, T.; Kondo, N.; Nozawa, Y. Increased activity of oleate-dependent type phospholipase D during actinomycin D-induced apoptosis in Jurkat T cells. J. Immunol. 1998, 61, 6469–6474. [Google Scholar]
- Caelles, C.; Helmberg, A.; Karin, M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 1994, 370, 220–223. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Zaika, A.; Moll, U.M. Death signal-induced localization of p53 protein to mitochondria: A potential role in apoptotic signaling. J. Biol. Chem. 2000, 275, 6202–6212. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934. [Google Scholar] [CrossRef]
- Morita, A.; Takahashi, I.; Sasatani, M.; Aoki, S.; Wang, B.; Ariyasu, S.; Tanaka, K.; Yamaguchi, T.; Sawa, A.; Nishi, Y.; et al. A chemical modulator of p53 transactivation that acts as a radioprotective agonist. Mol. Cancer Ther. 2018, 17, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.H.; Buckwalter, M.R.; Stephenson, A.C.; Hart, J.L.; Taylor, B.J.; O’Neill, K.L. Radiation-induced apoptosis in MOLT-4 cells requires de novo protein synthesis independent of de novo RNA synthesis. FEBS Lett. 2002, 514, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Morita, A.; Ohya, S.; Yamamoto, S.; Enomoto, A.; Ikekita, M. Cycloheximide suppresses radiation-induced apoptosis in MOLT-4 cells with Arg72 variant of p53 through translational inhibition of p53 accumulation. J. Radiat. Res. 2011, 52, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.L.; Johnston, B.F.; Mackay, S.P.; Breslin, C.J.; Robertson, M.N.; Harvey, A.L. The Drug Discovery Portal: A resource to enhance drug discovery from academia. Drug Discov. Today 2010, 15, 679–683. [Google Scholar] [CrossRef]
- Cheng, J.; Haas, M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol. Cell. Biol. 1990, 10, 5502–5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.Q.; Osada, M.; Ishioka, C.; Gamo, M.; Ikawa, S.; Suzuki, T.; Shimodaira, H.; Niitani, T.; Kudo, T.; Akiyama, M.; et al. Screening the p53 status of human cell lines using a yeast functional assay. Mol. Carcinog. 1997, 19, 243–253. [Google Scholar] [CrossRef]
- Oconnor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57, 4285–4300. [Google Scholar]
- Gong, B.D.; Chen, Q.; Endlich, B.; Mazumder, S.; Almasan, A. Ionizing radiation-induced, Bax-mediated cell death is dependent on activation of cysteine and serine proteases. Cell Growth Differ. 1999, 10, 491–502. [Google Scholar] [PubMed]
- Nakano, H.; Kohara, M.; Shinohara, K. Evaluation of the relative contribution of p53-mediated pathway in X-ray-induced apoptosis in human leukemic MOLT-4 cells by transfection with a mutant p53 gene at different expression levels. Cell Tissue Res. 2001, 306, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Zhu, J.; Suzuki, N.; Enomoto, A.; Matsumoto, Y.; Tomita, M.; Suzuki, T.; Ohtomo, K.; Hosoi, Y. Sodium orthovanadate suppresses DNA damage-induced caspase activation and apoptosis by inactivating p53. Cell Death Differ. 2006, 13, 499–511. [Google Scholar] [CrossRef]
- Morita, A.; Ariyasu, S.; Ohya, S.; Takahashi, I.; Wang, B.; Tanaka, K.; Uchida, T.; Okazaki, H.; Hanaya, K.; Enomoto, A.; et al. Evaluation of Zinc (II) chelators for inhibiting p53-mediated apoptosis. Oncotarget 2013, 4, 2439–2450. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K.; Toyoshima, H.; Sakai, R.; Miyagawa, K.; Hagiwara, K.; Ishikawa, F.; Takaku, F.; Yazaki, Y.; Hirai, H. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 1992, 79, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, Y.; Hayashi, S.; Takahashi, H.; Tsuneyama, K.; Takano, Y. Correlation between DNA alterations and p53 and p16 protein expression in cancer cell lines. Pathol. Res. Pract. 2005, 201, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Yamamoto, S.; Wang, B.; Tanaka, K.; Suzuki, N.; Aoki, S.; Ito, A.; Nanao, T.; Ohya, S.; Yoshino, M.; et al. Sodium orthovanadate inhibits p53-mediated apoptosis. Cancer Res. 2010, 70, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, A.; Ariyasu, S.; Wang, B.; Asanuma, T.; Onoda, T.; Sawa, A.; Tanaka, K.; Takahashi, I.; Togami, S.; Nenoi, M.; et al. AS-2, a novel inhibitor of p53-dependent apoptosis, prevents apoptotic mitochondrial dysfunction in a transcription-independent manner and protects mice from a lethal dose of ionizing radiation. Biochem. Biophys. Res. Commun. 2014, 450, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C., 3rd; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nature Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Pim, D.; Banks, L. p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int. J. Cancer 2004, 108, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Han, C.; Wan, G.; Huang, X.; Ivan, C.; Jiang, D.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Rao, P.H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Liu, X.; Lv, M.; Wu, Y.; Kuang, S.; Gong, J.; Yuan, P.; Zhong, Z.; Li, Q.; Jia, H.; et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012, 40, 5201–5214. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morita, A.; Ochi, S.; Satoh, H.; Ujita, S.; Matsushita, Y.; Tada, K.; Toyoda, M.; Nishiyama, Y.; Mizuno, K.; Deguchi, Y.; et al. A Novel RNA Synthesis Inhibitor, STK160830, Has Negligible DNA-Intercalating Activity for Triggering A p53 Response, and Can Inhibit p53-Dependent Apoptosis. Life 2021, 11, 1087. https://doi.org/10.3390/life11101087
Morita A, Ochi S, Satoh H, Ujita S, Matsushita Y, Tada K, Toyoda M, Nishiyama Y, Mizuno K, Deguchi Y, et al. A Novel RNA Synthesis Inhibitor, STK160830, Has Negligible DNA-Intercalating Activity for Triggering A p53 Response, and Can Inhibit p53-Dependent Apoptosis. Life. 2021; 11(10):1087. https://doi.org/10.3390/life11101087
Chicago/Turabian StyleMorita, Akinori, Shintaro Ochi, Hidetoshi Satoh, Shohei Ujita, Yosuke Matsushita, Kasumi Tada, Mihiro Toyoda, Yuichi Nishiyama, Kosuke Mizuno, Yuichi Deguchi, and et al. 2021. "A Novel RNA Synthesis Inhibitor, STK160830, Has Negligible DNA-Intercalating Activity for Triggering A p53 Response, and Can Inhibit p53-Dependent Apoptosis" Life 11, no. 10: 1087. https://doi.org/10.3390/life11101087
APA StyleMorita, A., Ochi, S., Satoh, H., Ujita, S., Matsushita, Y., Tada, K., Toyoda, M., Nishiyama, Y., Mizuno, K., Deguchi, Y., Suzuki, K., Tanaka, Y., Ueda, H., Inaba, T., Hosoi, Y., & Aoki, S. (2021). A Novel RNA Synthesis Inhibitor, STK160830, Has Negligible DNA-Intercalating Activity for Triggering A p53 Response, and Can Inhibit p53-Dependent Apoptosis. Life, 11(10), 1087. https://doi.org/10.3390/life11101087