
Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression


 , , ,
, , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
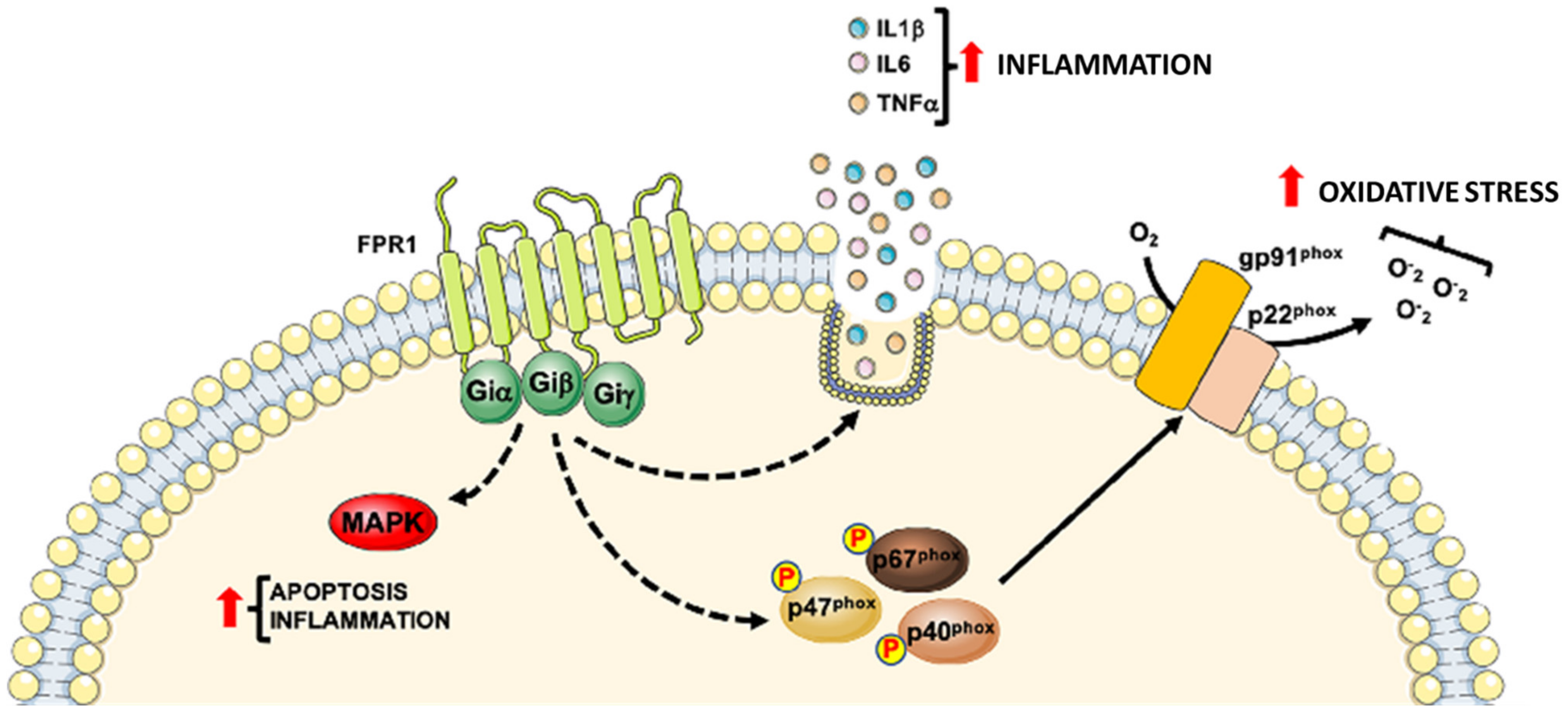
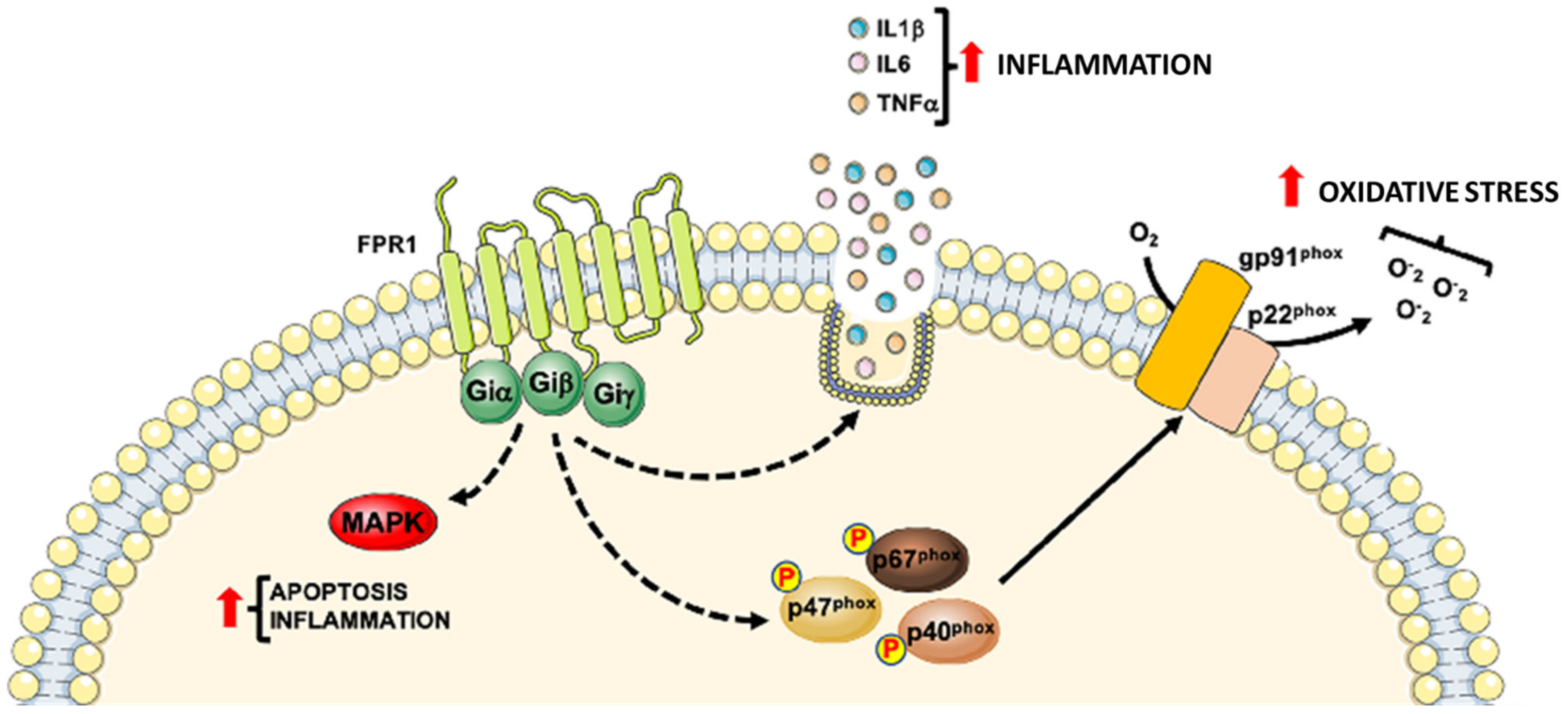
2. Formyl-Peptide Receptor 1
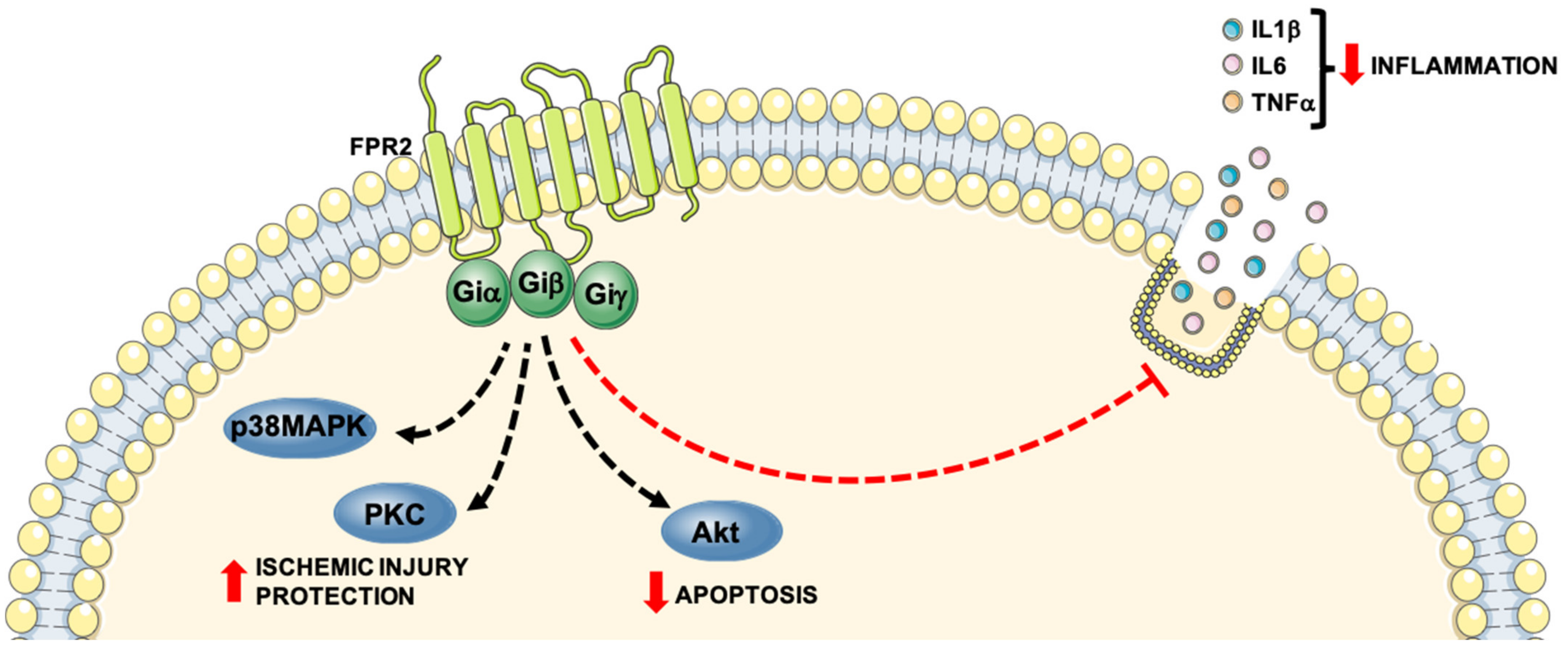
3. Formyl-Peptide Receptor 2

{kind=link}
{kind=link}
| Compound | Interaction | Therapeutic Effects |
|---|---|---|
| Cmpd17b | Small biased FPR1/FPR2 agonists | Reduction of necrosis in cardiomyocytes subjected to hypoxia–reoxygenation exerting a cardioprotective effect [69] |
| Reduction of inflammatory responses associated with reperfusion after an acute MI [69] | ||
| ZK-994 and ZK-142 | FPR2 | Inhibition of neutrophil accumulation in murine hind-limb IRI-induced second-organ lung injury [132] |
| ACT-389949 | FPR2 | Protection against heart failure [143] |
| CGEN-855A | FPR2 | Cardioprotective effects in rat and murine myocardial IRI, similar to those reported for the ANXA1 mimetic peptide Ac2-26 [130] |
| BMS-986235 | FPR2 | Protective properties in experimental heart failure [144] |
| compound 43 | Dual FPR1/FPR2 agonist | High degree of protection in a model of heart failure [146] |
| PLGA microspheres encapsulating WKYMVm | FPR2 | Induction of neovascularization in vivo hind limb ischemia model [109] |
4. Formyl-Peptide Receptor 3
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schiattarella, G.G.; Magliulo, F.; Cattaneo, F.; Gargiulo, G.; Sannino, A.; Franzone, A.; Oliveti, M.; Perrino, C.; Trimarco, B.; Esposito, G. Novel Molecular Approaches in Heart Failure: Seven Trans-Membrane Receptors Signaling in the Heart and Circulating Blood Leukocytes. Front. Cardiovasc. Med. 2015, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagerstrom, M.C.; Schioth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Perretti, M.; Godson, C. Formyl peptide receptor type 2 agonists to kick-start resolution pharmacology. Br. J. Pharmacol. 2020, 177, 4595–4600. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.; Perretti, M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacology 2010, 127, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Chen, K.; Gong, W.; Yoshimura, T.; Le, Y.; Wang, Y.; Wang, J.M. The Contribution of Chemoattractant GPCRs, Formylpeptide Receptors, to Inflammation and Cancer. Front. Endocrinol. 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirault, J.; Back, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharm. 2018, 9, 1273. [Google Scholar] [CrossRef] [PubMed]
- Trojan, E.; Bryniarska, N.; Leskiewicz, M.; Regulska, M.; Chamera, K.; Szuster-Gluszczak, M.; Leopoldo, M.; Lacivita, E.; Basta-Kaim, A. The Contribution of Formyl Peptide Receptor Dysfunction to the Course of Neuroinflammation: A Potential Role in the Brain Pathology. Curr. Neuropharmacol. 2020, 18, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Cattaneo, F.; Lippiello, P.; Cristiano, C.; Zurlo, F.; Castaldo, M.; Irace, C.; Borsello, T.; Santamaria, R.; Ammendola, R.; et al. Motor coordination and synaptic plasticity deficits are associated with increased cerebellar activity of NADPH oxidase, CAMKII, and PKC at preplaque stage in the TgCRND8 mouse model of Alzheimer’s disease. Neurobiol. Aging. 2018, 68, 123–133. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; Lucariello, A.; De Luca, A.; De Rosa, N.; Mazzarella, G.; Bianco, A.; Ammendola, R. Expression of Formyl-peptide Receptors in Human Lung Carcinoma. Anticancer Res. 2015, 35, 2769–2774. [Google Scholar]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Krepel, S.A.; Wang, J.M. Chemotactic Ligands that Activate G-Protein-Coupled Formylpeptide Receptors. Int. J. Mol. Sci. 2019, 20, 3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammendola, R.; Parisi, M.; Esposito, G.; Cattaneo, F. Pro-Resolving FPR2 Agonists Regulate NADPH Oxidase-Dependent Phosphorylation of HSP27, OSR1, and MARCKS and Activation of the Respective Upstream Kinases. Antioxidants 2021, 10, 134. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Fioretti, T.; Sarnataro, D.; Esposito, G.; Ammendola, R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch. Biochem. Biophys. 2016, 603, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Stama, M.L.; Slusarczyk, J.; Lacivita, E.; Kirpotina, L.N.; Schepetkin, I.A.; Chamera, K.; Riganti, C.; Perrone, R.; Quinn, M.T.; Basta-Kaim, A.; et al. Novel ureidopropanamide based N-formyl peptide receptor 2 (FPR2) agonists with potential application for central nervous system disorders characterized by neuroinflammation. Eur. J. Med. Chem. 2017, 141, 703–720. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavins, F.N.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef] [Green Version]
- Dahlgren, C.; Gabl, M.; Holdfeldt, A.; Winther, M.; Forsman, H. Basic characteristics of the neutrophil receptors that recognize formylated peptides, a danger-associated molecular pattern generated by bacteria and mitochondria. Biochem. Pharmacol. 2016, 114, 22–39. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Gulke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 Deficiency Promotes Mitochondrial Aberrations and Exacerbates Cardiac Injury Following Permanent Coronary Ligation via Enhanced Mitophagy and Apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Boccella, N.; Paolillo, R.; Cattaneo, F.; Trimarco, V.; Franzone, A.; D’Apice, S.; Giugliano, G.; Rinaldi, L.; Borzacchiello, D.; et al. Loss of Akap1 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure. Front. Physiol. 2018, 9, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Cattaneo, F.; Carrizzo, A.; Paolillo, R.; Boccella, N.; Ambrosio, M.; Damato, A.; Pironti, G.; Franzone, A.; Russo, G.; et al. Akap1 Regulates Vascular Function and Endothelial Cells Behavior. Hypertension 2018, 71, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Man, S.F. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation 2003, 107, 1514–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roversi, S.; Fabbri, L.M.; Sin, D.D.; Hawkins, N.M.; Agusti, A. Chronic Obstructive Pulmonary Disease and Cardiac Diseases. An Urgent Need for Integrated Care. Am. J. Respir. Crit. Care Med. 2016, 194, 1319–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, K.F.; Hurst, J.R.; Suissa, S. Cardiovascular disease and COPD: Dangerous liaisons? Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27. [Google Scholar] [CrossRef]
- Sievi, N.A.; Clarenbach, C.F.; Camen, G.; Rossi, V.A.; van Gestel, A.J.; Kohler, M. High prevalence of altered cardiac repolarization in patients with COPD. BMC Pulm. Med. 2014, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, S.D.; Wiersma, V.R.; Fokkema, I.E.; Berg, M.; Ten Hacken, N.H.T.; van den Berge, M.; Heijink, I.; Faiz, A. Acute cigarette smoke-induced eQTL affects formyl peptide receptor expression and lung function. Respirology 2021, 26, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lin, M.C.; Lee, C.H.; Liu, S.F.; Wang, C.C.; Fang, W.F.; Chao, T.Y.; Wu, C.C.; Wei, Y.F.; Chang, H.C.; et al. Defective formyl peptide receptor 2/3 and annexin A1 expressions associated with M2a polarization of blood immune cells in patients with chronic obstructive pulmonary disease. J. Transl. Med. 2018, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, J.; Kaur, G.; Gavins, F.N.E. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 1211. [Google Scholar] [CrossRef] [Green Version]
- Vital, S.A.; Senchenkova, E.Y.; Ansari, J.; Gavins, F.N.E. Targeting AnxA1/Formyl Peptide Receptor 2 Pathway Affords Protection against Pathological Thrombo-Inflammation. Cells 2020, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.S.; Wang, J.M.; Murphy, P.M.; Gao, J.L. Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formylpeptide receptor on mouse neutrophils. Biochem. Biophys. Res. Commun. 2000, 270, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getz, G.S.; Krishack, P.A.; Reardon, C.A. Serum amyloid A and atherosclerosis. Curr. Opin. Lipidol. 2016, 27, 531–535. [Google Scholar] [CrossRef]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Bairey Merz, C.N.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: The National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Misra, M.K.; Sarwat, M.; Bhakuni, P.; Tuteja, R.; Tuteja, N. Oxidative stress and ischemic myocardial syndromes. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2009, 15, RA209–RA219. [Google Scholar]
- Liu, Q.; Wang, S.; Cai, L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J. Diabetes Investig. 2014, 5, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Carsana, A. Metabolic Alterations in Cardiomyocytes of Patients with Duchenne and Becker Muscular Dystrophies. J. Clin. Med. 2019, 8, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattaneo, F.; Russo, R.; Castaldo, M.; Chambery, A.; Zollo, C.; Esposito, G.; Pedone, P.V.; Ammendola, R. Phosphoproteomic analysis sheds light on intracellular signaling cascades triggered by Formyl-Peptide Receptor 2. Sci. Rep. 2019, 9, 17894. [Google Scholar] [CrossRef]
- Annunziata, M.C.; Parisi, M.; Esposito, G.; Fabbrocini, G.; Ammendola, R.; Cattaneo, F. Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. Int. J. Mol. Sci. 2020, 21, 3818. [Google Scholar] [CrossRef]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Parisi, M.; Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013, 587, 1536–1542. [Google Scholar] [CrossRef] [Green Version]
- Castaldo, M.; Zollo, C.; Esposito, G.; Ammendola, R.; Cattaneo, F. NOX2-Dependent Reactive Oxygen Species Regulate Formyl-Peptide Receptor 1-Mediated TrkA Transactivation in SH-SY5Y Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2051235. [Google Scholar] [CrossRef] [Green Version]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, D.A.; Riccardi, A.; et al. VEGF-induced intracellular Ca(2+) oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [Green Version]
- Dorward, D.A.; Lucas, C.D.; Chapman, G.B.; Haslett, C.; Dhaliwal, K.; Rossi, A.G. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am. J. Pathol. 2015, 185, 1172–1184. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, T.; Wang, X.; Yuan, W.; Cheng, Y.; Zhang, H.; Xu, E.; Zhang, Y.; Shi, S.; Ma, D.; et al. FAM19A4 is a novel cytokine ligand of formyl peptide receptor 1 (FPR1) and is able to promote the migration and phagocytosis of macrophages. Cell. Mol. Immunol. 2015, 12, 615–624. [Google Scholar] [CrossRef]
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Le, Y.; Gao, J.L.; Zhao, J.; Wang, J.M.; Wang, A. Formylpeptide receptors mediate rapid neutrophil mobilization to accelerate wound healing. PLoS ONE 2014, 9, e90613. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Yao, X.; Ping, Y.; Jiang, T.; Liu, Q.; Xu, S.; Huang, J.; Mou, H.; Gong, W.; et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am. J. Pathol. 2011, 179, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Prevete, N.; Liotti, F.; Visciano, C.; Marone, G.; Melillo, R.M.; de Paulis, A. The formyl peptide receptor 1 exerts a tumor suppressor function in human gastric cancer by inhibiting angiogenesis. Oncogene 2015, 34, 3826–3838. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, K.; Xiang, Y.; Yoshimura, T.; Su, S.; Zhu, J.; Bian, X.W.; Wang, J.M. New development in studies of formyl-peptide receptors: Critical roles in host defense. J. Leukoc. Biol. 2016, 99, 425–435. [Google Scholar] [CrossRef]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011, 25, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Wu, M.S.; Lin, J.T.; Lin, M.T.; Shun, C.T.; Hua, K.T.; Kuo, M.L. Formyl Peptide receptor 1 expression is associated with tumor progression and survival in gastric cancer. Anticancer Res. 2014, 34, 2223–2229. [Google Scholar]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxidative Med. Cell. Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.C.; Kwon, Y.W.; Jang, I.H.; Jeong, G.O.; Lee, T.W.; Yoon, J.W.; Shin, H.J.; Jeong, H.C.; Ahn, Y.; Ko, T.H.; et al. Formyl Peptide Receptor 2 Is Involved in Cardiac Repair After Myocardial Infarction Through Mobilization of Circulating Angiogenic Cells. Stem Cells 2017, 35, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol. Sci. 2010, 31, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion--from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Gavins, F.N.; Kamal, A.M.; D’Amico, M.; Oliani, S.M.; Perretti, M. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 2005, 19, 100–102. [Google Scholar] [CrossRef]
- Zhou, Q.L.; Teng, F.; Zhang, Y.S.; Sun, Q.; Cao, Y.X.; Meng, G.W. FPR1 gene silencing suppresses cardiomyocyte apoptosis and ventricular remodeling in rats with ischemia/reperfusion injury through the inhibition of MAPK signaling pathway. Exp. Cell Res. 2018, 370, 506–518. [Google Scholar] [CrossRef]
- Honda, M.; Takeichi, T.; Hashimoto, S.; Yoshii, D.; Isono, K.; Hayashida, S.; Ohya, Y.; Yamamoto, H.; Sugawara, Y.; Inomata, Y. Intravital Imaging of Neutrophil Recruitment Reveals the Efficacy of FPR1 Blockade in Hepatic Ischemia-Reperfusion Injury. J. Immunol. 2017, 198, 1718–1728. [Google Scholar] [CrossRef] [Green Version]
- La, M.; Tailor, A.; D’Amico, M.; Flower, R.J.; Perretti, M. Analysis of the protection afforded by annexin 1 in ischaemia-reperfusion injury: Focus on neutrophil recruitment. Eur. J. Pharmacol. 2001, 429, 263–278. [Google Scholar] [CrossRef]
- Qin, C.X.; May, L.T.; Li, R.; Cao, N.; Rosli, S.; Deo, M.; Alexander, A.E.; Horlock, D.; Bourke, J.E.; Yang, Y.H.; et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017, 8, 14232. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Gordon, J.M.; Woodman, O.L.; Cao, A.H.; Dusting, G.J. Annexin-1 peptide Anx-1(2-26) protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br. J. Pharmacol. 2005, 145, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, R.H.; Sun, X.; Bilszta, J.L.; Gulluyan, L.M.; Dusting, G.J. Cardioprotective actions of an N-terminal fragment of annexin-1 in rat myocardium in vitro. Eur. J. Pharmacol. 2003, 461, 171–179. [Google Scholar] [CrossRef]
- Yang, Y.H.; Aeberli, D.; Dacumos, A.; Xue, J.R.; Morand, E.F. Annexin-1 regulates macrophage IL-6 and TNF via glucocorticoid-induced leucine zipper. J. Immunol. 2009, 183, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Perretti, M.; Getting, S.J.; Solito, E.; Murphy, P.M.; Gao, J.L. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am. J. Pathol. 2001, 158, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Buxton, K.D.; Pepe, S.; Cao, A.H.; Venardos, K.; Love, J.E.; Kaye, D.M.; Yang, Y.H.; Morand, E.F.; Ritchie, R.H. Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26). Br. J. Pharmacol. 2013, 168, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, R.; Li, Y.; Zhang, T.; Wu, N.; Zhang, J.; Guo, Z. Identification of Transcription Factor-Gene Regulatory Network in Acute Myocardial Infarction. Heart Lung Circ. 2017, 26, 343–353. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, W.; Liu, X.; Qi, J.; Deng, C. Biomarkers identification for acute myocardial infarction detection via weighted gene co-expression network analysis. Medicine 2017, 96, e8375. [Google Scholar] [CrossRef]
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Pan, D.; Chordia, M.D.; French, B.A.; Kron, I.L.; Yang, Z. The spleen contributes importantly to myocardial infarct exacerbation during post-ischemic reperfusion in mice via signaling between cardiac HMGB1 and splenic RAGE. Basic Res. Cardiol. 2016, 111, 62. [Google Scholar] [CrossRef] [Green Version]
- Marteau, J.B.; Zaiou, M.; Siest, G.; Visvikis-Siest, S. Genetic determinants of blood pressure regulation. J. Hypertens. 2005, 23, 2127–2143. [Google Scholar] [CrossRef]
- Cooper, R.S.; Luke, A.; Zhu, X.; Kan, D.; Adeyemo, A.; Rotimi, C.; Bouzekri, N.; Ward, R. Genome scan among Nigerians linking blood pressure to chromosomes 2, 3, and 19. Hypertension 2002, 40, 629–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathiresan, S.; Melander, O.; Guiducci, C.; Surti, A.; Burtt, N.P.; Rieder, M.J.; Cooper, G.M.; Roos, C.; Voight, B.F.; Havulinna, A.S.; et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008, 40, 189–197. [Google Scholar] [CrossRef]
- Benachour, H.; Zaiou, M.; Herbeth, B.; Lambert, D.; Lamont, J.V.; Pfister, M.; Siest, G.; Tiret, L.; Blankenberg, S.; Fitzgerald, P.S.; et al. Human formyl peptide receptor 1 (FPR1) c.32C>T SNP is associated with decreased soluble E-selectin levels. Pharmacogenomics 2009, 10, 951–959. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Wang, J.; Ribeiro, F.M.; Dixon, S.J.; Feldman, R.D.; Hegele, R.A.; Ferguson, S.S. Analysis of a missense variant of the human N-formyl peptide receptor that is associated with agonist-independent beta-arrestin association and indices of inflammation. Pharm. J. 2007, 7, 190–199. [Google Scholar] [CrossRef]
- Pravenec, M.; Kajiya, T.; Zidek, V.; Landa, V.; Mlejnek, P.; Simakova, M.; Silhavy, J.; Malinska, H.; Oliyarnyk, O.; Kazdova, L.; et al. Effects of human C-reactive protein on pathogenesis of features of the metabolic syndrome. Hypertension 2011, 57, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Shamieh, S.; Herbeth, B.; Azimi-Nezhad, M.; Benachour, H.; Masson, C.; Visvikis-Siest, S. Human formyl peptide receptor 1 C32T SNP interacts with age and is associated with blood pressure levels. Clin. Chim. Acta. 2012, 413, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Shimizu, K.; Mitchell, R.N.; Libby, P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Eliason, J.L.; Hannawa, K.K.; Ailawadi, G.; Sinha, I.; Ford, J.W.; Deogracias, M.P.; Roelofs, K.J.; Woodrum, D.T.; Ennis, T.L.; Henke, P.K.; et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 2005, 112, 232–240. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Fu, Y.; Deng, J.; Shen, Y.; Wang, Y.; Yu, F.; Xie, N.; Chen, Z.; Hong, T.; Peng, X.; et al. Deficiency of FAM3D (Family With Sequence Similarity 3, Member D), A Novel Chemokine, Attenuates Neutrophil Recruitment and Ameliorates Abdominal Aortic Aneurysm Development. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1616–1631. [Google Scholar] [CrossRef] [PubMed]
- Rizas, K.D.; Ippagunta, N.; Tilson, M.D., 3rd. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol. Rev. 2009, 17, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.H.; Chordia, M.D.; Spinosa, M.D.; Su, G.; Ladd, Z.; Pan, D.; Upchurch, G.R., Jr.; Sharma, A.K. Single-Photon Emission Computed Tomography Imaging Using Formyl Peptide Receptor 1 Ligand Can Diagnose Aortic Aneurysms in a Mouse Model. J. Surg. Res. 2020, 251, 239–247. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Czapiga, M.; Gao, J.L.; Kirk, A.; Lekstrom-Himes, J. Human platelets exhibit chemotaxis using functional N-formyl peptide receptors. Exp. Hematol. 2005, 33, 73–84. [Google Scholar] [CrossRef]
- Salamah, M.F.; Ravishankar, D.; Vaiyapuri, R.; Moraes, L.A.; Patel, K.; Perretti, M.; Gibbins, J.M.; Vaiyapuri, S. The formyl peptide fMLF primes platelet activation and augments thrombus formation. J. Thromb. Haemost. 2019, 17, 1120–1133. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.M.; et al. Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation 2019, 140, 319–335. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Gao, J.L. Leukocyte chemoattractant receptor FPR2 may accelerate atherogenesis. Med. Hypotheses 2012, 79, 101–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, M.J.; Galkina, E.V. wRAPping up early monocyte and neutrophil recruitment in atherogenesis via Annexin A1/FPR2 signaling. Circ. Res. 2015, 116, 774–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satish, M.; Agrawal, D.K. Pro-resolving lipid mediators in the resolution of neointimal hyperplasia pathogenesis in atherosclerotic diseases. Expert Rev. Cardiovasc. Ther. 2019, 17, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Schiattarella, G.G.; Perrino, C.; Cattaneo, F.; Pironti, G.; Franzone, A.; Gargiulo, G.; Magliulo, F.; Serino, F.; Carotenuto, G.; et al. Dermcidin: A skeletal muscle myokine modulating cardiomyocyte survival and infarct size after coronary artery ligation. Cardiovasc. Res. 2015, 107, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, M.H.; Laguna-Fernandez, A.; Gonzalez-Diez, M.; Paulsson-Berne, G.; Hansson, G.K.; Back, M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 2015, 105, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Back, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E(-/-) mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drechsler, M.; de Jong, R.; Rossaint, J.; Viola, J.R.; Leoni, G.; Wang, J.M.; Grommes, J.; Hinkel, R.; Kupatt, C.; Weber, C.; et al. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ. Res. 2015, 116, 827–835. [Google Scholar] [CrossRef]
- Kusters, D.H.; Chatrou, M.L.; Willems, B.A.; De Saint-Hubert, M.; Bauwens, M.; van der Vorst, E.; Bena, S.; Biessen, E.A.; Perretti, M.; Schurgers, L.J.; et al. Pharmacological Treatment with Annexin A1 Reduces Atherosclerotic Plaque Burden in LDLR-/- Mice on Western Type Diet. PLoS ONE 2015, 10, e0130484. [Google Scholar] [CrossRef] [Green Version]
- Back, M.; Weber, C.; Lutgens, E. Regulation of atherosclerotic plaque inflammation. J. Intern. Med. 2015, 278, 462–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredman, G.; Kamaly, N.; Spolitu, S.; Milton, J.; Ghorpade, D.; Chiasson, R.; Kuriakose, G.; Perretti, M.; Farokzhad, O.; Tabas, I. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 2015, 7, 275ra20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.C.; Kwon, Y.W.; Jang, I.H.; Jeong, G.O.; Yoon, J.W.; Kim, C.D.; Kwon, S.M.; Bae, Y.S.; Kim, J.H. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem Cells 2014, 32, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Heo, S.C.; Kwon, Y.W.; Kim, H.D.; Kim, S.H.; Jang, I.H.; Kim, J.H.; Hwang, N.S. Injectable PLGA microspheres encapsulating WKYMVM peptide for neovascularization. Acta Biomater. 2015, 25, 76–85. [Google Scholar] [CrossRef]
- Jang, E.J.; Bae, I.H.; Park, D.S.; Lee, S.Y.; Lim, K.S.; Park, J.K.; Shim, J.W.; Sim, D.S.; Jeong, M.H. Effect of a novel peptide, WKYMVm- and sirolimus-coated stent on re-endothelialization and anti-restenosis. J. Mater. Sci. Mater. Med. 2015, 26, 251. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, I.H.; Heo, S.C.; Kim, J.H.; Hwang, N.S. Biomedical therapy using synthetic WKYMVm hexapeptide. Organogenesis 2016, 12, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Lakota, K.; Mrak-Poljsak, K.; Bozic, B.; Tomsic, M.; Sodin-Semrl, S. Serum amyloid A activation of human coronary artery endothelial cells exhibits a neutrophil promoting molecular profile. Microvasc. Res. 2013, 90, 55–63. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, S.D.; Baek, S.H.; Choi, J.H.; Bae, Y.S. Role of formyl peptide receptor 2 on the serum amyloid A-induced macrophage foam cell formation. Biochem. Biophys. Res. Commun. 2013, 433, 255–259. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, E.; Kim, S.D.; Seo, J.K.; Bae, Y.S. Oxidized low-density lipoprotein-induced foam cell formation is mediated by formyl peptide receptor 2. Biochem. Biophys. Res. Commun. 2014, 443, 1003–1007. [Google Scholar] [CrossRef]
- Ding, Y.; Feng, Y.; Zou, Y.; Wang, F.; Liu, H.; Liu, C.; Zhang, Y. [Gly14]-humanin restores cathepsin D function via FPRL1 and promotes autophagic degradation of Ox-LDL in HUVECs. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 2406–2416. [Google Scholar] [CrossRef]
- Dong, Z.; An, F.; Wu, T.; Zhang, C.; Zhang, M.; Zhang, Y.; An, G.; An, F. PTX3, a key component of innate immunity, is induced by SAA via FPRL1-mediated signaling in HAECs. J. Cell. Biochem. 2011, 112, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Edfeldt, K.; Agerberth, B.; Rottenberg, M.E.; Gudmundsson, G.H.; Wang, X.B.; Mandal, K.; Xu, Q.; Yan, Z.Q. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [Green Version]
- Salamah, M.F.; Ravishankar, D.; Kodji, X.; Moraes, L.A.; Williams, H.F.; Vallance, T.M.; Albadawi, D.A.; Vaiyapuri, R.; Watson, K.; Gibbins, J.M.; et al. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. 2018, 2, 2973–2985. [Google Scholar] [CrossRef]
- D’Amico, M.; Di Filippo, C.; La, M.; Solito, E.; McLean, P.G.; Flower, R.J.; Oliani, S.M.; Perretti, M. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000, 14, 1867–1869. [Google Scholar] [CrossRef] [PubMed]
- La, M.; D’Amico, M.; Bandiera, S.; Di Filippo, C.; Oliani, S.M.; Gavins, F.N.; Flower, R.J.; Perretti, M. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: Analysis of their mechanism of action. FASEB. Exp. Biol. 2001, 15, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Relton, J.K.; Strijbos, P.J.; O’Shaughnessy, C.T.; Carey, F.; Forder, R.A.; Tilders, F.J.; Rothwell, N.J. Lipocortin-1 is an endogenous inhibitor of ischemic damage in the rat brain. J. Exp. Med. 1991, 174, 305–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.K.; Gil, C.D.; Oliani, S.M.; Gavins, F.N. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. FASEB J. 2015, 29, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Komshian, S.; Sansbury, B.E.; Wu, B.; Mottola, G.; Chen, M.; Spite, M.; Conte, M.S. Biosynthesis of proresolving lipid mediators by vascular cells and tissues. FASEB J. 2017, 31, 3393–3402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Shu, H.H.; Chang, L.; Ye, F.; Xu, K.Q.; Huang, W.Q. Resolvin D1 protects against hepatic ischemia/reperfusion injury in rats. Int. Immunopharmacol. 2015, 28, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Lee, S.M. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim. Biophys. Acta 2016, 1861, 1025–1035. [Google Scholar] [CrossRef]
- Liu, W.; Huang, J.; Doycheva, D.; Gamdzyk, M.; Tang, J.; Zhang, J.H. RvD1binding with FPR2 attenuates inflammation via Rac1/NOX2 pathway after neonatal hypoxic-ischemic injury in rats. Exp. Neurol. 2019, 320, 112982. [Google Scholar] [CrossRef] [PubMed]
- Van Lookeren Campagne, M.; Thomas, G.R.; Thibodeaux, H.; Palmer, J.T.; Williams, S.P.; Lowe, D.G.; van Bruggen, N. Secondary reduction in the apparent diffusion coefficient of water, increase in cerebral blood volume, and delayed neuronal death after middle cerebral artery occlusion and early reperfusion in the rat. J. Cereb. Blood Flow Metab. 1999, 19, 1354–1364. [Google Scholar] [CrossRef] [Green Version]
- Gavins, F.N.; Yona, S.; Kamal, A.M.; Flower, R.J.; Perretti, M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood 2003, 101, 4140–4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, M.O.; Hannan, K.; Burne, M.J.; Lappin, D.W.; Doran, P.; Coleman, P.; Stenson, C.; Taylor, C.T.; Daniels, F.; Godson, C.; et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J. Am. Soc. Nephrol. Jasn 2002, 13, 1657–1662. [Google Scholar] [CrossRef] [Green Version]
- Bannenberg, G.; Moussignac, R.L.; Gronert, K.; Devchand, P.R.; Schmidt, B.A.; Guilford, W.J.; Bauman, J.G.; Subramanyam, B.; Perez, H.D.; Parkinson, J.F.; et al. Lipoxins and novel 15-epi-lipoxin analogs display potent anti-inflammatory actions after oral administration. Br. J. Pharmacol. 2004, 143, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Houard, X.; Ollivier, V.; Louedec, L.; Michel, J.B.; Back, M. Differential inflammatory activity across human abdominal aortic aneurysms reveals neutrophil-derived leukotriene B4 as a major chemotactic factor released from the intraluminal thrombus. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1376–1383. [Google Scholar] [CrossRef]
- Pillai, P.S.; Leeson, S.; Porter, T.F.; Owens, C.D.; Kim, J.M.; Conte, M.S.; Serhan, C.N.; Gelman, S. Chemical mediators of inflammation and resolution in post-operative abdominal aortic aneurysm patients. Inflammation 2012, 35, 98–113. [Google Scholar] [CrossRef]
- Pope, N.H.; Salmon, M.; Davis, J.P.; Chatterjee, A.; Su, G.; Conte, M.S.; Ailawadi, G.; Upchurch, G.R., Jr. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016, 30, 4192–4201. [Google Scholar] [CrossRef] [Green Version]
- Petri, M.H.; Thul, S.; Andonova, T.; Lindquist-Liljeqvist, M.; Jin, H.; Skenteris, N.T.; Arnardottir, H.; Maegdefessel, L.; Caidahl, K.; Perretti, M.; et al. Resolution of Inflammation Through the Lipoxin and ALX/FPR2 Receptor Pathway Protects Against Abdominal Aortic Aneurysms. Basic Transl. Sci. 2018, 3, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 9999. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Jadapalli, J.K.; Tourki, B.; Halade, G.V. Inhibition of FPR2 impaired leukocytes recruitment and elicited non-resolving inflammation in acute heart failure. Pharmacol. Res. 2019, 146, 104295. [Google Scholar] [CrossRef]
- Tourki, B.; Kain, V.; Pullen, A.B.; Norris, P.C.; Patel, N.; Arora, P.; Leroy, X.; Serhan, C.N.; Halade, G.V. Lack of resolution sensor drives age-related cardiometabolic and cardiorenal defects and impedes inflammation-resolution in heart failure. Mol. Metab. 2020, 31, 138–149. [Google Scholar] [CrossRef]
- Corminboeuf, O.; Leroy, X. FPR2/ALXR agonists and the resolution of inflammation. J. Med. Chem. 2015, 58, 537–559. [Google Scholar] [CrossRef]
- De Gaetano, M.; Butler, E.; Gahan, K.; Zanetti, A.; Marai, M.; Chen, J.; Cacace, A.; Hams, E.; Maingot, C.; McLoughlin, A.; et al. Asymmetric synthesis and biological evaluation of imidazole- and oxazole-containing synthetic lipoxin A4 mimetics (sLXms). Eur. J. Med. Chem. 2019, 162, 80–108. [Google Scholar] [CrossRef]
- Stalder, A.K.; Lott, D.; Strasser, D.S.; Cruz, H.G.; Krause, A.; Groenen, P.M.; Dingemanse, J. Biomarker-guided clinical development of the first-in-class anti-inflammatory FPR2/ALX agonist ACT-389949. Br. J. Clin. Pharmacol. 2017, 83, 476–486. [Google Scholar] [CrossRef]
- Asahina, Y.; Wurtz, N.R.; Arakawa, K.; Carson, N.; Fujii, K.; Fukuchi, K.; Garcia, R.; Hsu, M.Y.; Ishiyama, J.; Ito, B.; et al. Discovery of BMS-986235/LAR-1219: A Potent Formyl Peptide Receptor 2 (FPR2) Selective Agonist for the Prevention of Heart Failure. J. Med. Chem. 2020, 63, 9003–9019. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, B.; Leoni, G.; Hinkel, R.; Ormanns, S.; Paulin, N.; Ortega-Gomez, A.; Viola, J.R.; de Jong, R.; Bongiovanni, D.; Bozoglu, T.; et al. Pro-Angiogenic Macrophage Phenotype to Promote Myocardial Repair. J. Am. Coll. Cardiol. 2019, 73, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Ito, B.R.; Lupisella, J.A.; Carson, N.A.; Hsu, M.Y.; Fernando, G.; Heroux, M.; Bouvier, M.; Dierks, E.; Kick, E.K.; et al. Preservation of Post-Infarction Cardiac Structure and Function via Long-Term Oral Formyl Peptide Receptor Agonist Treatment. Basic Transl. Sci. 2019, 4, 905–920. [Google Scholar] [CrossRef]
- Migeotte, I.; Riboldi, E.; Franssen, J.D.; Gregoire, F.; Loison, C.; Wittamer, V.; Detheux, M.; Robberecht, P.; Costagliola, S.; Vassart, G.; et al. Identification and characterization of an endogenous chemotactic ligand specific for FPRL2. J. Exp. Med. 2005, 201, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiet, M.J.; Macari, L.; Dahlgren, C.; Boulay, F. N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J. Biol. Chem. 2011, 286, 26718–26731. [Google Scholar] [CrossRef] [Green Version]

| CVDs | Expression and/or Stimulation | Role |
|---|---|---|
| Ischemia reperfusion injury (IRI) | FPR1 deficiency m | Protective effects by reducing the risk of heart injury induced by IRI [65] |
| FPR1 silencing r | Protective effects mediated by depression of inflammation, cardiomyocyte apoptosis and ventricular remodeling in rats with I/R injury through the suppression of the MAPK signaling pathway activation [66] | |
| FPR1 antagonist CSH m | Protective effects by reducing hepatocyte necrosis/apoptosis, and diminishing inflammatory cytokine, chemokine, and oxidative stress levels as well as accumulation of neutrophils in the necrotic area [67] | |
| FPR1 stimulation with ANXA1 m and r | Cardioprotective role by preserving inotropic responsiveness at the level of ventricular muscle and contractile function of cardiac muscle in vitro [70,71,72,73] | |
| FPR1 blockade m | Beneficial effects mediated by abrogation of reperfusion-induced exacerbation of infarct size [78] | |
| Blood pressure (BP) levels | FPR1 C32T (rs5030878) single nuclear polymorphism (SNP) h | Negative prognostic factor and detrimental effects associated with increased C-reactive protein (CRP) levels linearly related with BP [83,84,85] |
| Abdominal aortic aneurysm (AAA) | FPR1 involvement m | Detrimental effects since FPR1 results involved in neutrophil recruitment and aggravated AAA development [91] |
| Acute myocardial infarction (AMI) | FPR1 as a differentially expressed gene (DEG) h | Role has to be determined even if FPR1 results a DEG in human AMI blood tissue, compared with normal blood tissue using microarray data [75] |
| Potential beneficial role for AMI prevention [76] | ||
| Platelet-mediated complications | FPR1 inhibition or gene deletion m and h | Detrimental effects by impairing agonist-induced platelet activation in Fpr1-deficient mice or in pharmacologically FPR1 inhibited human platelets [94] |
| Endothelial cell function and HUVECs | FPR1 stimulation with NfMLF hc | Beneficial effects by promoting proliferation and capillary network formation |
| Angiogenesis | FPR1 stimulation with ANXA1 hc | Beneficial effects by inducing angiogenesis and/or production of angiogenic factors [56,57,58] |
| CVDs | Expression and/or Stimulation | Effects |
|---|---|---|
| Atherosclerotic lesions | FPR2 stimulation with SAA hc and mc | Detrimental effects contributing to atherosclerosis progression in human aortic endothelial cells (HAECs) [116] |
| Detrimental effects contributing to atherosclerosis progression upregulating the secretion of long pentraxin 3 (PTX3) in human aortic endothelial cells [116] | ||
| Detrimental effects by upregulating oxidized low-density lipoprotein (oxLDL) contributing to macrophages differentiation into foam cells and in turn inflammatory cytokine production and plaque formation [114] | ||
| FPR2 mRNA levels h and m (up-regulated expression) | Dual role by promoting both disease progression (detrimental) and plaque stability (beneficial) [101] | |
| FPR2 stimulation with AnxA1 m | Protective role by reducing sizes and macrophage accumulation in the atherosclerotic lesion [103] | |
| Protective effects by reducing the progression of existing plaques of aortic arch and subclavian artery by FPR2 dependent reduction of neutrophil rolling and adhesion to activated endothelial cells [104] | ||
| Protective effects exerted by proresolving ANXA1 mimicking peptide Ac2-26 reduces experimental atherosclerosis in presence of a functional FPR2 [106] | ||
| FPR2 stimulation with lipoxinA4 m | Protective effects by reducing macrophage infiltration and apoptotic cells in atherosclerotic lesions [102] | |
| FPR2 stimulation with LL-37 m | Detrimental effects by contributing to plaque formation by priming circulating platelet and inducing thromboinflammation [118,119] | |
| Neovascularization | FPR2 stimulation with WKYMVm hc, m and rb | Beneficial effects by recruiting endothelial progenitor cells, contributing to neovascularization and promoting re-endothelialization [108,109] |
| Protective effects by inhibiting restenosis [110] | ||
| Ischemia reperfusion injury (IRI) | FPR2 stimulation with LXA4 or AnxaA1 m | Protective effects by counter regulating the inflammatory response during IRI [123,124] |
| FPR2 antagonist Boc2 m | Detrimental effects exerted by pre-ischemia Boc2 administration resulting in LXA4 abrogated production and impaired vascular reactivity [124] | |
| FPR2 stimulation with Ac2-26 r | Protective effects by preserving cardiomyocyte contractility related to the activation of PKC, p38, and KATP channels [70] | |
| FPR2 stimulation with SAA hc | Detrimental effects, contributing to atherosclerosis progression by upregulating the secretion of long pentraxin 3 (PTX3) in human aortic endothelial cells [116] | |
| FPR2 stimulation with RvD1 r | Protective effects by inhibiting inflammatory cascades; reducing IL-6, TNF-α and MPO levels; diminishing apoptosis by Akt phosphorylation, and attenuating IR-induced damage [126] | |
| Protective effects by reducing percent of infarction area, ameliorating short- and long-term neurological deficits trough the activation of Rac1/NOX2 signaling pathway [128] | ||
| FPR2 stimulation with CGEN-855A r and m | Cardioprotective effects in rat and murine myocardial IRI [130] | |
| FPR2 stimulation with 15-epi-16-(p-fluorophenoxy)-LXA4-methyl ester m | Protective effects in renal IRI models, by modulating cytokine and chemokine expression and neutrophil recruitment [131] | |
| FPR2 stimulation with compound 17b m and mc | Protective effects by reducing inflammatory responses associated with reperfusion after an acute MI [69] | |
| FPR2 stimulation with ZK-994 and ZK-142 m | Protective effects by inhibition of neutrophil accumulation in murine hind-limb IRI-induced second-organ lung injury [132] | |
| Abdominal aortic aneurysm (AAA) | FPR2 stimulation with LXA4 m | Protective role by limiting neutrophil inflammation [135,136] |
| Myocardial infarction (MI) | FPR2 stimulation with WKYMVm m | Cardiac protection by mobilizing circulating angiogenic cells, contributing to their homing to ischemic heart [62] |
| FPR2 stimulation with 15-epimer-LXA4 m | Protective effects by triggering early activation of the resolving phase and improving left ventricular function post-MI [138] | |
| FPR2 inactivation by WRW4 m | Detrimental effects leading leukocytes to nonresolving inflammation in acute MI [139] | |
| Fpr2 gene deletion m | Detrimental effects by impairing biosynthesis of specialized proresolving mediators amplifying unresolved inflammation after cardiac injury [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caso, V.M.; Manzo, V.; Pecchillo Cimmino, T.; Conti, V.; Caso, P.; Esposito, G.; Russo, V.; Filippelli, A.; Ammendola, R.; Cattaneo, F. Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life 2021, 11, 243. https://doi.org/10.3390/life11030243
Caso VM, Manzo V, Pecchillo Cimmino T, Conti V, Caso P, Esposito G, Russo V, Filippelli A, Ammendola R, Cattaneo F. Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life. 2021; 11(3):243. https://doi.org/10.3390/life11030243
Chicago/Turabian StyleCaso, Valentina Maria, Valentina Manzo, Tiziana Pecchillo Cimmino, Valeria Conti, Pio Caso, Gabriella Esposito, Vincenzo Russo, Amelia Filippelli, Rosario Ammendola, and Fabio Cattaneo. 2021. "Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression" Life 11, no. 3: 243. https://doi.org/10.3390/life11030243
APA StyleCaso, V. M., Manzo, V., Pecchillo Cimmino, T., Conti, V., Caso, P., Esposito, G., Russo, V., Filippelli, A., Ammendola, R., & Cattaneo, F. (2021). Regulation of Inflammation and Oxidative Stress by Formyl Peptide Receptors in Cardiovascular Disease Progression. Life, 11(3), 243. https://doi.org/10.3390/life11030243