
Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
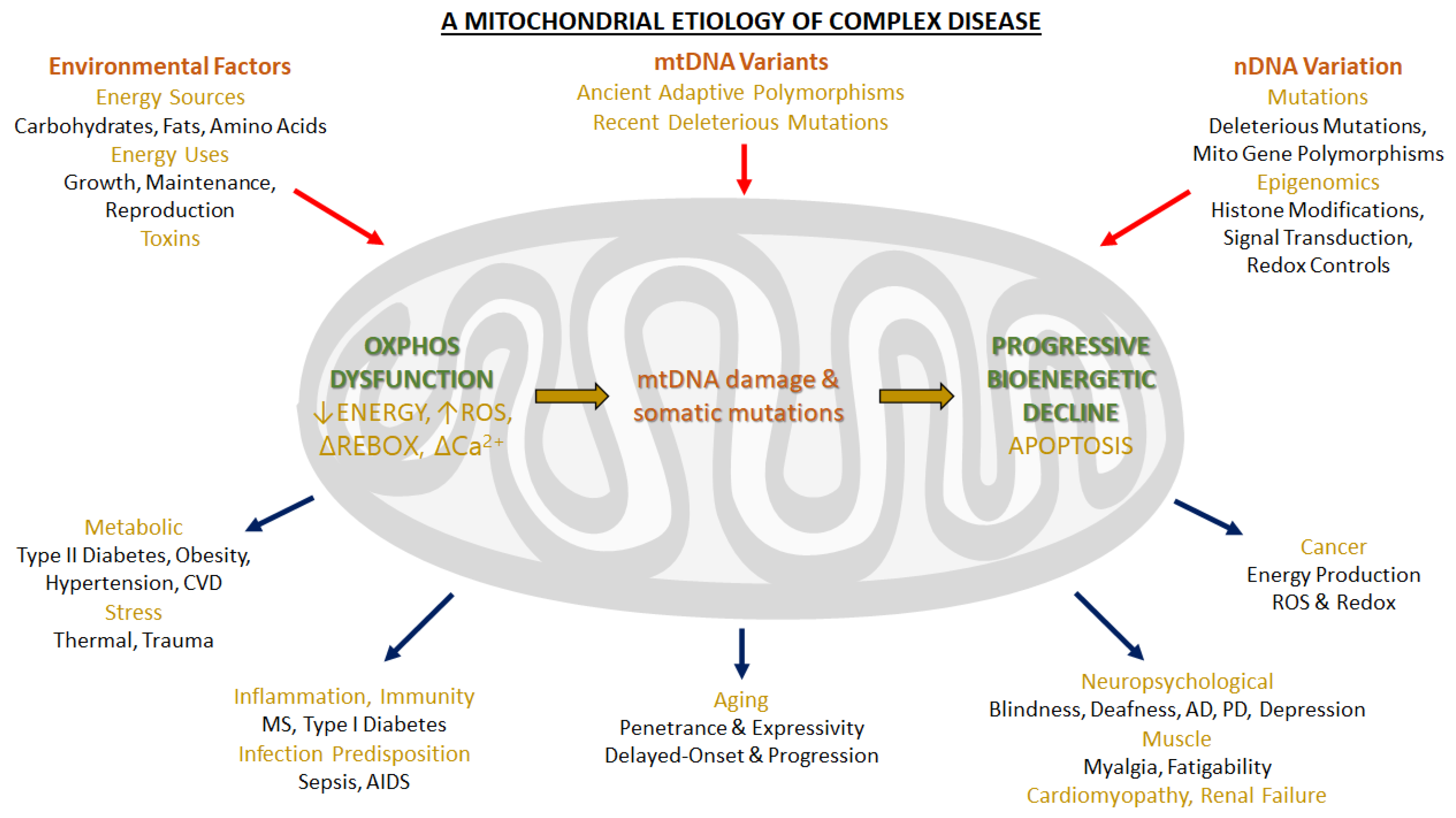
:1. Introduction: Functions of Mitochondria. The Oxidative Phosphorylation System
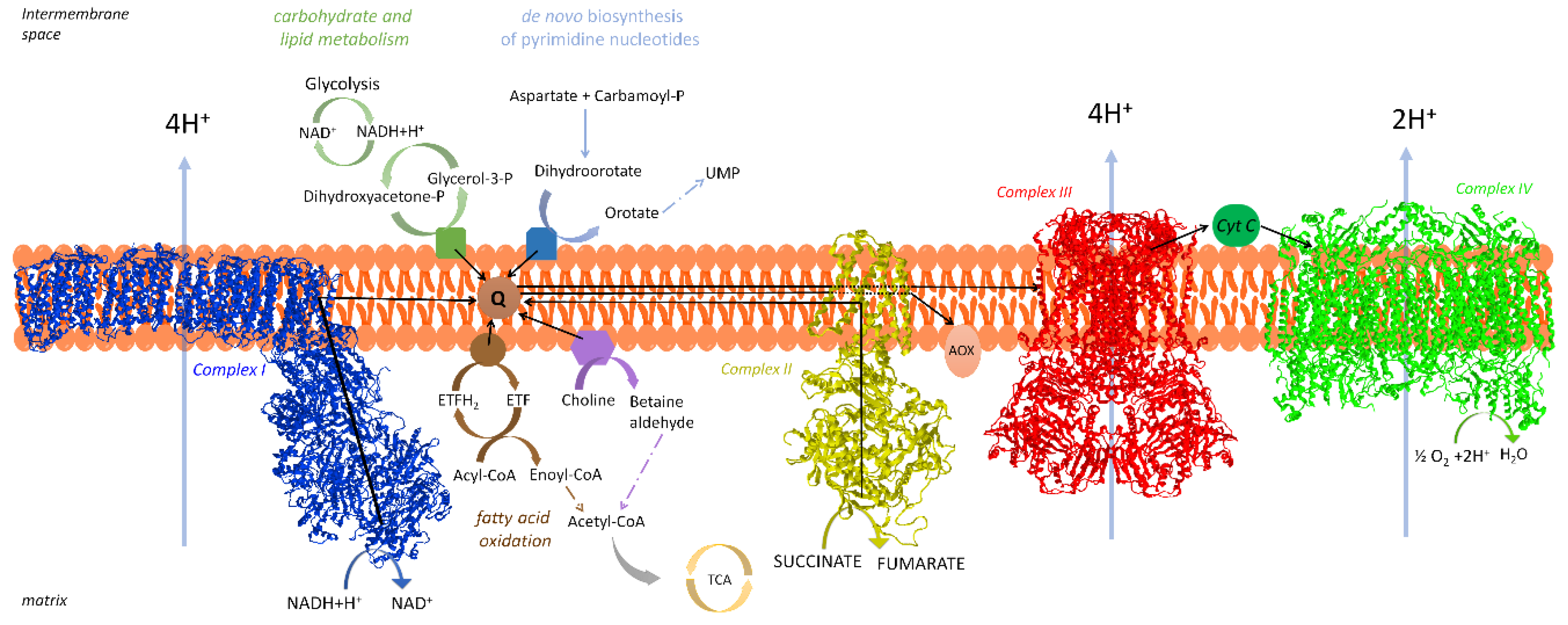
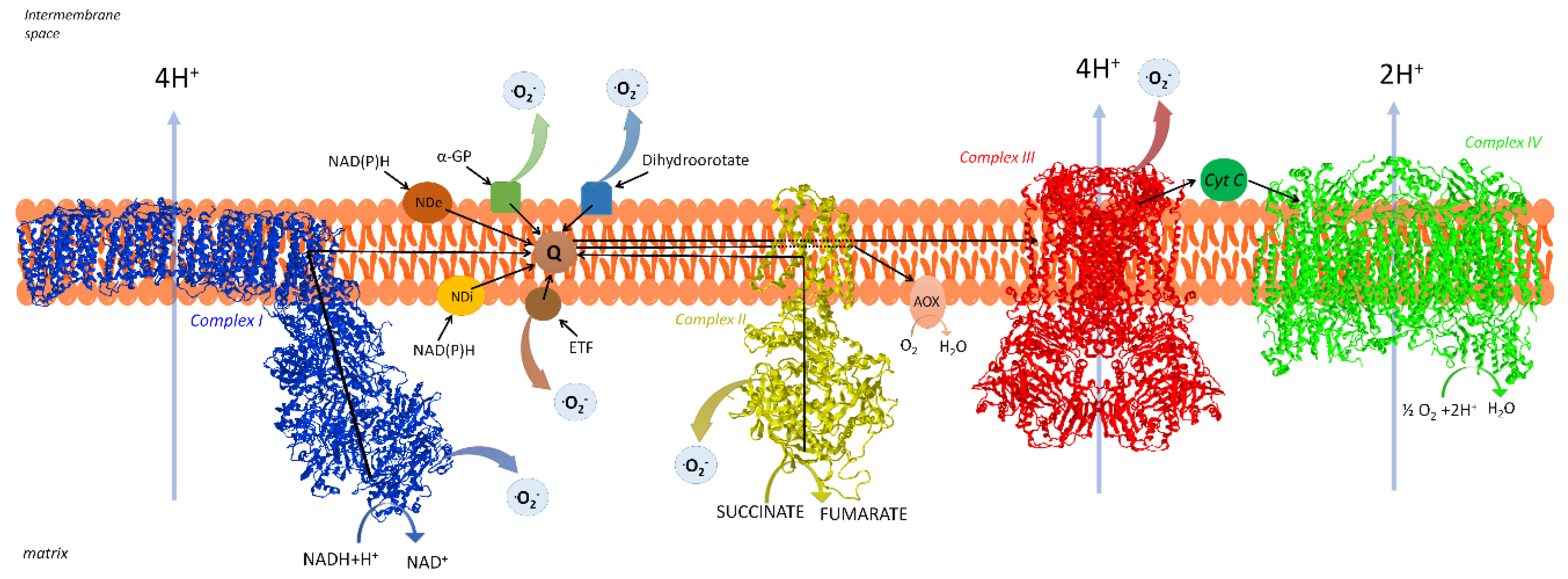
2. The Respiratory Chain of Mitochondria
2.1. Organization of the Respiratory Chain: Historical Outline
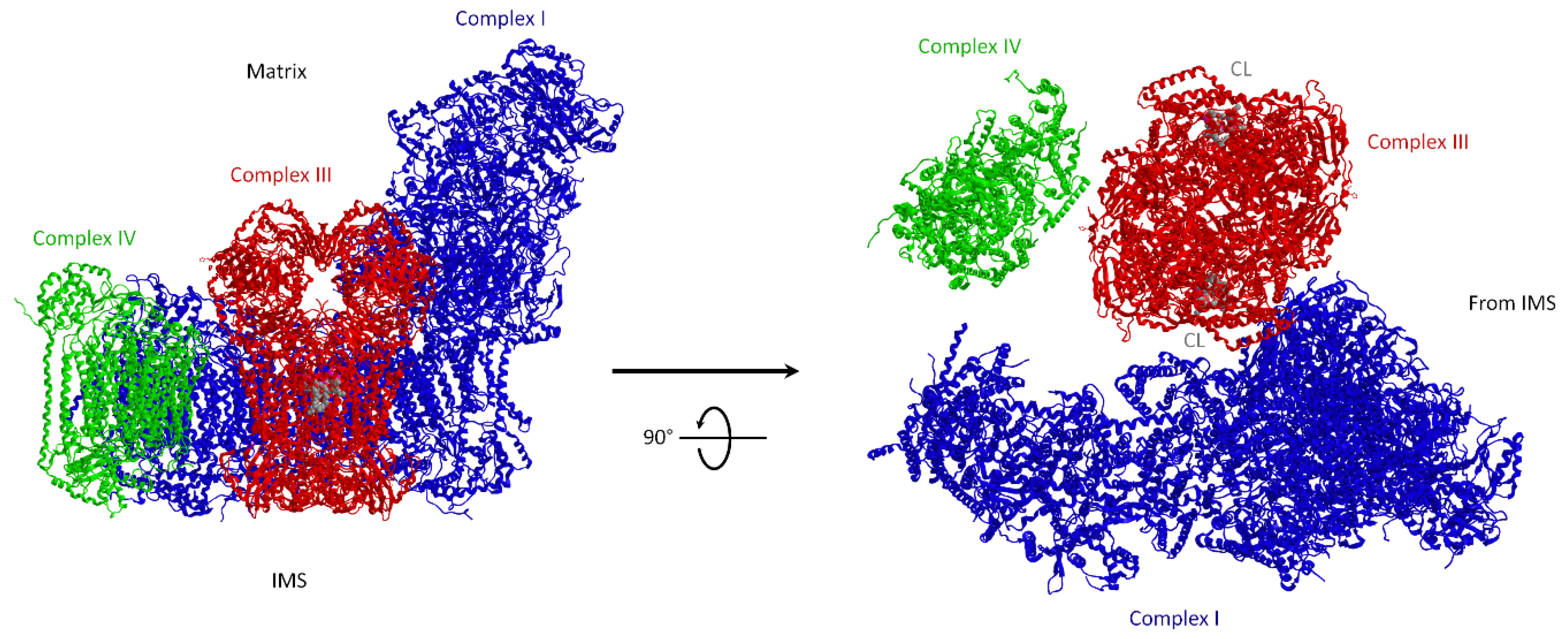
2.2. Distribution and Composition of Respiratory Supercomplexes
3. Supercomplexes May Provide a Kinetic Advantage to Electron Transfer
3.1. Structural Evidence Suggesting Channeling
3.1.1. Molecular Structure of Supercomplexes

3.1.2. Supercomplexes Are Dynamic Structures: The Plasticity Model
3.1.3. The Role of Lipids: Cardiolipin in Supercomplexes
3.2. Kinetic Evidence for Channeling in the Coenzyme Q Region
3.2.1. Rate Advantage Imposed by SCs
3.2.2. Metabolic Flux Control Analysis
3.2.3. Coenzyme Q Compartmentalization
3.3. If Electron Transfer Occurs via Channeling, What Is the Role of Coenzyme Q Pool?
3.3.1. Dissociation Equilibrium of Bound Coenzyme Q
3.3.2. Electron Transfer between Free Complexes
3.4. Electron Transfer through Cytochrome c: Is There Evidence for Channeling?
3.5. Supercomplexes and Regulation of Metabolic Fluxes
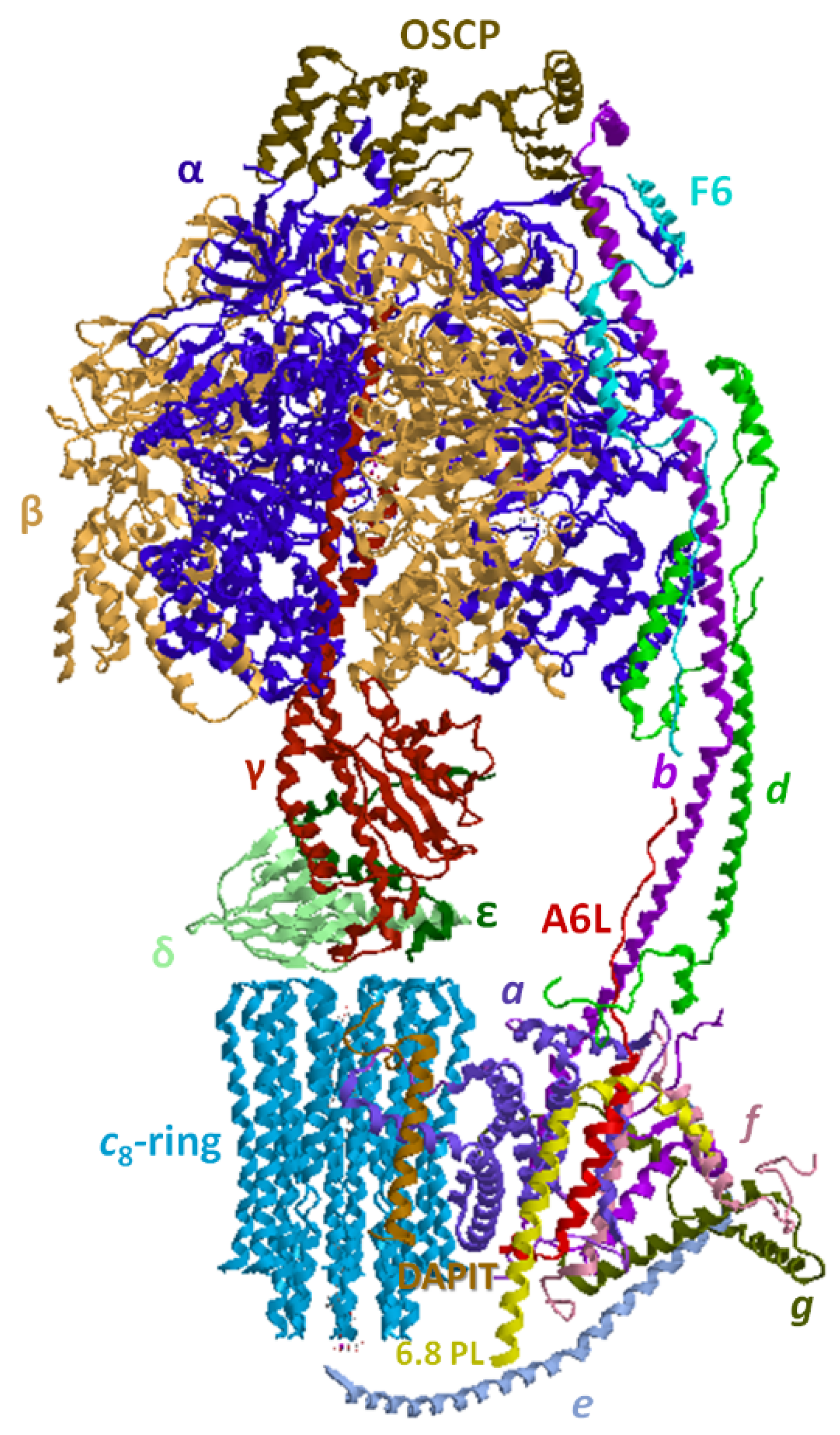
4. Overview of the F1FO-ATP Synthase/Hydrolase and Its Supramolecular Structure
4.1. The F1FO-ATP Synthase from the Energy Production to Mitochondrial Morphology
4.2. Structural Basis of the Bioenergetic Mechanism
4.3. The ATP Synthase Supramolecular Arrangement and the Mitochondrial Shape
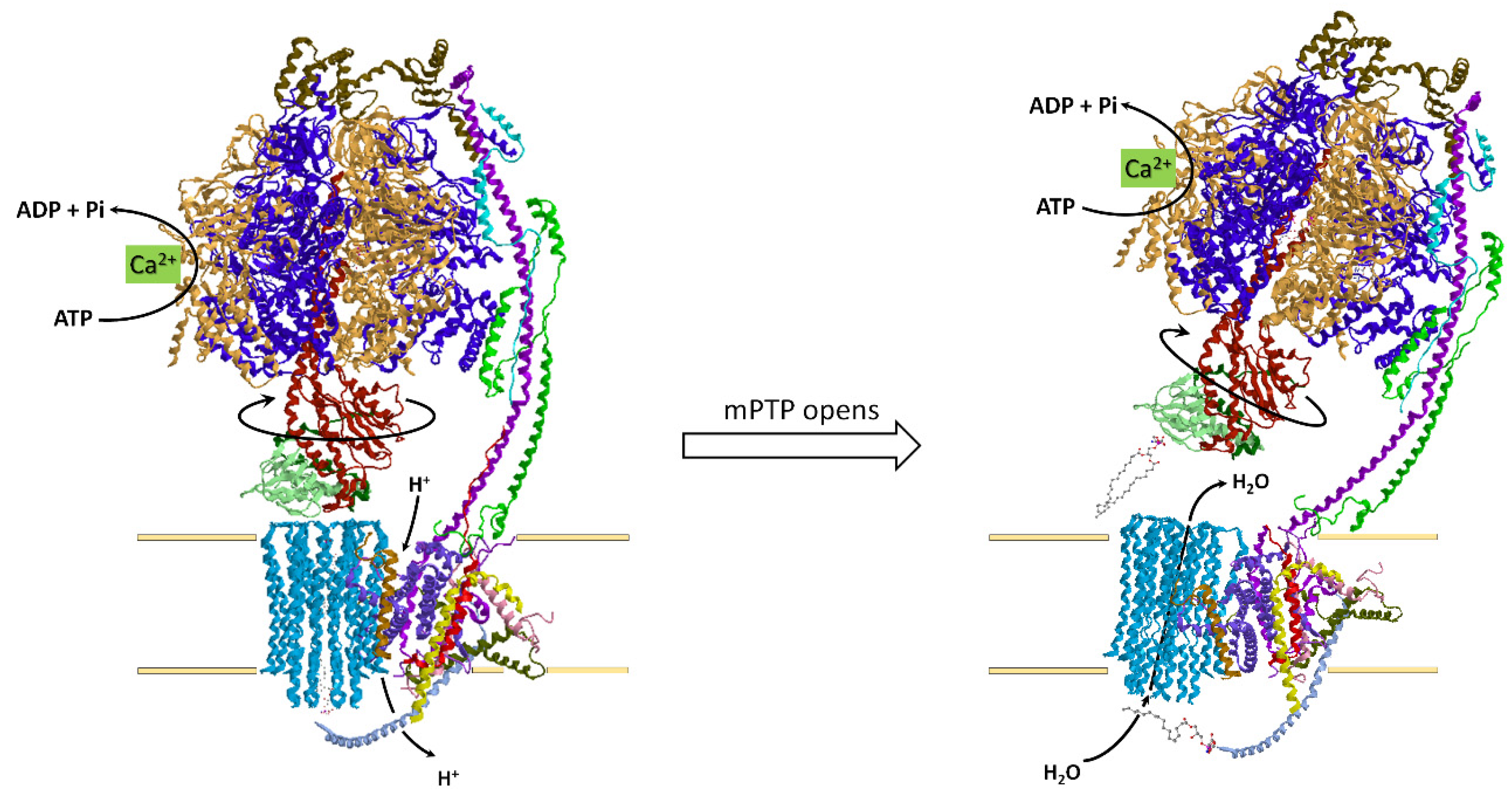
4.4. How the ATP Synthase/Hydrolase Is Involved in the Mitochondrial Permeability Transition Pore
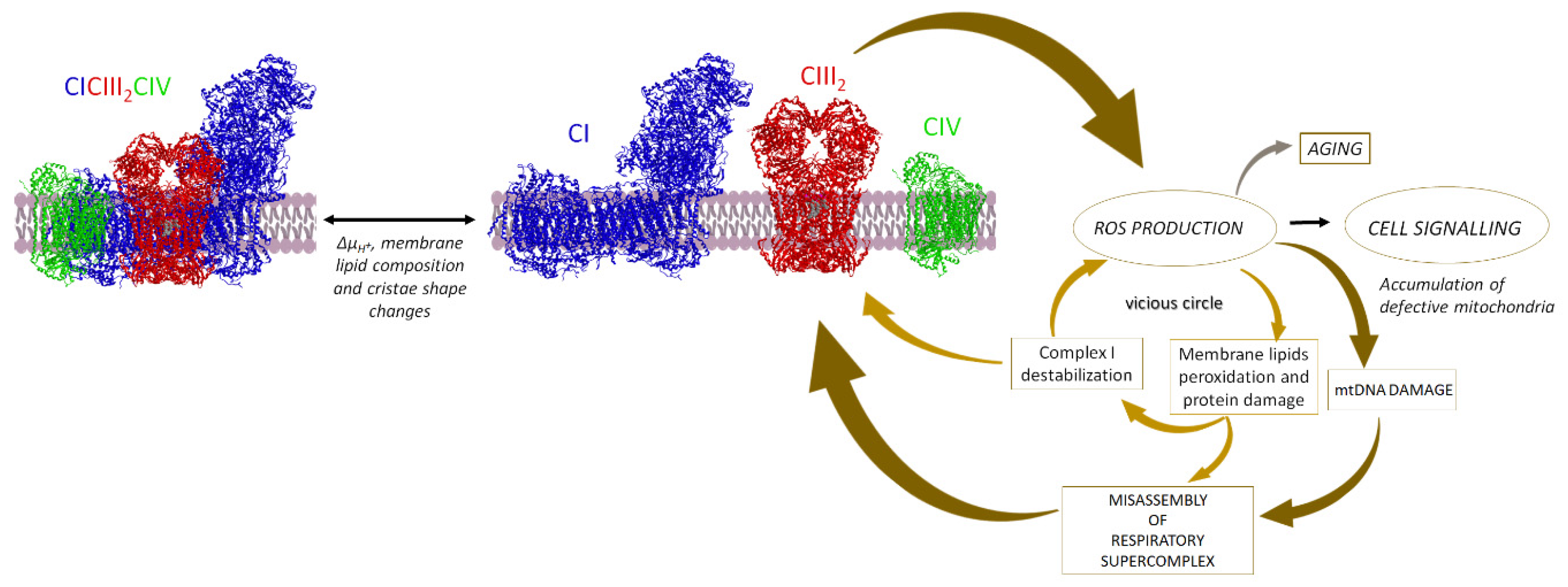
5. SCs Association Protects from ROS Damage
5.1. Mitochondrial Sources of ROS
ROS from the Respiratory Chain
5.2. Control of ROS Production
5.3. ROS as Signals
5.3.1. Regulation of Mitochondrial ROS in Cell Signaling
5.3.2. Hypoxia and ROS Production
5.4. ROS and Mitochondrial Quality Control
5.5. Supercomplexes Protect Complex I from ROS Damage and Limit ROS Generation
5.5.1. Supercomplex Association Protects from ROS Damage
5.5.2. Supercomplex Association Limits ROS Generation
6. Physiological and Pathological Implications
6.1. Uncoupling: When Proton Movement and ATP Synthesis Are Disjointed
6.1.1. Mitochondrial Uncoupling Due to Dissipative Pathways
6.1.2. Intrinsic ATP Synthase Uncoupling Due by Amino Acid Changes in the a Subunit
6.2. Supercomplexes and ROS Signaling
6.3. Supercomplexes in Pathology and Aging
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondrial Genetic Medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Wallace, D.C.; Burelle, Y. The Rise of Mitochondria in Medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetic Origins of Complexity and Disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef] [Green Version]
- DiMauro, S.; Hirano, M.; Schon, E.A. Approaches to the Treatment of Mitochondrial Diseases. Muscle Nerve 2006, 34, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Mitochondrial Medicine for Aging and Neurodegenerative Diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Isaev, N.K.; Plotnikov, E.Y.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Morosanova, M.A.; Jankauskas, S.S.; Zorov, S.D.; Babenko, V.A. Perspectives of Mitochondrial Medicine. Biochemistry 2013, 78, 979–990. [Google Scholar] [CrossRef]
- Luft, R. The Development of Mitochondrial Medicine. Proc. Natl. Acad. Sci. USA 1994, 91, 8731–8738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelos, I.; Shadiack, E.; Ganetzky, R.D.; Falk, M.J. Mitochondrial Medicine Therapies: Rationale, Evidence, and Dosing Guidelines. Curr. Opin. Pediatr. 2020, 32, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Ferguson, S.J. 1-Chemiosmotic Energy Transduction. In Bioenergetics, 4th ed.; Academic Press: Boston, MA, USA, 2013; pp. 3–12. ISBN 978-0-12-388425-1. [Google Scholar]
- Lenaz, G.; Genova, M.L. Structure and Organization of Mitochondrial Respiratory Complexes: A New Understanding of an Old Subject. Antioxid. Redox Signal. 2010, 12, 961–1008. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, Y.; Haavik, A.G.; Fowler, L.R.; Griffiths, D.E. Studies on the Electron Transfer System. XLII. Reconstitution of the Electron Transfer System. J. Biol. Chem. 1962, 237, 2661–2669. [Google Scholar] [CrossRef]
- Green, D.E.; Tzagoloff, A. The Mitochondrial Electron Transfer Chain. Arch. Biochem. Biophys. 1966, 116, 293–304. [Google Scholar] [CrossRef]
- Houstĕk, J.; Cannon, B.; Lindberg, O. Gylcerol-3-Phosphate Shuttle and Its Function in Intermediary Metabolism of Hamster Brown-Adipose Tissue. Eur. J. Biochem. 1975, 54, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.D.; Frerman, F.E. Reaction of Electron-Transfer Flavoprotein with Electron-Transfer Flavoprotein-Ubiquinone Oxidoreductase. Biochemistry 1985, 24, 3922–3925. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [Green Version]
- Salvi, F.; Gadda, G. Human Choline Dehydrogenase: Medical Promises and Biochemical Challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef]
- Poole, L.B. The Basics of Thiols and Cysteines in Redox Biology and Chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Subcell. Biochem. 2018, 87, 167–227. [Google Scholar] [CrossRef]
- Parey, K.; Wirth, C.; Vonck, J.; Zickermann, V. Respiratory Complex I—Structure, Mechanism and Evolution. Curr. Opin. Struct. Biol. 2020, 63, 1–9. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Xia, D.; Esser, L.; Tang, W.-K.; Zhou, F.; Zhou, Y.; Yu, L.; Yu, C.-A. Structural Analysis of Cytochrome Bc1 Complexes: Implications to the Mechanism of Function. Biochim. Biophys. Acta 2013, 1827, 1278–1294. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the Intact 14-Subunit Human Cytochrome c Oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kröger, A.; Klingenberg, M. The Kinetics of the Redox Reactions of Ubiquinone Related to the Electron-Transport Activity in the Respiratory Chain. Eur. J. Biochem. 1973, 34, 358–368. [Google Scholar] [CrossRef]
- Kröger, A.; Klingenberg, M. Further Evidence for the Pool Function of Ubiquinone as Derived from the Inhibition of the Electron Transport by Antimycin. Eur. J. Biochem. 1973, 39, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The Random Collision Model and a Critical Assessment of Diffusion and Collision in Mitochondrial Electron Transport. J. Bioenergy Biomembr. 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial Respiratory Supercomplexes in Mammalian Cells: Structural versus Functional Role. J. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Chance, B.; Williams, G.R. The Respiratory Chain and Oxidative Phosphorylation. Adv. Enzymol. Relat. Subj. Biochem. 1956, 17, 65–134. [Google Scholar] [CrossRef]
- Hatefi, Y.; Haavik, A.G.; Griffiths, D.E. Studies on the Electron Transfer System. XL. Preparation and Properties of Mitochondrial DPNH-Coenzyme Q Reductase. J. Biol. Chem. 1962, 237, 1676–1680. [Google Scholar] [CrossRef]
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Ozawa, T.; Papa, S. Bioenergetics: Structure and Function of Energy Transducing Systems; Springer: Berlin/Heidelberg, Germany, 1987; ISBN 978-3-540-18007-4. [Google Scholar]
- Hochman, J.; Ferguson-Miller, S.; Schindler, M. Mobility in the Mitochondrial Electron Transport Chain. Biochemistry 1985, 24, 2509–2516. [Google Scholar] [CrossRef]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the Respiratory Chains of Yeast and Mammalian Mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Schägger, H. Advantages and Limitations of Clear-Native PAGE. Proteomics 2005, 5, 4338–4346. [Google Scholar] [CrossRef]
- Lenaz, G.; Tioli, G.; Falasca, A.I.; Genova, M.L. Complex i Function in Mitochondrial Supercomplexes. Biochim. Biophys. Acta Bioenergy 2016, 1857, 991–1000. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Enriquez, J.A. The Function of the Respiratory Supercomplexes: The Plasticity Model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, M.L.; Lenaz, G. A Critical Appraisal of the Role of Respiratory Supercomplexes in Mitochondria. Biol. Chem. 2013, 394, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.-G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Brave, F.; Becker, T. Supercomplex Formation Boosts Respiration. EMBO Rep. 2020, e51830. [Google Scholar] [CrossRef]
- Vonck, J.; Schäfer, E. Supramolecular Organization of Protein Complexes in the Mitochondrial Inner Membrane. Biochim. Biophys. Acta 2009, 1793, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genova, M.L.; Lenaz, G. Functional Role of Mitochondrial Respiratory Supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Schägger, H.; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexes I-V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Respiratory Cytochrome Supercomplexes: Cytochrome Complexes: Evolution, Structures, Energy Transduction, and Signaling; Advances in Photosynthesis and Respiration; Springer: Dordrecht, The Netherlands, 2016; ISBN 978-94-017-7479-6. [Google Scholar]
- Wang, Y.; Mohsen, A.-W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for Physical Association of Mitochondrial Fatty Acid Oxidation and Oxidative Phosphorylation Complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsiah-Sefaa, A.; McKenzie, M. Combined Defects in Oxidative Phosphorylation and Fatty Acid β-Oxidation in Mitochondrial Disease. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaz, G.; Tioli, G.; Falasca, A.I.; Genova, M.L. Coenzyme Q and Respiratory Supercomplexes: Physiological and Pathological Implications. Rend. Fis. Acc. Lincei 2018, 29, 383–395. [Google Scholar] [CrossRef]
- Fang, J.; Uchiumi, T.; Yagi, M.; Matsumoto, S.; Amamoto, R.; Takazaki, S.; Yamaza, H.; Nonaka, K.; Kang, D. Dihydro-Orotate Dehydrogenase Is Physically Associated with the Respiratory Complex and Its Loss Leads to Mitochondrial Dysfunction. Biosci. Rep. 2013, 33, e00021. [Google Scholar] [CrossRef]
- Hildebrandt, T.M. Modulation of Sulfide Oxidation and Toxicity in Rat Mitochondria by Dehydroascorbic Acid. Biochim. Biophys. Acta 2011, 1807, 1206–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovádi, J. Physiological Significance of Metabolic Channelling. J. Theor. Biol. 1991, 152, 1–22. [Google Scholar] [CrossRef]
- Gupte, S.S.; Hackenbrock, C.R. The Role of Cytochrome c Diffusion in Mitochondrial Electron Transport. J. Biol. Chem. 1988, 263, 5248–5253. [Google Scholar] [CrossRef]
- Althoff, T.; Mills, D.J.; Popot, J.-L.; Kühlbrandt, W. Arrangement of Electron Transport Chain Components in Bovine Mitochondrial Supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef] [Green Version]
- Dudkina, N.V.; Kudryashev, M.; Stahlberg, H.; Boekema, E.J. Interaction of Complexes I, III, and IV within the Bovine Respirasome by Single Particle Cryoelectron Tomography. Proc. Natl. Acad. Sci. USA 2011, 108, 15196–15200. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The Architecture of Respiratory Supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Letts, J.A.; Sazanov, L.A. Clarifying the Supercomplex: The Higher-Order Organization of the Mitochondrial Electron Transport Chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609.e10. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.S.; Mills, D.J.; Vonck, J.; Kühlbrandt, W. Functional Asymmetry and Electron Flow in the Bovine Respirasome. eLife 2016, 5. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-Dependent Formation of Mitochondrial Respiratory Supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. The Protonmotive Q Cycle: A General Formulation. FEBS Lett. 1975, 59, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Sarewicz, M.; Osyczka, A. Electronic Connection between the Quinone and Cytochrome C Redox Pools and Its Role in Regulation of Mitochondrial Electron Transport and Redox Signaling. Physiol. Rev. 2015, 95, 219–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covian, R.; Zwicker, K.; Rotsaert, F.A.; Trumpower, B.L. Asymmetric and Redox-Specific Binding of Quinone and Quinol at Center N of the Dimeric Yeast Cytochrome Bc1 Complex. Consequences for Semiquinone Stabilization. J. Biol. Chem. 2007, 282, 24198–24208. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Degliesposti, G.; Skehel, M.; Sazanov, L.A. Structures of Respiratory Supercomplex I+III2 Reveal Functional and Conformational Crosstalk. Mol. Cell 2019, 75, 1131–1146.e6. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, C.; Genova, M.L.; Parenti Castelli, G.; Lenaz, G. The Mitochondrial Respiratory Chain Is Partially Organized in a Supercomplex Assembly: Kinetic Evidence Using Flux Control Analysis. J. Biol. Chem. 2004, 279, 36562–36569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Enríquez, J.A. Supramolecular Organization of Respiratory Complexes. Annu. Rev. Physiol. 2016, 78, 533–561. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M. Protein Crowding in the Inner Mitochondrial Membrane. Biochim. Biophys. Acta Bioenergy 2021, 1862, 148305. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, M.; Lazarou, M.; Thorburn, D.R.; Ryan, M.T. Mitochondrial Respiratory Chain Supercomplexes Are Destabilized in Barth Syndrome Patients. J. Mol. Biol. 2006, 361, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.-P.; Landes, T.; Arnauné-Pelloquin, L.; Vial, G.; Taleux, N.; Slomianny, C.; et al. Barth Syndrome: Cellular Compensation of Mitochondrial Dysfunction and Apoptosis Inhibition Due to Changes in Cardiolipin Remodeling Linked to Tafazzin (TAZ) Gene Mutation. Biochim. Biophys. Acta 2013, 1832, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmurs, M.; Foti, M.; Raemy, E.; Vaz, F.M.; Martinou, J.-C.; Bairoch, A.; Lane, L. C11orf83, a Mitochondrial Cardiolipin-Binding Protein Involved in Bc1 Complex Assembly and Supercomplex Stabilization. Mol. Cell. Biol. 2015, 35, 1139–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, C.D.; Basu Ball, W.; Pryce, E.N.; Gohil, V.M. Specific Requirements of Nonbilayer Phospholipids in Mitochondrial Respiratory Chain Function and Formation. Mol. Biol. Cell 2016, 27, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Baracca, A.; Biondi, A.; Casalena, G.; Faccioli, M.; Falasca, A.I.; Formiggini, G.; Sgarbi, G.; Solaini, G.; Lenaz, G. Is Supercomplex Organization of the Respiratory Chain Required for Optimal Electron Transfer Activity? Biochim. Biophys. Acta 2008, 1777, 740–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Oxidative Stress, Mitochondrial Bioenergetics, and Cardiolipin in Aging. Free Radic. Biol. Med. 2010, 48, 1286–1295. [Google Scholar] [CrossRef]
- Wasmus, C.; Dudek, J. Metabolic Alterations Caused by Defective Cardiolipin Remodeling in Inherited Cardiomyopathies. Life 2020, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Rieger, B.; Krajčová, A.; Duwe, P.; Busch, K.B. ALCAT1 Overexpression Affects Supercomplex Formation and Increases ROS in Respiring Mitochondria. Oxid. Med. Cell Longev. 2019, 2019, 9186469. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Kubota-Sakashita, M.; Nagatsuka, Y.; Hirabayashi, Y.; Hanasaka, T.; Tohyama, K.; Kato, T. Cardiolipin Is Essential for Early Embryonic Viability and Mitochondrial Integrity of Neurons in Mammals. FASEB J. 2020, 34, 1465–1480. [Google Scholar] [CrossRef] [Green Version]
- Ragan, C.I.; Heron, C. The Interaction between Mitochondrial NADH-Ubiquinone Oxidoreductase and Ubiquinol-Cytochrome c Oxidoreductase. Evidence for Stoicheiometric Association. Biochem. J. 1978, 174, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heron, C.; Ragan, C.I.; Trumpower, B.L. The Interaction between Mitochondrial NADH-Ubiquinone Oxidoreductase and Ubiquinol-Cytochrome c Oxidoreductase. Restoration of Ubiquinone-Pool Behaviour. Biochem. J. 1978, 174, 791–800. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Fato, R.; Di Bernardo, S.; Jarreta, D.; Costa, A.; Genova, M.L.; Parenti Castelli, G. Localization and Mobility of Coenzyme Q in Lipid Bilayers and Membranes. Biofactors 1999, 9, 87–93. [Google Scholar] [CrossRef] [PubMed]
- García-Poyatos, C.; Cogliati, S.; Calvo, E.; Hernansanz-Agustín, P.; Lagarrigue, S.; Magni, R.; Botos, M.; Langa, X.; Amati, F.; Vázquez, J.; et al. Scaf1 Promotes Respiratory Supercomplexes and Metabolic Efficiency in Zebrafish. EMBO Rep. 2020, 21, e50287. [Google Scholar] [CrossRef]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol Enrichment in Liver Mitochondria Impairs Oxidative Phosphorylation and Disrupts the Assembly of Respiratory Supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Tomková, V.; Sandoval-Acuña, C.; Torrealba, N.; Truksa, J. Mitochondrial Fragmentation, Elevated Mitochondrial Superoxide and Respiratory Supercomplexes Disassembly Is Connected with the Tamoxifen-Resistant Phenotype of Breast Cancer Cells. Free Radic. Biol. Med. 2019, 143, 510–521. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-EIF2α Axis. Mol. Cell 2019, 74, 877–890.e6. [Google Scholar] [CrossRef]
- Kholodenko, B.N.; Westerhoff, H.V. Metabolic Channelling and Control of the Flux. FEBS Lett. 1993, 320, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Blaza, J.N.; Serreli, R.; Jones, A.J.Y.; Mohammed, K.; Hirst, J. Kinetic Evidence against Partitioning of the Ubiquinone Pool and the Catalytic Relevance of Respiratory-Chain Supercomplexes. Proc. Natl. Acad. Sci. USA 2014, 111, 15735–15740. [Google Scholar] [CrossRef] [Green Version]
- Fato, R.; Estornell, E.; Di Bernardo, S.; Pallotti, F.; Parenti Castelli, G.; Lenaz, G. Steady-State Kinetics of the Reduction of Coenzyme Q Analogs by Complex I (NADH:Ubiquinone Oxidoreductase) in Bovine Heart Mitochondria and Submitochondrial Particles. Biochemistry 1996, 35, 2705–2716. [Google Scholar] [CrossRef]
- Quarato, G.; Piccoli, C.; Scrima, R.; Capitanio, N. Variation of Flux Control Coefficient of Cytochrome c Oxidase and of the Other Respiratory Chain Complexes at Different Values of Protonmotive Force Occurs by a Threshold Mechanism. Biochim. Biophys. Acta 2011, 1807, 1114–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaambre, T.; Chekulayev, V.; Shevchuk, I.; Karu-Varikmaa, M.; Timohhina, N.; Tepp, K.; Bogovskaja, J.; Kütner, R.; Valvere, V.; Saks, V. Metabolic Control Analysis of Cellular Respiration in Situ in Intraoperational Samples of Human Breast Cancer. J. Bioenergy Biomembr. 2012, 44, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Kaambre, T.; Chekulayev, V.; Shevchuk, I.; Tepp, K.; Timohhina, N.; Varikmaa, M.; Bagur, R.; Klepinin, A.; Anmann, T.; Koit, A.; et al. Metabolic Control Analysis of Respiration in Human Cancer Tissue. Front. Physiol. 2013, 4, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutman, M.; Silman, N. Mutual Inhibition between NADH Oxidase and Succinoxidase Activities in Respiring Submitochondrial Particles. FEBS Lett. 1972, 26, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Gutman, M.; Kearney, E.B.; Singer, T.P. Control of Succinate Dehydrogenase in Mitochondria. Biochemistry 1971, 10, 4763–4770. [Google Scholar] [CrossRef]
- Gutman, M. Kinetic Analysis of Electron Flux through the Quinones in the Mitochondrial System. In Coenzyme Q; Lenaz, G., Ed.; Wiley: Hoboken, NJ, USA, 1985. [Google Scholar]
- Ragan, C.I.; Cottingham, I.R. The Kinetics of Quinone Pools in Electron Transport. Biochim. Biophys. Acta 1985, 811, 13–31. [Google Scholar] [CrossRef]
- Jørgensen, B.M.; Rasmussen, H.N.; Rasmussen, U.F. Ubiquinone Reduction Pattern in Pigeon Heart Mitochondria. Identification of Three Distinct Ubiquinone Pools. Biochem. J. 1985, 229, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Lenaz, G.; Genova, M.L. Kinetics of Integrated Electron Transfer in the Mitochondrial Respiratory Chain: Random Collisions vs. Solid State Electron Channeling. Am. J. Physiol. Cell Physiol. 2007, 292, C1221–C1239. [Google Scholar] [CrossRef] [Green Version]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531.e4. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J. Open Questions: Respiratory Chain Supercomplexes-Why Are They There and What Do They Do? BMC Biol. 2018, 16, 111. [Google Scholar] [CrossRef] [Green Version]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic Consequences from Xenotopic Expression of a Tunicate AOX in Mouse Mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenergy 2020, 1861, 148137. [Google Scholar] [CrossRef]
- Calvo, E.; Cogliati, S.; Hernansanz-Agustín, P.; Loureiro-López, M.; Guarás, A.; Casuso, R.A.; García-Marqués, F.; Acín-Pérez, R.; Martí-Mateos, Y.; Silla-Castro, J.C.; et al. Functional Role of Respiratory Supercomplexes in Mice: SCAF1 Relevance and Segmentation of the Qpool. Sci. Adv. 2020, 6, eaba7509. [Google Scholar] [CrossRef]
- Estornell, E.; Fato, R.; Castelluccio, C.; Cavazzoni, M.; Parenti Castelli, G.; Lenaz, G. Saturation Kinetics of Coenzyme Q in NADH and Succinate Oxidation in Beef Heart Mitochondria. FEBS Lett. 1992, 311, 107–109. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Lemasters, J.J.; Hackenbrock, C.R. Lateral Diffusion of Ubiquinone during Electron Transfer in Phospholipid- and Ubiquinone-Enriched Mitochondrial Membranes. J. Biol. Chem. 1982, 257, 10789–10793. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac Mitochondria in Heart Failure: Decrease in Respirasomes and Oxidative Phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Van Raam, B.J.; Sluiter, W.; de Wit, E.; Roos, D.; Verhoeven, A.J.; Kuijpers, T.W. Mitochondrial Membrane Potential in Human Neutrophils Is Maintained by Complex III Activity in the Absence of Supercomplex Organisation. PLoS ONE 2008, 3, e2013. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.J.Y.; Blaza, J.N.; Bridges, H.R.; May, B.; Moore, A.L.; Hirst, J. A Self-Assembled Respiratory Chain That Catalyzes NADH Oxidation by Ubiquinone-10 Cycling between Complex I and the Alternative Oxidase. Angew. Chem. Int. Ed. Engl. 2016, 55, 728–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoner, C.D. Steady-State Kinetics of the Overall Oxidative Phosphorylation Reaction in Heart Mitochondria. Determination of the Coupling Relationships between the Respiratory Reactions and Miscellaneous Observations Concerning Rate-Limiting Steps. J. Bioenergy Biomembr. 1984, 16, 115–141. [Google Scholar] [CrossRef] [PubMed]
- Rauchová, H.; Fato, R.; Drahota, Z.; Lenaz, G. Steady-State Kinetics of Reduction of Coenzyme Q Analogs by Glycerol-3-Phosphate Dehydrogenase in Brown Adipose Tissue Mitochondria. Arch. Biochem. Biophys. 1997, 344, 235–241. [Google Scholar] [CrossRef]
- Mráček, T.; Holzerová, E.; Drahota, Z.; Kovářová, N.; Vrbacký, M.; Ješina, P.; Houštěk, J. ROS Generation and Multiple Forms of Mammalian Mitochondrial Glycerol-3-Phosphate Dehydrogenase. Biochim. Biophys. Acta 2014, 1837, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesci, S.; Lenaz, G. The Mitochondrial Energy Conversion Involves Cytochrome c Diffusion into the Respiratory Supercomplexes. Biochim. Biophys. Acta Bioenergy 2021, 1862, 148394. [Google Scholar] [CrossRef] [PubMed]
- Boumans, H.; Grivell, L.A.; Berden, J.A. The Respiratory Chain in Yeast Behaves as a Single Functional Unit. J. Biol. Chem. 1998, 273, 4872–4877. [Google Scholar] [CrossRef] [Green Version]
- Heinemeyer, J.; Braun, H.-P.; Boekema, E.J.; Kouril, R. A Structural Model of the Cytochrome C Reductase/Oxidase Supercomplex from Yeast Mitochondria. J. Biol. Chem. 2007, 282, 12240–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mileykovskaya, E.; Penczek, P.A.; Fang, J.; Mallampalli, V.K.P.S.; Sparagna, G.C.; Dowhan, W. Arrangement of the Respiratory Chain Complexes in Saccharomyces Cerevisiae Supercomplex III2IV2 Revealed by Single Particle Cryo-Electron Microscopy. J. Biol. Chem. 2012, 287, 23095–23103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trouillard, M.; Meunier, B.; Rappaport, F. Questioning the Functional Relevance of Mitochondrial Supercomplexes by Time-Resolved Analysis of the Respiratory Chain. Proc. Natl. Acad. Sci. USA 2011, 108, E1027–E1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydström Lundin, C.; von Ballmoos, C.; Ott, M.; Ädelroth, P.; Brzezinski, P. Regulatory Role of the Respiratory Supercomplex Factors in Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 2016, 113, E4476–E4485. [Google Scholar] [CrossRef] [Green Version]
- Stuchebrukhov, A.; Schäfer, J.; Berg, J.; Brzezinski, P. Kinetic Advantage of Forming Respiratory Supercomplexes. Biochim. Biophys. Acta Bioenergy 2020, 1861, 148193. [Google Scholar] [CrossRef] [PubMed]
- Berndtsson, J.; Aufschnaiter, A.; Rathore, S.; Marin-Buera, L.; Dawitz, H.; Diessl, J.; Kohler, V.; Barrientos, A.; Büttner, S.; Fontanesi, F.; et al. Respiratory Supercomplexes Enhance Electron Transport by Decreasing Cytochrome c Diffusion Distance. EMBO Rep. 2020, e51015. [Google Scholar] [CrossRef]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vögtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 Mediates Cytochrome Oxidase Assembly and Respirasome Formation, Revealing Heterogeneity of the Enzyme Complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Loshuertos, R.; Enríquez, J.A. Respiratory Supercomplexes and the Functional Segmentation of the CoQ Pool. Free Radic. Biol. Med. 2016, 100, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Speijer, D. Oxygen Radicals Shaping Evolution: Why Fatty Acid Catabolism Leads to Peroxisomes While Neurons Do without It: FADH₂/NADH Flux Ratios Determining Mitochondrial Radical Formation Were Crucial for the Eukaryotic Invention of Peroxisomes and Catabolic Tissue Differentiation. Bioessays 2011, 33, 88–94. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schertl, P.; Braun, H.-P. Respiratory Electron Transfer Pathways in Plant Mitochondria. Front. Plant Sci. 2014, 5, 163. [Google Scholar] [CrossRef] [Green Version]
- Muench, S.P.; Trinick, J.; Harrison, M.A. Structural Divergence of the Rotary ATPases. Q. Rev. Biophys. 2011, 44, 311–356. [Google Scholar] [CrossRef]
- Okuno, D.; Iino, R.; Noji, H. Rotation and Structure of FoF1-ATP Synthase. J. Biochem. 2011, 149, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pagliarani, A. The a Subunit Asymmetry Dictates the Two Opposite Rotation Directions in the Synthesis and Hydrolysis of ATP by the Mitochondrial ATP Synthase. Med. Hypotheses 2015, 84, 53–57. [Google Scholar] [CrossRef]
- Boyer, P.D. The ATP Synthase—A Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Muneyuki, E.; Hisabori, T. ATP Synthase—A Marvellous Rotary Engine of the Cell. Nat. Rev. Mol. Cell Biol. 2001, 2, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Zhang, L.; Zong, S.; Guo, R.; Liu, T.; Yi, J.; Wang, P.; Zhuo, W.; Yang, M. Cryo-EM Structure of the Mammalian ATP Synthase Tetramer Bound with Inhibitory Protein IF1. Science 2019, 364, 1068–1075. [Google Scholar] [CrossRef]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM Structure of the Entire Mammalian F-Type ATP Synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Algieri, C.; Pagliarani, A. A Therapeutic Role for the F1FO-ATP Synthase. SLAS Discov. 2019, 24, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Pagliarani, A.; Algieri, C.; Trombetti, F. Mitochondrial F-Type ATP Synthase: Multiple Enzyme Functions Revealed by the Membrane-Embedded FO Structure. Crit. Rev. Biochem. Mol. Biol. 2020, 1–13. [Google Scholar] [CrossRef]
- Junge, W.; Lill, H.; Engelbrecht, S. ATP Synthase: An Electrochemical Transducer with Rotatory Mechanics. Trends Biochem. Sci. 1997, 22, 420–423. [Google Scholar] [CrossRef]
- Junge, W.; Sielaff, H.; Engelbrecht, S. Torque Generation and Elastic Power Transmission in the Rotary F(O)F(1)-ATPase. Nature 2009, 459, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pagliarani, A. Opposite Rotation Directions in the Synthesis and Hydrolysis of ATP by the ATP Synthase: Hints from a Subunit Asymmetry. J. Membr. Biol. 2015, 248, 163–169. [Google Scholar] [CrossRef]
- Lippe, G.; Coluccino, G.; Zancani, M.; Baratta, W.; Crusiz, P. Mitochondrial F-ATP Synthase and Its Transition into an Energy-Dissipating Molecular Machine. Oxid. Med. Cell Longev. 2019, 2019, 8743257. [Google Scholar] [CrossRef] [PubMed]
- Symersky, J.; Osowski, D.; Walters, D.E.; Mueller, D.M. Oligomycin Frames a Common Drug-Binding Site in the ATP Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, 13961–13965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549. [Google Scholar] [CrossRef]
- Hahn, A.; Parey, K.; Bublitz, M.; Mills, D.J.; Zickermann, V.; Vonck, J.; Kühlbrandt, W.; Meier, T. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Mol. Cell 2016, 63, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Boyer, P.D. Catalytic Site Occupancy during ATP Synthase Catalysis. FEBS Lett. 2002, 512, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Murataliev, M.B.; Boyer, P.D. The Mechanism of Stimulation of MgATPase Activity of Chloroplast F1-ATPase by Non-Catalytic Adenine-Nucleotide Binding. Acceleration of the ATP-Dependent Release of Inhibitory ADP from a Catalytic Site. Eur. J. Biochem. 1992, 209, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding Structure, Function, and Mutations in the Mitochondrial ATP Synthase. Microb. Cell 2015, 2, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Pogoryelov, D.; Klyszejko, A.L.; Krasnoselska, G.O.; Heller, E.-M.; Leone, V.; Langer, J.D.; Vonck, J.; Müller, D.J.; Faraldo-Gómez, J.D.; Meier, T. Engineering Rotor Ring Stoichiometries in the ATP Synthase. Proc. Natl. Acad. Sci. USA 2012, 109, E1599–E1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Ballmoos, C.; Cook, G.M.; Dimroth, P. Unique Rotary ATP Synthase and Its Biological Diversity. Annu. Rev. Biophys. 2008, 37, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Ventrella, V.; Trombetti, F.; Pirini, M.; Pagliarani, A. Mussel and Mammalian ATP Synthase Share the Same Bioenergetic Cost of ATP. J. Bioenergy Biomembr. 2013, 45, 289–300. [Google Scholar] [CrossRef]
- Hahn, A.; Vonck, J.; Mills, D.J.; Meier, T.; Kühlbrandt, W. Structure, Mechanism, and Regulation of the Chloroplast ATP Synthase. Science 2018, 360, eaat4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Suzuki, T.; Rubinstein, J.L. Structure of a Bacterial ATP Synthase. eLife 2019, 8. [Google Scholar] [CrossRef]
- Allegretti, M.; Klusch, N.; Mills, D.J.; Vonck, J.; Kühlbrandt, W.; Davies, K.M. Horizontal Membrane-Intrinsic α-Helices in the Stator a-Subunit of an F-Type ATP Synthase. Nature 2015, 521, 237–240. [Google Scholar] [CrossRef]
- Kühlbrandt, W.; Davies, K.M. Rotary ATPases: A New Twist to an Ancient Machine. Trends Biochem. Sci. 2016, 41, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Klusch, N.; Murphy, B.J.; Mills, D.J.; Yildiz, Ö.; Kühlbrandt, W. Structural Basis of Proton Translocation and Force Generation in Mitochondrial ATP Synthase. eLife 2017, 6, e33274. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer Ribbons of ATP Synthase Shape the Inner Mitochondrial Membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual Cristae within the Same Mitochondrion Display Different Membrane Potentials and Are Functionally Independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef]
- Srivastava, A.P.; Luo, M.; Zhou, W.; Symersky, J.; Bai, D.; Chambers, M.G.; Faraldo-Gómez, J.D.; Liao, M.; Mueller, D.M. High-Resolution Cryo-EM Analysis of the Yeast ATP Synthase in a Lipid Membrane. Science 2018, 360, eaas9699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, P.D. Bioenergetic Coupling to Protonmotive Force: Should We Be Considering Hydronium Ion Coordination and Not Group Protonation? Trends Biochem. Sci. 1988, 13, 5–7. [Google Scholar] [CrossRef]
- Von Ballmoos, C.; Dimroth, P. Two Distinct Proton Binding Sites in the ATP Synthase Family. Biochemistry 2007, 46, 11800–11809. [Google Scholar] [CrossRef] [PubMed]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef] [PubMed]
- Pogoryelov, D.; Krah, A.; Langer, J.D.; Yildiz, Ö.; Faraldo-Gómez, J.D.; Meier, T. Microscopic Rotary Mechanism of Ion Translocation in the F(o) Complex of ATP Synthases. Nat. Chem. Biol. 2010, 6, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.D. New Perspective on the Reversibility of ATP Synthesis and Hydrolysis by Fo·F1-ATP Synthase (Hydrolase). Biochem. Moscow 2019, 84, 1247–1255. [Google Scholar] [CrossRef]
- Mitome, N.; Ono, S.; Sato, H.; Suzuki, T.; Sone, N.; Yoshida, M. Essential Arginine Residue of the F(o)-a Subunit in F(o)F(1)-ATP Synthase Has a Role to Prevent the Proton Shortcut without c-Ring Rotation in the F(o) Proton Channel. Biochem. J. 2010, 430, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, B.J.; Klusch, N.; Langer, J.; Mills, D.J.; Yildiz, Ö.; Kühlbrandt, W. Rotary Substates of Mitochondrial ATP Synthase Reveal the Basis of Flexible F1-Fo Coupling. Science 2019, 364. [Google Scholar] [CrossRef]
- Blum, T.B.; Hahn, A.; Meier, T.; Davies, K.M.; Kühlbrandt, W. Dimers of Mitochondrial ATP Synthase Induce Membrane Curvature and Self-Assemble into Rows. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [Green Version]
- Arnold, I.; Pfeiffer, K.; Neupert, W.; Stuart, R.A.; Schägger, H. Yeast Mitochondrial F1F0-ATP Synthase Exists as a Dimer: Identification of Three Dimer-Specific Subunits. EMBO J. 1998, 17, 7170–7178. [Google Scholar] [CrossRef]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the Yeast F1Fo-ATP Synthase Dimer and Its Role in Shaping the Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eydt, K.; Davies, K.M.; Behrendt, C.; Wittig, I.; Reichert, A.S. Cristae Architecture Is Determined by an Interplay of the MICOS Complex and the F1FO ATP Synthase via Mic27 and Mic10. Microb. Cell 2017, 4, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampelt, H.; van der Laan, M. The Yin & Yang of Mitochondrial Architecture—Interplay of MICOS and F1Fo-ATP Synthase in Cristae Formation. Microb. Cell 2017, 4, 236–239. [Google Scholar] [CrossRef]
- Colina-Tenorio, L.; Horten, P.; Pfanner, N.; Rampelt, H. Shaping the Mitochondrial Inner Membrane in Health and Disease. J. Intern. Med. 2020, 287, 645–664. [Google Scholar] [CrossRef]
- Faccenda, D.; Tan, C.H.; Seraphim, A.; Duchen, M.R.; Campanella, M. IF1 Limits the Apoptotic-Signalling Cascade by Preventing Mitochondrial Remodelling. Cell Death Differ. 2013, 20, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Esparza-Moltó, P.B.; Cuezva, J.M. The Role of Mitochondrial H+-ATP Synthase in Cancer. Front. Oncol. 2018, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, J.J.; Morales-Ríos, E.; Cortés-Hernandez, P.; Rodríguez-Zavala, J.S. The Inhibitor Protein (IF1) Promotes Dimerization of the Mitochondrial F1F0-ATP Synthase. Biochemistry 2006, 45, 12695–12703. [Google Scholar] [CrossRef]
- Nakamura, J.; Fujikawa, M.; Yoshida, M. IF1, a Natural Inhibitor of Mitochondrial ATP Synthase, Is Not Essential for the Normal Growth and Breeding of Mice. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, C.; Davies, K.M.; Faraldo-Gómez, J.D. Mitochondrial ATP Synthase Dimers Spontaneously Associate Due to a Long-Range Membrane-Induced Force. J. Gen. Physiol. 2018, 150, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesci, S.; Pagliarani, A. Emerging Roles for the Mitochondrial ATP Synthase Supercomplexes. Trends Biochem. Sci. 2019, 44, 821–823. [Google Scholar] [CrossRef]
- Arselin, G.; Giraud, M.-F.; Dautant, A.; Vaillier, J.; Brèthes, D.; Coulary-Salin, B.; Schaeffer, J.; Velours, J. The GxxxG Motif of the Transmembrane Domain of Subunit e Is Involved in the Dimerization/Oligomerization of the Yeast ATP Synthase Complex in the Mitochondrial Membrane. Eur. J. Biochem. 2003, 270, 1875–1884. [Google Scholar] [CrossRef]
- Bustos, D.M.; Velours, J. The Modification of the Conserved GXXXG Motif of the Membrane-Spanning Segment of Subunit g Destabilizes the Supramolecular Species of Yeast ATP Synthase. J. Biol. Chem. 2005, 280, 29004–29010. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Bueler, S.A.; Rubinstein, J.L. Atomic Model for the Dimeric FO Region of Mitochondrial ATP Synthase. Science 2017, 358, 936–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, F.K.; Sanchez, C.G.; Hurd, T.R.; Seifert, J.R.K.; Czech, B.; Preall, J.B.; Hannon, G.J.; Lehmann, R. ATP Synthase Promotes Germ Cell Differentiation Independent of Oxidative Phosphorylation. Nat. Cell. Biol. 2015, 17, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Faccenda, D.; Campanella, M. Molecular Regulation of the Mitochondrial F(1)F(o)-ATPsynthase: Physiological and Pathological Significance of the Inhibitory Factor 1 (IF(1)). Int. J. Cell. Biol. 2012, 2012, 367934. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Jonas, E.A. The New Role of F1Fo ATP Synthase in Mitochondria-Mediated Neurodegeneration and Neuroprotection. Exp. Neurol. 2020, 332, 113400. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-Dependent Dissociation of ATP Synthase Dimers and Loss of Inner-Membrane Cristae in Mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c Subunit of the FO ATP Synthase in Mitochondrial Permeability Transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.-A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An Uncoupling Channel within the C-Subunit Ring of the F1FO ATP Synthase Is the Mitochondrial Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Jonas, E.A. ATP Synthase C-Subunit Ring as the Channel of Mitochondrial Permeability Transition: Regulator of Metabolism in Development and Degeneration. J. Mol. Cell. Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Nesci, S.; Trombetti, F.; Fabbri, M.; Ventrella, V.; Pagliarani, A. Mitochondrial F1FO-ATPase and Permeability Transition Pore Response to Sulfide in the Midgut Gland of Mytilus Galloprovincialis. Biochimie 2021, 180, 222–228. [Google Scholar] [CrossRef]
- Torrezan-Nitao, E.; Figueiredo, R.C.B.Q.; Marques-Santos, L.F. Mitochondrial Permeability Transition Pore in Sea Urchin Female Gametes. Mech. Dev. 2018, 154, 208–218. [Google Scholar] [CrossRef]
- Liu, Y.; Zhi, D.; Li, M.; Liu, D.; Wang, X.; Wu, Z.; Zhang, Z.; Fei, D.; Li, Y.; Zhu, H.; et al. Shengmai Formula Suppressed Over-Activated Ras/MAPK Pathway in C. Elegans by Opening Mitochondrial Permeability Transition Pore via Regulating Cyclophilin D. Sci. Rep. 2016, 6, 38934. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, J.; Tu, C.; Wang, Z.; Xin, W. The Protective Effect of Blueberry Anthocyanins against Perfluorooctanoic Acid-Induced Disturbance in Planarian (Dugesia Japonica). Ecotoxicol. Environ. Saf. 2016, 127, 170–174. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, L.; Zhang, Y.; Mao, L.; Jiang, H. Mitochondria Response to Camptothecin and Hydroxycamptothecine-Induced Apoptosis in Spodoptera Exigua Cells. Pestic. Biochem. Physiol. 2017, 140, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Hu, Y.-F.; Song, J.; Yi, H.-S.; Wang, L.; Yang, Y.-Y.; Wang, Y.-P.; Zhang, M.; Pan, M.-H.; Lu, C. Effects of 10-Hydroxycamptothecin on Intrinsic Mitochondrial Pathway in Silkworm BmN-SWU1 Cells. Pestic. Biochem. Physiol. 2016, 127, 15–20. [Google Scholar] [CrossRef]
- Konràd, C.; Kiss, G.; Töröcsik, B.; Lábár, J.L.; Gerencser, A.A.; Mándi, M.; Adam-Vizi, V.; Chinopoulos, C. A Distinct Sequence in the Adenine Nucleotide Translocase from Artemia Franciscana Embryos Is Associated with Insensitivity to Bongkrekate and Atypical Effects of Adenine Nucleotides on Ca2+ Uptake and Sequestration. FEBS J. 2011, 278, 822–836. [Google Scholar] [CrossRef]
- Konrad, C.; Kiss, G.; Torocsik, B.; Adam-Vizi, V.; Chinopoulos, C. Absence of Ca2+-Induced Mitochondrial Permeability Transition but Presence of Bongkrekate-Sensitive Nucleotide Exchange in C. Crangon and P. Serratus. PLoS ONE 2012, 7, e39839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menze, M.A.; Fortner, G.; Nag, S.; Hand, S.C. Mechanisms of Apoptosis in Crustacea: What Conditions Induce versus Suppress Cell Death? Apoptosis 2010, 15, 293–312. [Google Scholar] [CrossRef] [Green Version]
- Nesci, S.; Pagliarani, A. Incoming News on the F-Type ATPase Structure and Functions in Mammalian Mitochondria. BBA Adv. 2021, 1, 100001. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Ventrella, V.; Pirini, M.; Pagliarani, A. Kinetic Properties of the Mitochondrial F1FO-ATPase Activity Elicited by Ca(2+) in Replacement of Mg(2+). Biochimie 2017, 140, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, S.; Melandri, A.B.; Solaini, G. Relevance of Divalent Cations to ATP-Driven Proton Pumping in Beef Heart Mitochondrial F0F1-ATPase. J. Bioenergy Biomembr. 1998, 30, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Pagliarani, A. Ca2+ as Cofactor of the Mitochondrial H+ -Translocating F1 FO -ATP(Hydrol)Ase. Proteins 2020. [Google Scholar] [CrossRef]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Bernardini, C.; Fabbri, M.; Forni, M.; Nesci, S. Mitochondrial Ca2+-Activated F1 FO -ATPase Hydrolyzes ATP and Promotes the Permeability Transition Pore. Ann. N. Y. Acad. Sci. 2019, 1457, 142–157. [Google Scholar] [CrossRef]
- Algieri, V.; Algieri, C.; Maiuolo, L.; De Nino, A.; Pagliarani, A.; Tallarida, M.A.; Trombetti, F.; Nesci, S. 1,5-Disubstituted-1,2,3-Triazoles as Inhibitors of the Mitochondrial Ca2+ -Activated F1 FO -ATP(Hydrol)Ase and the Permeability Transition Pore. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca(2+) Binding to F-ATP Synthase β Subunit Triggers the Mitochondrial Permeability Transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S. Mitochondrial Permeability Transition, F1FO-ATPase and Calcium: An Enigmatic Triangle. EMBO Rep. 2017, 18, 1265–1267. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Algieri, C.; Trombetti, F.; Ventrella, V.; Fabbri, M.; Pagliarani, A. Sulfide Affects the Mitochondrial Respiration, the Ca2+-Activated F1FO-ATPase Activity and the Permeability Transition Pore but Does Not Change the Mg2+-Activated F1FO-ATPase Activity in Swine Heart Mitochondria. Pharmacol. Res. 2021, 166, 105495. [Google Scholar] [CrossRef] [PubMed]
- Ader, N.R.; Hoffmann, P.C.; Ganeva, I.; Borgeaud, A.C.; Wang, C.; Youle, R.J.; Kukulski, W. Molecular and Topological Reorganizations in Mitochondrial Architecture Interplay during Bax-Mediated Steps of Apoptosis. eLife 2019, 8. [Google Scholar] [CrossRef]
- Carraro, M.; Carrer, A.; Urbani, A.; Bernardi, P. Molecular Nature and Regulation of the Mitochondrial Permeability Transition Pore(s), Drug Target(s) in Cardioprotection. J. Mol. Cell. Cardiol. 2020, 144, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M. Mitochondrial Channels: Ion Fluxes and More. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, J.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of Mitochondrial Permeability Transition by Deletion of the ANT Family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial Permeability Transition Involves Dissociation of F1FO ATP Synthase Dimers and C-Ring Conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Morciano, G.; Preti, D.; Pedriali, G.; Aquila, G.; Missiroli, S.; Fantinati, A.; Caroccia, N.; Pacifico, S.; Bonora, M.; Talarico, A.; et al. Discovery of Novel 1,3,8-Triazaspiro[4.5]Decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J. Med. Chem. 2018, 61, 7131–7143. [Google Scholar] [CrossRef] [PubMed]
- Algieri, C.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Nesci, S. Phenylglyoxal Inhibition of the Mitochondrial F1FO-ATPase Activated by Mg2+ or by Ca2+ Provides Clues on the Mitochondrial Permeability Transition Pore. Arch. Biochem. Biophys. 2020, 681, 108258. [Google Scholar] [CrossRef] [PubMed]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papu John, A.S.; Kundu, S.; Pushpakumar, S.; Amin, M.; Tyagi, S.C.; Sen, U. Hydrogen Sulfide Inhibits Ca2+-Induced Mitochondrial Permeability Transition Pore Opening in Type-1 Diabetes. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E269–E283. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, X.; Zhang, L.; Liu, B.; Zhu, Z.; He, Y.; Chen, J.; Zhu, J.; Yu, H. CypD-MPTP Axis Regulates Mitochondrial Functions Contributing to Osteogenic Dysfunction of MC3T3-E1 Cells in Inflammation. J. Physiol. Biochem. 2018, 74, 395–402. [Google Scholar] [CrossRef]
- Visalli, G.; Facciolà, A.; Currò, M.; Laganà, P.; La Fauci, V.; Iannazzo, D.; Pistone, A.; Di Pietro, A. Mitochondrial Impairment Induced by Sub-Chronic Exposure to Multi-Walled Carbon Nanotubes. Int. J. Environ. Res. Public Health 2019, 16, 792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Kreuzer, J.; Kumsta, C.; Wu, L.; Kamer, K.J.; Cedillo, L.; Zhang, Y.; Li, S.; Kacergis, M.C.; Webster, C.M.; et al. Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell 2019, 177, 299–314.e16. [Google Scholar] [CrossRef] [Green Version]
- Shares, B.H.; Smith, C.O.; Sheu, T.-J.; Sautchuk, R.; Schilling, K.; Shum, L.C.; Paine, A.; Huber, A.; Gira, E.; Brown, E.; et al. Inhibition of the Mitochondrial Permeability Transition Improves Bone Fracture Repair. Bone 2020, 137, 115391. [Google Scholar] [CrossRef]
- Nesci, S. The Mitochondrial Permeability Transition Pore in Cell Death: A Promising Drug Binding Bioarchitecture. Med. Res. Rev. 2020, 40, 811–817. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Cui, Y.; Pan, M.; Ma, J.; Song, X.; Cao, W.; Zhang, P. Recent Progress in the Use of Mitochondrial Membrane Permeability Transition Pore in Mitochondrial Dysfunction-Related Disease Therapies. Mol. Cell Biochem. 2020. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015; ISBN 978-0-19-180213-3. [Google Scholar]
- Lenaz, G. Mitochondria and Reactive Oxygen Species. Which Role in Physiology and Pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar] [CrossRef]
- Lenaz, G.; Strocchi, P. Reactive Oxygen Species in the Induction of Toxicity. In General, Applied and Systems Toxicology; American Cancer Society: Atlanta, GA, USA, 2011; ISBN 978-0-470-74430-7. [Google Scholar]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Galkin, A.; Brandt, U. Superoxide Radical Formation by Pure Complex I (NADH:Ubiquinone Oxidoreductase) from Yarrowia Lipolytica. J. Biol. Chem. 2005, 280, 30129–30135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kussmaul, L.; Hirst, J. The Mechanism of Superoxide Production by NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Heart Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Esterházy, D.; King, M.S.; Yakovlev, G.; Hirst, J. Production of Reactive Oxygen Species by Complex I (NADH:Ubiquinone Oxidoreductase) from Escherichia Coli and Comparison to the Enzyme from Mitochondria. Biochemistry 2008, 47, 3964–3971. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.J.; Brand, M.D. Inhibitors of the Quinone-Binding Site Allow Rapid Superoxide Production from Mitochondrial NADH:Ubiquinone Oxidoreductase (Complex I). J. Biol. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.J.; Buckingham, J.A.; Boysen, H.M.; Brand, M.D. Diphenyleneiodonium Acutely Inhibits Reactive Oxygen Species Production by Mitochondrial Complex I during Reverse, but Not Forward Electron Transport. Biochim. Biophys. Acta 2008, 1777, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Ventura, B.; Giuliano, G.; Bovina, C.; Formiggini, G.; Parenti Castelli, G.; Lenaz, G. The Site of Production of Superoxide Radical in Mitochondrial Complex I Is Not a Bound Ubisemiquinone but Presumably Iron-Sulfur Cluster N2. FEBS Lett. 2001, 505, 364–368. [Google Scholar] [CrossRef]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Fato, R.; Genova, M.L.; Bergamini, C.; Bianchi, C.; Biondi, A. Mitochondrial Complex I: Structural and Functional Aspects. Biochim. Biophys. Acta 2006, 1757, 1406–1420. [Google Scholar] [CrossRef] [Green Version]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial Production of Reactive Oxygen Species. Biochem. Mosc. 2013, 78, 1490–1511. [Google Scholar] [CrossRef]
- Ralph, S.J.; Moreno-Sánchez, R.; Neuzil, J.; Rodríguez-Enríquez, S. Inhibitors of Succinate: Quinone Reductase/Complex II Regulate Production of Mitochondrial Reactive Oxygen Species and Protect Normal Cells from Ischemic Damage but Induce Specific Cancer Cell Death. Pharm. Res. 2011, 28, 2695–2730. [Google Scholar] [CrossRef]
- Vinogradov, A.D.; Grivennikova, V.G. Generation of Superoxide-Radical by the NADH:Ubiquinone Oxidoreductase of Heart Mitochondria. Biochemistry 2005, 70, 120–127. [Google Scholar] [CrossRef]
- Ohnishi, S.T.; Ohnishi, T.; Muranaka, S.; Fujita, H.; Kimura, H.; Uemura, K.; Yoshida, K.; Utsumi, K. A Possible Site of Superoxide Generation in the Complex I Segment of Rat Heart Mitochondria. J. Bioenergy Biomembr. 2005, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-Mediated Reactive Oxygen Species Generation: Modulation by Cytochrome c and NAD(P)+ Oxidation-Reduction State. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkov, A.A.; Fiskum, G. Regulation of Brain Mitochondrial H2O2 Production by Membrane Potential and NAD(P)H Redox State. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Crofts, A.R. The Cytochrome Bc1 Complex: Function in the Context of Structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Roberts, A.G.; Bowman, M.K.; Kramer, D.M. Architecture of the Qo Site of the Cytochrome Bc1 Complex Probed by Superoxide Production. Biochemistry 2003, 42, 6493–6499. [Google Scholar] [CrossRef]
- Jezek, P.; Hlavatá, L. Mitochondria in Homeostasis of Reactive Oxygen Species in Cell, Tissues, and Organism. Int. J. Biochem. Cell. Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Casteilla, L.; Rigoulet, M.; Pénicaud, L. Mitochondrial ROS Metabolism: Modulation by Uncoupling Proteins. IUBMB Life 2001, 52, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Brandt, U. The Mechanism of Mitochondrial Superoxide Production by the Cytochrome Bc1 Complex. J. Biol. Chem. 2008, 283, 21649–21654. [Google Scholar] [CrossRef] [Green Version]
- Sarewicz, M.; Borek, A.; Cieluch, E.; Swierczek, M.; Osyczka, A. Discrimination between Two Possible Reaction Sequences That Create Potential Risk of Generation of Deleterious Radicals by Cytochrome Bc1. Implications for the Mechanism of Superoxide Production. Biochim. Biophys. Acta 2010, 1797, 1820–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarewicz, M.; Bujnowicz, Ł.; Bhaduri, S.; Singh, S.K.; Cramer, W.A.; Osyczka, A. Metastable Radical State, Nonreactive with Oxygen, Is Inherent to Catalysis by Respiratory and Photosynthetic Cytochromes Bc1/B6f. Proc. Natl. Acad. Sci. USA 2017, 114, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujnowicz, Ł.; Borek, A.; Kuleta, P.; Sarewicz, M.; Osyczka, A. Suppression of Superoxide Production by a Spin-Spin Coupling between Semiquinone and the Rieske Cluster in Cytochrome Bc1. FEBS Lett. 2019, 593, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaz, G. Role of Mitochondria in the Generation of Reactive Oxygen Species. In Handbook on Reactive Oxygen Species (ROS); Suzuki, M., Yamamoto, S., Eds.; Nova Biomedical: Waltham, MA, USA, 2014. [Google Scholar]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Hemendinger, R.; Greenamyre, J.T.; Rosenfeld, J. Species- and Tissue-Specific Relationships between Mitochondrial Permeability Transition and Generation of ROS in Brain and Liver Mitochondria of Rats and Mice. Am. J. Physiol. Cell Physiol. 2007, 292, C708–C718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, E.B.; Navarete, F.D.T.; Kowaltowski, A.J. Tissue-, Substrate-, and Site-Specific Characteristics of Mitochondrial Reactive Oxygen Species Generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Role of Uncoupled and Non-Coupled Oxidations in Maintenance of Safely Low Levels of Oxygen and Its One-Electron Reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef]
- Brand, M.D. Uncoupling to Survive? The Role of Mitochondrial Inefficiency in Ageing. Exp. Gerontol. 2000, 35, 811–820. [Google Scholar] [CrossRef]
- Wojtczak, L.; Lebiedzińska, M.; Suski, J.M.; Więckowski, M.R.; Schönfeld, P. Inhibition by Purine Nucleotides of the Release of Reactive Oxygen Species from Muscle Mitochondria: Indication for a Function of Uncoupling Proteins as Superoxide Anion Transporters. Biochem. Biophys. Res. Commun. 2011, 407, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Raha, S.; Myint, A.T.; Johnstone, L.; Robinson, B.H. Control of Oxygen Free Radical Formation from Mitochondrial Complex I: Roles for Protein Kinase A and Pyruvate Dehydrogenase Kinase. Free Radic. Biol. Med. 2002, 32, 421–430. [Google Scholar] [CrossRef]
- Maj, M.C.; Raha, S.; Myint, T.; Robinson, B.H. Regulation of NADH/CoQ Oxidoreductase: Do Phosphorylation Events Affect Activity? Protein J. 2004, 23, 25–32. [Google Scholar] [CrossRef]
- Scacco, S.; Petruzzella, V.; Bertini, E.; Luso, A.; Papa, F.; Bellomo, F.; Signorile, A.; Torraco, A.; Papa, S. Mutations in Structural Genes of Complex I Associated with Neurological Diseases. Ital. J. Biochem. 2006, 55, 254–262. [Google Scholar] [PubMed]
- Kadenbach, B.; Ramzan, R.; Vogt, S. High Efficiency versus Maximal Performance—The Cause of Oxidative Stress in Eukaryotes: A Hypothesis. Mitochondrion 2013, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.C.; Liu, J.; Manfredi, G.; Mühlschlegel, F.A.; Buck, J.; Levin, L.R.; Barrientos, A. A Mitochondrial CO2-Adenylyl Cyclase-CAMP Signalosome Controls Yeast Normoxic Cytochrome c Oxidase Activity. FASEB J. 2014, 28, 4369–4380. [Google Scholar] [CrossRef] [Green Version]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-Specific Regulation of Cytochrome c by Post-Translational Modifications: Respiration, the Mitochondrial Membrane Potential, ROS, and Apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Vaishnav, A.; Liu, J.; Varughese, A.; Wan, J.; Turner, A.A.; Ji, Q.; Zurek, M.P.; Kapralov, A.A.; Kagan, V.E.; et al. Serine-47 Phosphorylation of Cytochrome c in the Mammalian Brain Regulates Cytochrome c Oxidase and Caspase-3 Activity. FASEB J. 2019, 33, 13503–13514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Loscalzo, J. Redox Regulation of Mitochondrial Function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.W.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-Based Regulation of Signal Transduction: Principles, Pitfalls, and Promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of Reactive Oxygen Species Generation in Cell Signaling. Mol. Cell. 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS Generation and Its Regulation: Mechanisms Involved in H(2)O(2) Signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Gough, D.R.; Cotter, T.G. Hydrogen Peroxide: A Jekyll and Hyde Signalling Molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, J.; Yi, J. Redox Sensing by Proteins: Oxidative Modifications on Cysteines and the Consequent Events. Antioxid. Redox Signal. 2012, 16, 649–657. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial Thiols in Antioxidant Protection and Redox Signaling: Distinct Roles for Glutathionylation and Other Thiol Modifications. Antioxid. Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS-Dependent Signal Transduction. Curr. Opin. Cell. Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell. Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Wu, W.-S. The Signaling Mechanism of ROS in Tumor Progression. Cancer Metast. Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Bak, D.W.; Weerapana, E. Cysteine-Mediated Redox Signalling in the Mitochondria. Mol. BioSyst. 2015, 11, 678–697. [Google Scholar] [CrossRef]
- Haddad, J.J. Antioxidant and Prooxidant Mechanisms in the Regulation of Redox(y)-Sensitive Transcription Factors. Cell Signal 2002, 14, 879–897. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.-D.V.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.P.; Zuckerbraun, B.S. Mitochondrial Signaling: Forwards, Backwards, and in Between. Oxid. Med. Cell Longev. 2013, 2013, 351613. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. ROS and Redox Signaling in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Finkel, T. Signal Transduction by Mitochondrial Oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, M.; Lepow, T.S.; Billingham, L.K.; Murphy, M.P.; Siegel, R.M. New Tricks from an Old Dog: Mitochondrial Redox Signaling in Cellular Inflammation. Semin. Immunol. 2012, 24, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sho, T.; Xu, J. Role and Mechanism of ROS Scavengers in Alleviating NLRP3-Mediated Inflammation. Biotechnol. Appl. Biochem. 2019, 66, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of Autophagy by ROS: Physiology and Pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Hoffman, D.L.; Brookes, P.S. Oxygen Sensitivity of Mitochondrial Reactive Oxygen Species Generation Depends on Metabolic Conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-Alpha-Induced ROS Production Triggering Apoptosis Is Directly Linked to Romo1 and Bcl-X(L). Cell Death Differ 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ns, C.; Ds, M.; Ce, F.; Tm, W.; Ja, M.; Am, R.; Pt, S. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1alpha during Hypoxia: A Mechanism of O2 Sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Kim, J.J.; Kim, T.W.; Kim, B.S.; Lee, M.-S.; Yoo, Y.D. Serum Deprivation-Induced Reactive Oxygen Species Production Is Mediated by Romo1. Apoptosis 2010, 15, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-Induced ROS Release: An Update and Review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikalov, S. Crosstalk between Mitochondria and NADPH Oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.-J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef] [PubMed]
- Koju, N.; Taleb, A.; Zhou, J.; Lv, G.; Yang, J.; Cao, X.; Lei, H.; Ding, Q. Pharmacological Strategies to Lower Crosstalk between Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Mitochondria. Biomed. Pharmacother. 2019, 111, 1478–1498. [Google Scholar] [CrossRef]
- Fourquet, S.; Huang, M.-E.; D’Autreaux, B.; Toledano, M.B. The Dual Functions of Thiol-Based Peroxidases in H2O2 Scavenging and Signaling. Antioxid. Redox Signal. 2008, 10, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kil, I.S. Multiple Functions and Regulation of Mammalian Peroxiredoxins. Annu. Rev. Biochem. 2017, 86, 749–775. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A Historical Overview and Speculative Preview of Novel Mechanisms and Emerging Concepts in Cell Signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules—From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial Peroxiredoxin Involvement in Antioxidant Defence and Redox Signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Colombrita, C.; Spadaro, F.; Butterfield, D.A.; Giuffrida Stella, A.M. Increased Expression of Heat Shock Proteins in Rat Brain during Aging: Relationship with Mitochondrial Function and Glutathione Redox State. Mech. Ageing Dev. 2004, 125, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Leso, V.; Trovato-Salinaro, A.; Ventimiglia, B.; Cavallaro, M.; Scuto, M.; Rizza, S.; Zanoli, L.; Neri, S.; et al. Oxidative Stress, Glutathione Status, Sirtuin and Cellular Stress Response in Type 2 Diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, G.; Morgan, B.; Riemer, J. Mitochondrial Glutathione: Regulation and Functions. Antioxid. Redox Signal. 2017, 27, 1162–1177. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redox Potential of GSH/GSSG Couple: Assay and Biological Significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial Thiols in the Regulation of Cell Death Pathways. Antioxid. Redox Signal. 2012, 17, 1714–1727. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Colombo, G.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R.; Milzani, A. S-Glutathiolation in Life and Death Decisions of the Cell. Free Radic. Res. 2011, 45, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.M.G.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Gill, R.; Mailloux, R.J. Protein S-Glutathionylation: The Linchpin for the Transmission of Regulatory Information on Redox Buffering Capacity in Mitochondria. Chem. Biol. Interact. 2019, 299, 151–162. [Google Scholar] [CrossRef]
- Smith, K.A.; Waypa, G.B.; Schumacker, P.T. Redox Signaling during Hypoxia in Mammalian Cells. Redox Biol. 2017, 13, 228–234. [Google Scholar] [CrossRef]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Inducible Factor-Dependent Extension of the Replicative Life Span during Hypoxia. Mol. Cell. Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef] [Green Version]
- Grishko, V.; Solomon, M.; Breit, J.F.; Killilea, D.W.; Ledoux, S.P.; Wilson, G.L.; Gillespie, M.N. Hypoxia Promotes Oxidative Base Modifications in the Pulmonary Artery Endothelial Cell VEGF Gene. FASEB J. 2001, 15, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kietzmann, T.; Görlach, A. Reactive Oxygen Species in the Control of Hypoxia-Inducible Factor-Mediated Gene Expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Kaluz, S.; Kaluzová, M.; Stanbridge, E.J. Rational Design of Minimal Hypoxia-Inducible Enhancers. Biochem. Biophys. Res. Commun. 2008, 370, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Jones, D.P. Superoxide in Apoptosis. Mitochondrial Generation Triggered by Cytochrome c Loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The Role of Oxidized Cytochrome c in Regulating Mitochondrial Reactive Oxygen Species Production and Its Perturbation in Ischaemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Ischemic Defects in the Electron Transport Chain Increase the Production of Reactive Oxygen Species from Isolated Rat Heart Mitochondria. Am. J. Physiol. Cell Physiol. 2008, 294, C460–C466. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.; Budinger, G.R.; Kemp, R.A.; Schumacker, P.T. Inhibition of Cytochrome-c Oxidase Activity during Prolonged Hypoxia. Am. J. Physiol. 1995, 268, L918–L925. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; de Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon Monoxide Signals via Inhibition of Cytochrome c Oxidase and Generation of Mitochondrial Reactive Oxygen Species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial Dysfunction Resulting from Loss of Cytochrome c Impairs Cellular Oxygen Sensing and Hypoxic HIF-Alpha Activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial Complex III Is Required for Hypoxia-Induced ROS Production and Cellular Oxygen Sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen Sensing by Mitochondria at Complex III: The Paradox of Increased Reactive Oxygen Species during Hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Korde, A.S.; Yadav, V.R.; Zheng, Y.-M.; Wang, Y.-X. Primary Role of Mitochondrial Rieske Iron-Sulfur Protein in Hypoxic ROS Production in Pulmonary Artery Myocytes. Free Radic. Biol. Med. 2011, 50, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustín, P.; Ramos, E.; Navarro, E.; Parada, E.; Sánchez-López, N.; Peláez-Aguado, L.; Cabrera-García, J.D.; Tello, D.; Buendia, I.; Marina, A.; et al. Mitochondrial Complex I Deactivation Is Related to Superoxide Production in Acute Hypoxia. Redox Biol. 2017, 12, 1040–1051. [Google Scholar] [CrossRef]
- Mittal, M.; Gu, X.Q.; Pak, O.; Pamenter, M.E.; Haag, D.; Fuchs, D.B.; Schermuly, R.T.; Ghofrani, H.A.; Brandes, R.P.; Seeger, W.; et al. Hypoxia Induces Kv Channel Current Inhibition by Increased NADPH Oxidase-Derived Reactive Oxygen Species. Free Radic. Biol. Med. 2012, 52, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef]
- Tomko, R.J.; Hochstrasser, M. Molecular Architecture and Assembly of the Eukaryotic Proteasome. Annu. Rev. Biochem. 2013, 82, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luce, K.; Weil, A.C.; Osiewacz, H.D. Mitochondrial Protein Quality Control Systems in Aging and Disease. Adv. Exp. Med. Biol. 2010, 694, 108–125. [Google Scholar] [CrossRef]
- Taylor, E.B.; Rutter, J. Mitochondrial Quality Control by the Ubiquitin-Proteasome System. Biochem. Soc. Trans. 2011, 39, 1509–1513. [Google Scholar] [CrossRef] [Green Version]
- Bragoszewski, P.; Turek, M.; Chacinska, A. Control of Mitochondrial Biogenesis and Function by the Ubiquitin-Proteasome System. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Zhang, Y.; Li, P.-F. Mitochondrial Ubiquitin Ligase in Cardiovascular Disorders. Adv. Exp. Med. Biol. 2017, 982, 327–333. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, S.; Lee, J.; Singh, K.; Lee, I.; Suzuki, C.K. Multitasking in the Mitochondrion by the ATP-Dependent Lon Protease. Biochim. Biophys. Acta 2012, 1823, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.-L.; Szweda, L.I.; Friguet, B. Mitochondrial Protein Oxidation and Degradation in Response to Oxidative Stress and Aging. Exp. Gerontol. 2006, 41, 653–657. [Google Scholar] [CrossRef]
- Ugarte, N.; Petropoulos, I.; Friguet, B. Oxidized Mitochondrial Protein Degradation and Repair in Aging and Oxidative Stress. Antioxid. Redox Signal. 2010, 13, 539–549. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Bergamini, E. Autophagy: A Cell Rep.air Mechanism That Retards Ageing and Age-Associated Diseases and Can Be Intensified Pharmacologically. Mol. Asp. Med. 2006, 27, 403–410. [Google Scholar] [CrossRef]
- Chen, Y.; Gibson, S.B. Is Mitochondrial Generation of Reactive Oxygen Species a Trigger for Autophagy? Autophagy 2008, 4, 246–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, Mitochondria and Oxidative Stress: Cross-Talk and Redox Signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, P.; Xie, X.-H.; Chen, C.-H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.-T. Molecular Regulation Mechanisms and Interactions Between Reactive Oxygen Species and Mitophagy. DNA Cell Biol. 2019, 38, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy Is Triggered by Mild Oxidative Stress in a Mitochondrial Fission Dependent Manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Schägger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of Respirasomes for the Assembly/Stability of Human Respiratory Chain Complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [Green Version]
- Acín-Pérez, R.; Bayona-Bafaluy, M.P.; Fernández-Silva, P.; Moreno-Loshuertos, R.; Pérez-Martos, A.; Bruno, C.; Moraes, C.T.; Enríquez, J.A. Respiratory Complex III Is Required to Maintain Complex I in Mammalian Mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Gajewski, C.D.; Lenaz, G.; Manfredi, G. Respiratory Chain Supercomplexes Set the Threshold for Respiration Defects in Human MtDNA Mutant Cybrids. Hum. Mol. Genet. 2006, 15, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A Defect in the Mitochondrial Complex III, but Not Complex IV, Triggers Early ROS-Dependent Damage in Defined Brain Regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory Supercomplexes Act as a Platform for Complex III-Mediated Maturation of Human Mitochondrial Complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Enríquez, J.A.; Moraes, C.T. Cells Lacking Rieske Iron-Sulfur Protein Have a Reactive Oxygen Species-Associated Decrease in Respiratory Complexes I and IV. Mol. Cell Biol. 2012, 32, 415–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Affect Mitochondrial Electron Transport Complex I Activity through Oxidative Cardiolipin Damage. Gene 2002, 286, 135–141. [Google Scholar] [CrossRef]
- Jang, S.; Lewis, T.S.; Powers, C.; Khuchua, Z.; Baines, C.P.; Wipf, P.; Javadov, S. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid. Redox Signal. 2017, 27, 57–69. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Ruggiero, F.M. Reactive Oxygen Species Generated by the Mitochondrial Respiratory Chain Affect the Complex III Activity via Cardiolipin Peroxidation in Beef-Heart Submitochondrial Particles. Mitochondrion 2001, 1, 151–159. [Google Scholar] [CrossRef]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial Dysfunction in Rat Brain with Aging Involvement of Complex I, Reactive Oxygen Species and Cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef]
- Petrosillo, G.; Matera, M.; Moro, N.; Ruggiero, F.M.; Paradies, G. Mitochondrial Complex I Dysfunction in Rat Heart with Aging: Critical Role of Reactive Oxygen Species and Cardiolipin. Free Radic. Biol. Med. 2009, 46, 88–94. [Google Scholar] [CrossRef]
- Dencher, N.A.; Frenzel, M.; Reifschneider, N.H.; Sugawa, M.; Krause, F. Proteome Alterations in Rat Mitochondria Caused by Aging. Ann. N. Y. Acad. Sci. 2007, 1100, 291–298. [Google Scholar] [CrossRef]
- Schäfer, E.; Dencher, N.A.; Vonck, J.; Parcej, D.N. Three-Dimensional Structure of the Respiratory Chain Supercomplex I1III2IV1 from Bovine Heart Mitochondria. Biochemistry 2007, 46, 12579–12585. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Dudkina, N.V.; Jänsch, L.; Braun, H.-P.; Boekema, E.J. A Structural Investigation of Complex I and I+III2 Supercomplex from Zea Mays at 11-13 A Resolution: Assignment of the Carbonic Anhydrase Domain and Evidence for Structural Heterogeneity within Complex I. Biochim. Biophys. Acta 2008, 1777, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Radermacher, M.; Ruiz, T.; Clason, T.; Benjamin, S.; Brandt, U.; Zickermann, V. The Three-Dimensional Structure of Complex I from Yarrowia Lipolytica: A Highly Dynamic Enzyme. J. Struct. Biol. 2006, 154, 269–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex i. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Gutiérrez, C.; Jiménez-Blasco, D.; Quintana-Cabrera, R. Intertwined ROS and Metabolic Signaling at the Neuron-Astrocyte Interface. Neurochem. Res. 2020. [Google Scholar] [CrossRef]
- Reyes-Galindo, M.; Suarez, R.; Esparza-Perusquía, M.; de Lira-Sánchez, J.; Pardo, J.P.; Martínez, F.; Flores-Herrera, O. Mitochondrial Respirasome Works as a Single Unit and the Cross-Talk between Complexes I, III2 and IV Stimulates NADH Dehydrogenase Activity. Biochim. Biophys. Acta Bioenergy 2019, 1860, 618–627. [Google Scholar] [CrossRef]
- Lenaz, G.; Baracca, A.; Barbero, G.; Bergamini, C.; Dalmonte, M.E.; Del Sole, M.; Faccioli, M.; Falasca, A.; Fato, R.; Genova, M.L.; et al. Mitochondrial Respiratory Chain Super-Complex I-III in Physiology and Pathology. Biochim. Biophys. Acta 2010, 1797, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Huertas, J.R.; Al Fazazi, S.; Hidalgo-Gutierrez, A.; López, L.C.; Casuso, R.A. Antioxidant Effect of Exercise: Exploring the Role of the Mitochondrial Complex I Superassembly. Redox Biol. 2017, 13, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Taylor, E.B.; Dephoure, N.; Heo, J.-M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a Protein Mediating Respiratory Supercomplex Stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strogolova, V.; Furness, A.; Robb-McGrath, M.; Garlich, J.; Stuart, R.A. Rcf1 and Rcf2, Members of the Hypoxia-Induced Gene 1 Protein Family, Are Critical Components of the Mitochondrial Cytochrome Bc1-Cytochrome c Oxidase Supercomplex. Mol. Cell Biol. 2012, 32, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Winge, D.R. Sealing the Mitochondrial Respirasome. Mol. Cell Biol. 2012, 32, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Ikeda, K.; Inoue, S. Functional Mechanisms of Mitochondrial Respiratory Chain Supercomplex Assembly Factors and Their Involvement in Muscle Quality. Int. J. Mol. Sci. 2020, 21, 3182. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Gluing the Respiratory Chain Together. Cardiolipin Is Required for Supercomplex Formation in the Inner Mitochondrial Membrane. J. Biol. Chem. 2002, 277, 43553–43556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, C.S.; Jadiya, P.; Zhang, X.; McLendon, J.M.; Abouassaly, G.M.; Witmer, N.H.; Anderson, E.J.; Elrod, J.W.; Boudreau, R.L. Mitoregulin: A LncRNA-Encoded Microprotein That Supports Mitochondrial Supercomplexes and Respiratory Efficiency. Cell Rep. 2018, 23, 3710–3720.e8. [Google Scholar] [CrossRef]
- Hou, T.; Zhang, R.; Jian, C.; Ding, W.; Wang, Y.; Ling, S.; Ma, Q.; Hu, X.; Cheng, H.; Wang, X. NDUFAB1 Confers Cardio-Protection by Enhancing Mitochondrial Bioenergetics through Coordination of Respiratory Complex and Supercomplex Assembly. Cell Res. 2019, 29, 754–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Javadov, S. Elucidating the Contribution of ETC Complexes I and II to the Respirasome Formation in Cardiac Mitochondria. Sci. Rep. 2018, 8, 17732. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Camacho, I.; Correa, F.; El Hafidi, M.; Silva-Palacios, A.; Ostolga-Chavarría, M.; Esparza-Perusquía, M.; Olvera-Sánchez, S.; Flores-Herrera, O.; Zazueta, C. Cardioprotective Strategies Preserve the Stability of Respiratory Chain Supercomplexes and Reduce Oxidative Stress in Reperfused Ischemic Hearts. Free Radic. Biol. Med. 2018, 129, 407–417. [Google Scholar] [CrossRef]
- Ramírez-Camacho, I.; García-Niño, W.R.; Flores-García, M.; Pedraza-Chaverri, J.; Zazueta, C. Alteration of Mitochondrial Supercomplexes Assembly in Metabolic Diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165935. [Google Scholar] [CrossRef] [PubMed]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.-P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S.J. 4-The Chemiosmotic Proton Circuit in Isolated Organelles: Theory and Practice. In Bioenergetics, 4th ed.; Academic Press: Boston, MA, USA, 2013; pp. 53–87. ISBN 978-0-12-388425-1. [Google Scholar]
- Nicholls, D.G. The Physiological Regulation of Uncoupling Proteins. Biochim. Biophys. Acta 2006, 1757, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ Transport Is an Integral Function of the Mitochondrial ADP/ATP Carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Feniouk, B.A.; Mulkidjanian, A.Y.; Junge, W. Proton Slip in the ATP Synthase of Rhodobacter Capsulatus: Induction, Proton Conduction, and Nucleotide Dependence. Biochim. Biophys. Acta 2005, 1706, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uziel, G.; Moroni, I.; Lamantea, E.; Fratta, G.M.; Ciceri, E.; Carrara, F.; Zeviani, M. Mitochondrial Disease Associated with the T8993G Mutation of the Mitochondrial ATPase 6 Gene: A Clinical, Biochemical, and Molecular Study in Six Families. J. Neurol. Neurosurg. Psychiatry 1997, 63, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Trounce, I.; Neill, S.; Wallace, D.C. Cytoplasmic Transfer of the MtDNA Nt 8993 T-->G (ATP6) Point Mutation Associated with Leigh Syndrome into MtDNA-Less Cells Demonstrates Cosegregation with a Decrease in State III Respiration and ADP/O Ratio. Proc. Natl. Acad. Sci. USA 1994, 91, 8334–8338. [Google Scholar] [CrossRef] [Green Version]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient Coupling between Proton Transport and ATP Synthesis May Be the Pathogenic Mechanism for NARP and Leigh Syndrome Resulting from the T8993G Mutation in MtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Dautant, A.; Godard, F.; Tribouillard-Tanvier, D.; di Rago, J.-P. Functional Investigation of an Universally Conserved Leucine Residue in Subunit a of ATP Synthase Targeted by the Pathogenic m.9176 T>G Mutation. Biochim. Biophys. Acta Bioenergy 2019, 1860, 52–59. [Google Scholar] [CrossRef]
- Kucharczyk, R.; Rak, M.; di Rago, J.-P. Biochemical Consequences in Yeast of the Human Mitochondrial DNA 8993T>C Mutation in the ATPase6 Gene Found in NARP/MILS Patients. Biochim. Biophys. Acta 2009, 1793, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical Phenotypes Associated with the Mitochondrial ATP6 Gene Mutations at Nt8993. Biochim. Biophys. Acta 2007, 1767, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.; Situ, A.J.; Ulmer, T.S. Structural and Thermodynamic Basis of Proline-Induced Transmembrane Complex Stabilization. Sci. Rep. 2016, 6, 29809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrage, L.C.; Tang, S.; Wang, J.; Donti, T.R.; Walkiewicz, M.; Luchak, J.M.; Chen, L.-C.; Schmitt, E.S.; Niu, Z.; Erana, R.; et al. Mitochondrial Myopathy, Lactic Acidosis, and Sideroblastic Anemia (MLASA) plus Associated with a Novel de Novo Mutation (m.8969G>A) in the Mitochondrial Encoded ATP6 Gene. Mol. Genet. Metab. 2014, 113, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoczeń, N.; Dautant, A.; Binko, K.; Godard, F.; Bouhier, M.; Su, X.; Lasserre, J.-P.; Giraud, M.-F.; Tribouillard-Tanvier, D.; Chen, H.; et al. Molecular Basis of Diseases Caused by the MtDNA Mutation m.8969G>A in the Subunit a of ATP Synthase. Biochim. Biophys. Acta 2018, 1859, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Speijer, D. Being Right on Q: Shaping Eukaryotic Evolution. Biochem. J. 2016, 473, 4103–4127. [Google Scholar] [CrossRef] [Green Version]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E. Mitochondrial Role in Cell Aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA Mutations as an Important Contributor to Ageing and Degenerative Diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Barja, G. Updating the Mitochondrial Free Radical Theory of Aging: An Integrated View, Key Aspects, and Confounding Concepts. Antioxid. Redox Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G. Role of Mitochondria in Oxidative Stress and Ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Webb, M.; Sideris, D.P. Intimate Relations-Mitochondria and Ageing. Int. J. Mol. Sci. 2020, 21, 7580. [Google Scholar] [CrossRef]
- Barja, G. Towards a Unified Mechanistic Theory of Aging. Exp. Gerontol. 2019, 124, 110627. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Genova, M.L.; Lenaz, G. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid. Redox Signal. 2015, 23, 208–238. [Google Scholar] [CrossRef]
- Barrientos, A.; Moraes, C.T. Titrating the Effects of Mitochondrial Complex I Impairment in the Cell Physiology. J. Biol. Chem. 1999, 274, 16188–16197. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; D’Aurelio, M.; Merlo Pich, M.; Genova, M.L.; Ventura, B.; Bovina, C.; Formiggini, G.; Parenti Castelli, G. Mitochondrial Bioenergetics in Aging. Biochim. Biophys. Acta 2000, 1459, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Ventura, B.; Genova, M.L.; Bovina, C.; Formiggini, G.; Lenaz, G. Control of Oxidative Phosphorylation by Complex I in Rat Liver Mitochondria: Implications for Aging. Biochim. Biophys. Acta 2002, 1553, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Gómez, L.A.; Monette, J.S.; Chavez, J.D.; Maier, C.S.; Hagen, T.M. Supercomplexes of the Mitochondrial Electron Transport Chain Decline in the Aging Rat Heart. Arch. Biochem. Biophys. 2009, 490, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, L.A.; Hagen, T.M. Age-Related Decline in Mitochondrial Bioenergetics: Does Supercomplex Destabilization Determine Lower Oxidative Capacity and Higher Superoxide Production? Semin. Cell Dev. Biol. 2012, 23, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Frenzel, M.; Rommelspacher, H.; Sugawa, M.D.; Dencher, N.A. Ageing Alters the Supramolecular Architecture of OxPhos Complexes in Rat Brain Cortex. Exp. Gerontol. 2010, 45, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Edgar, D.; Shabalina, I.; Camara, Y.; Wredenberg, A.; Calvaruso, M.A.; Nijtmans, L.; Nedergaard, J.; Cannon, B.; Larsson, N.-G.; Trifunovic, A. Random Point Mutations with Major Effects on Protein-Coding Genes Are the Driving Force behind Premature Aging in MtDNA Mutator Mice. Cell Metab. 2009, 10, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]


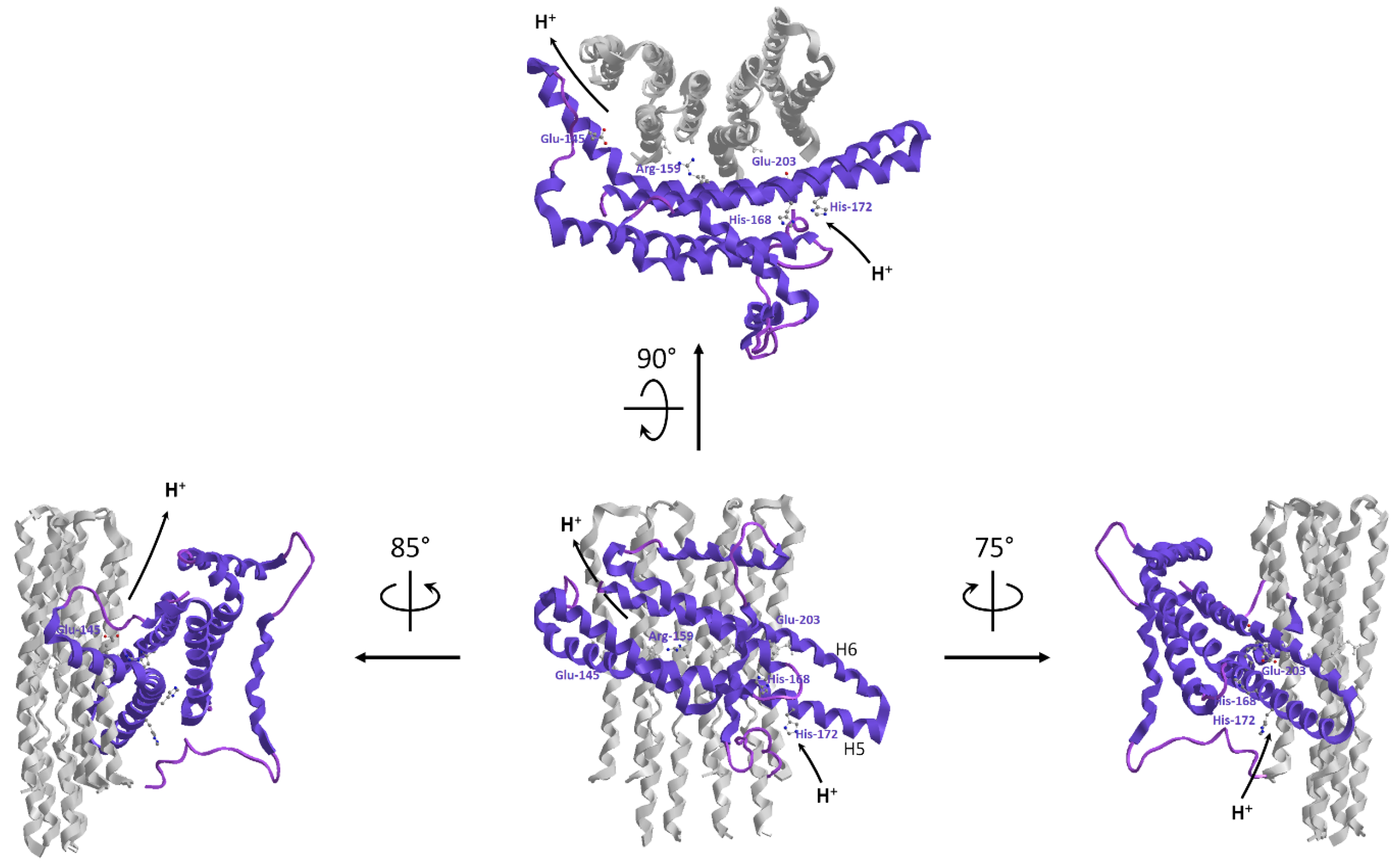
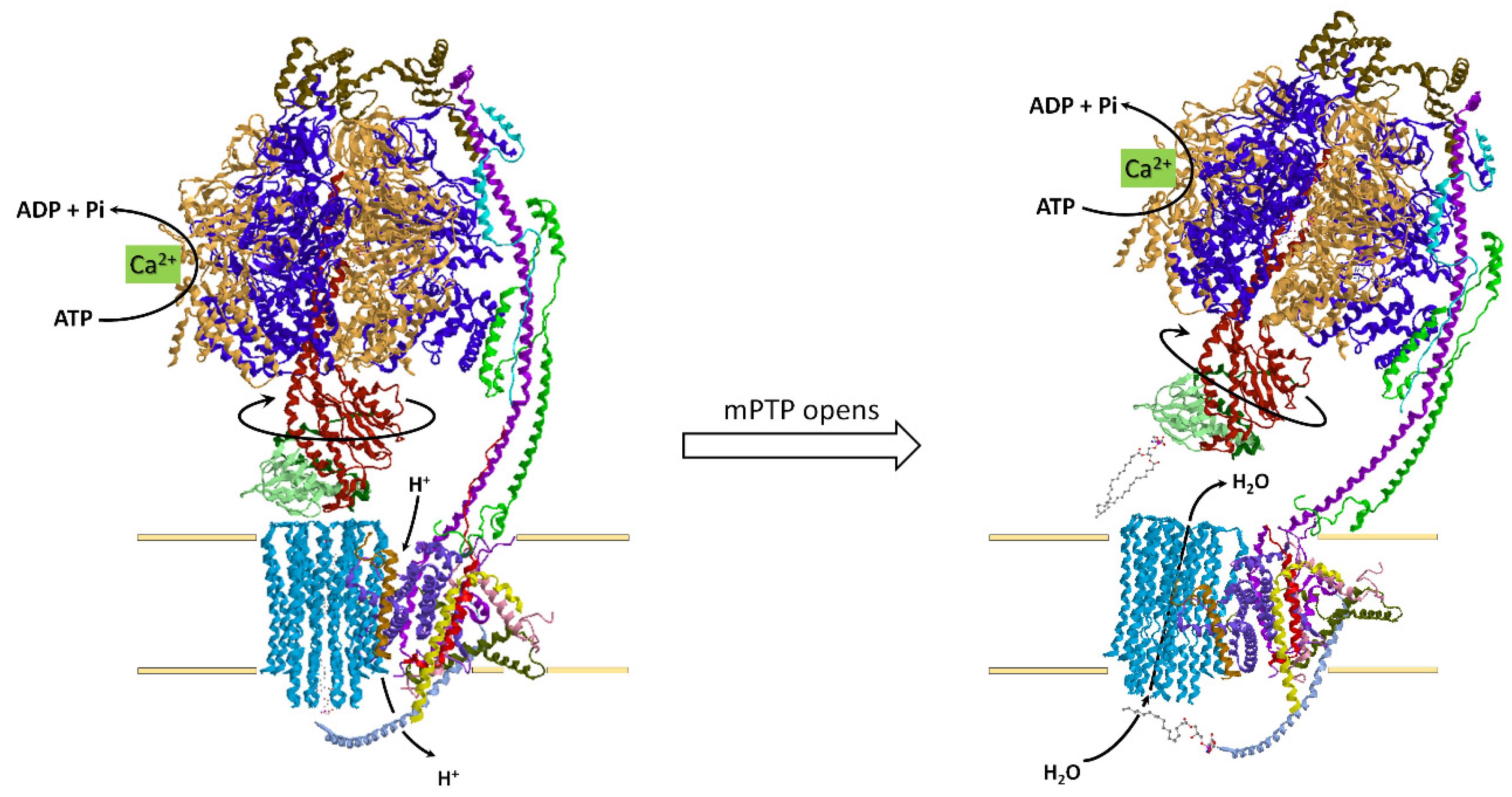
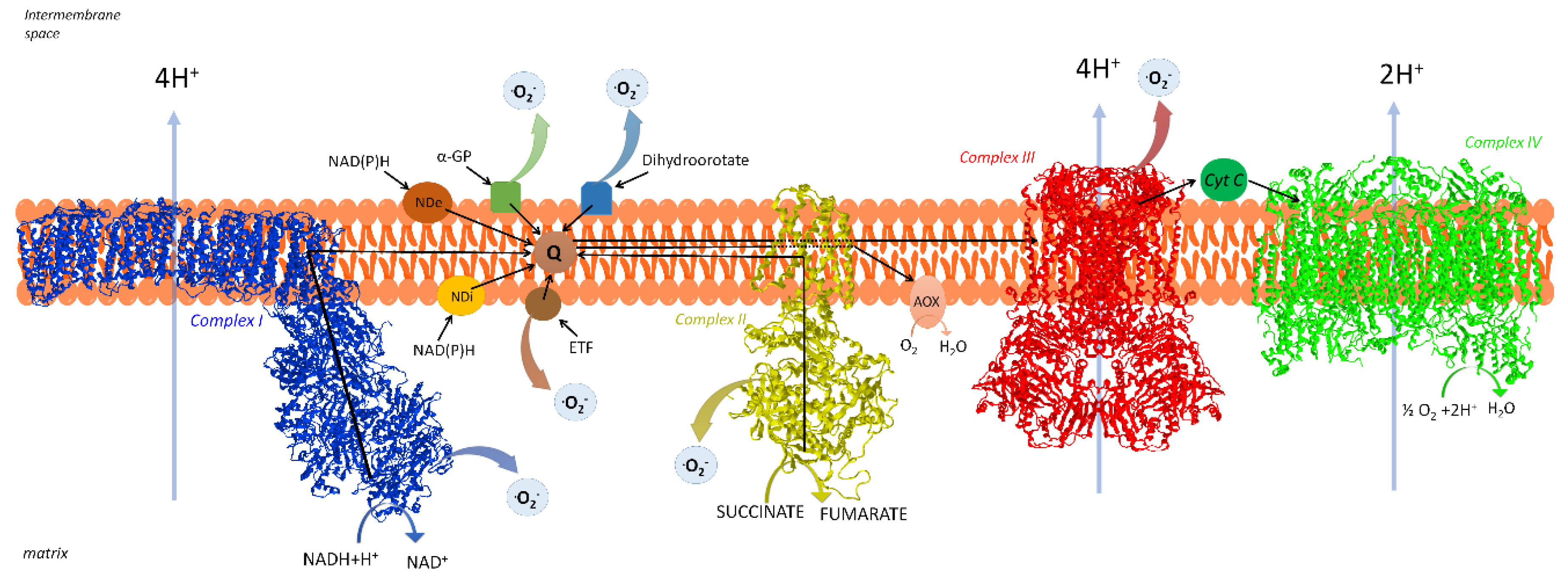
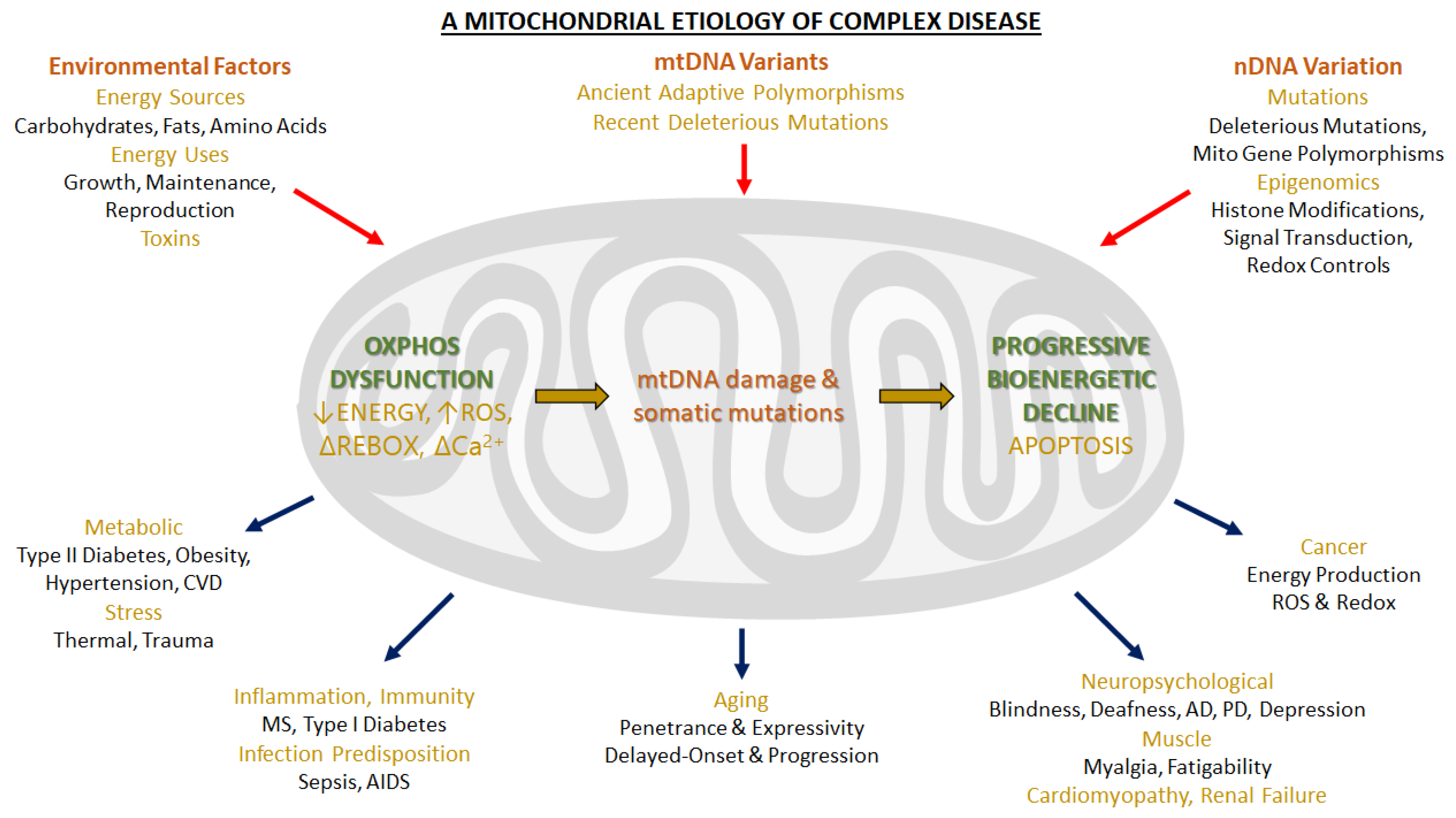
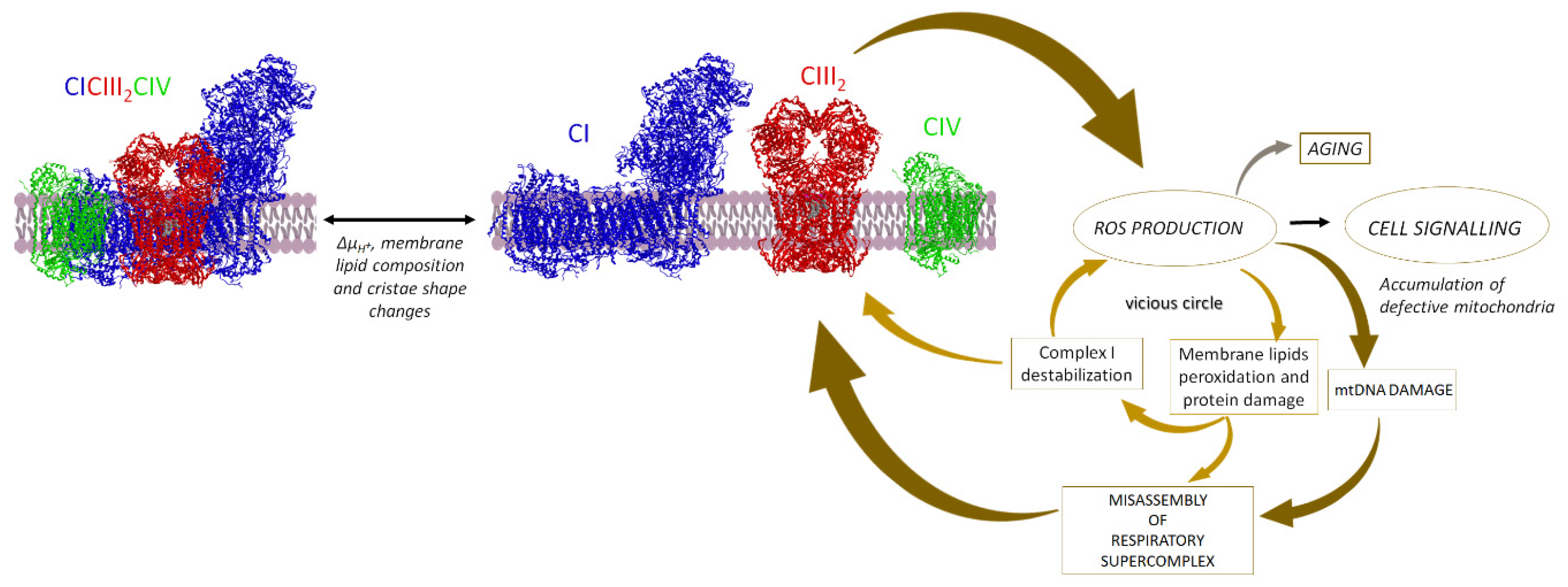
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. https://doi.org/10.3390/life11030242
Nesci S, Trombetti F, Pagliarani A, Ventrella V, Algieri C, Tioli G, Lenaz G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life. 2021; 11(3):242. https://doi.org/10.3390/life11030242
Chicago/Turabian StyleNesci, Salvatore, Fabiana Trombetti, Alessandra Pagliarani, Vittoria Ventrella, Cristina Algieri, Gaia Tioli, and Giorgio Lenaz. 2021. "Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology" Life 11, no. 3: 242. https://doi.org/10.3390/life11030242
APA StyleNesci, S., Trombetti, F., Pagliarani, A., Ventrella, V., Algieri, C., Tioli, G., & Lenaz, G. (2021). Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life, 11(3), 242. https://doi.org/10.3390/life11030242