1. Introduction
Amoebiasis in humans is caused by
Entamoeba histolytica whose cytotoxic activity on a variety of target cells has been widely demonstrated [
1]. The process is contact dependent and involves adherence of the parasite through its surface lectin, known as Gal/GalNAc lectin (Gal lectin), which is composed of a 260 KDa heterodimer of disulfide-linked heavy (170 KDa) and light (35/31 KDa) subunits, which is noncovalently associated with an intermediate subunit of 150 KDa. The carbohydrate recognition domain (CRD) is localized in a cysteine-rich region in the heavy subunit from amino acids 895–998 [
2]. The precise cytotoxic mechanism is unknown, but after adherence, host intracellular calcium becomes dramatically elevated, and host proteins become dephosphorylated contributing both events to cell death [
3]. Although there has not been identified a specific receptor to which the Gal lectin binds, there are in the literature some reports related to pattern recognition receptors (PRRs). Chadee et al. [
4] demonstrated that the amoebic lectin induced transcription of the Toll-like receptor 2 (TLR-2) gene in a murine macrophages cell line and that in TLR-2 gene regulation a mitogen-activated protein kinase (MAPK or MAP kinase) was involved; however, in that study, binding of the Gal lectin to the TLR-2 was not demonstrated. A later study showed that the recombinant CRD of the Gal lectin binds to TLR-2 and TLR-4 in human colonic cells and activates the classic signaling pathway of these receptors, concluding that the CRD of the amoebic Gal lectin worked similar to a pathogen-associated molecular pattern (PAMP) inducing expression of TLRs and inflammatory cytokines by binding to TLR receptors in the colonic cells [
5].
When analyzing the CRD’s sequence, Dodson et al. [
2] found there was some limited identity in sequence to the receptor-binding domain of hepatocyte growth factor (HGF). Specifically, the region from amino acids 913–939 of CRD had 52% sequence identity with amino acids 59–85 of HGF, which forms part of the receptor-binding domain sufficient for high-affinity HGF binding. The similarity was such that the recombinant CRD and even the purified Gal lectin competed with HGF for binding to the c-Met receptor in competition binding assays. The competition was not due to the carbohydrate-binding activity since the presence of
N-acetylgalactosamine (GalNAc) in the assay did not modify the result. The biological consequences of this structural trait of Gal lectin have not yet been studied in in vitro cell coculture models. In the present study, we analyzed the interaction of trophozoites from
E. histolytica HM-1:IMSS with HepG2 cells (a cell line derived from human hepatocarcinoma) through the amoebic Gal lectin (in the trophozoites’ membrane) and the c-Met receptor (at the HepG2 cells’ surface). The results obtained by immunoprecipitation with anti-c-Met antibodies after coculture revealed a band with an approximate molecular weight of 60 KDa recognized by anti-Gal lectin antibodies. We also found that both molecules, i.e., Gal lectin and c-Met, colocalized in cocultures of trophozoites and HepG2 cells, as shown by confocal fluorescence microscopy images. Finally, our results from a parallel microscopic study indicated that the cytotoxic effect caused by trophozoites to HepG2 cells was prevented by pretreatment of HepG2 cells with HGF before coculture with amoebic trophozoites, but the adherence of the trophozoites was still observed; the latter seems to involve the c-Met receptor in the cytotoxic effect.
2. Materials and Methods
2.1. Cell Culture
Entamoeba histolytica trophozoites, HM-1:IMSS, were axenically cultured in TYI-S-33 medium in culture flasks at 37 °C according to standard protocols [
6]. Virulence was defined as the ability of 5 × 10
5 trophozoites to produce multiple liver abscesses in hamsters 7 days after intraportal injection. Such virulence was maintained by passing axenic amoebic cultures through hamsters’ livers twice a month, recovering the parasites from 7 days old abscesses and again growing them axenically. Cultures with 72 h expansion were used to perform total lysate of amoebas, cocultures, and in vitro assays.
HepG2 cells from a human-liver-derived cell line were cultured in high glucose DMEM medium supplemented with (10%) fetal bovine serum and (1%) antibiotic cocktail penicillin–streptomycin at 37 °C. HepG2 cells obtained this way were used to perform total cell lysates, cocultures with amoebic trophozoites, and in vitro assays.
2.2. Cocultures of Entamoeba histolytica: HepG2
HepG2 cells 3 × 106 were seeded in 10 cm diameter Petri dishes until reaching 60% confluence; subsequently, HepG2 cells were washed with sterile PBS and coincubated with 6 × 106 amoebic trophozoites in 3.5 mL of serum-free TYI medium at 37 °C for 15 min. Elapsed time, cells were recovered with a cell scraper and lysed with lysis buffer (RIPA) supplemented with 10 mM EDTA and 100 mM iodoacetamide.
For microscopic studies, cocultures were performed on sterile coverslips placed in a 12-well multichamber; 6 × 104 HepG2 cells were seeded per well. Subsequently, cell cultures were washed with sterile PBS and coincubated with 6 × 104 (for immunofluorescence staining) or 2 × 104 (for hematoxylin and eosin staining) amoebic trophozoites suspension in 0.5 mL of serum-free TYI medium at 37 °C for 10 or 15 min.
2.3. Immunoprecipitation
For each assay, 1 mg of protein, (from coculture) in a final volume of 0.5 mL of lysis buffer supplemented with 10 mM EDTA and 100 mM iodoacetamide was used as the starting sample.
The preclearing of the sample was performed with 60 µL Agarose Protein g (APG) for 2 h at 4 °C under constant stirring; after that time, samples were centrifuged at 12,000 rpm for 5 min at 4 °C, and the supernate was recovered. The supernate (precleared sample) was incubated for 2 h at 4 °C under constant stirring with 10 μL goat anti-c-Met receptor antibody (SIGMA H9786). In parallel, 60 µL APG were incubated with 14 µL of 10% bovine serum albumin (BSA) to prevent unspecific binding, APG-BSA was centrifuged at 3000 rpm, and the supernate was discarded. Subsequently, the precleared sample treated with antibody and BSA–APG were mixed and incubated overnight at 4 °C under constant stirring. Samples were centrifuged at 12,000 rpm for one minute at 4 °C and washed 6 times with 200 μL wash Buffer.
2.4. Western Blot
Western blot was performed as described elsewhere [
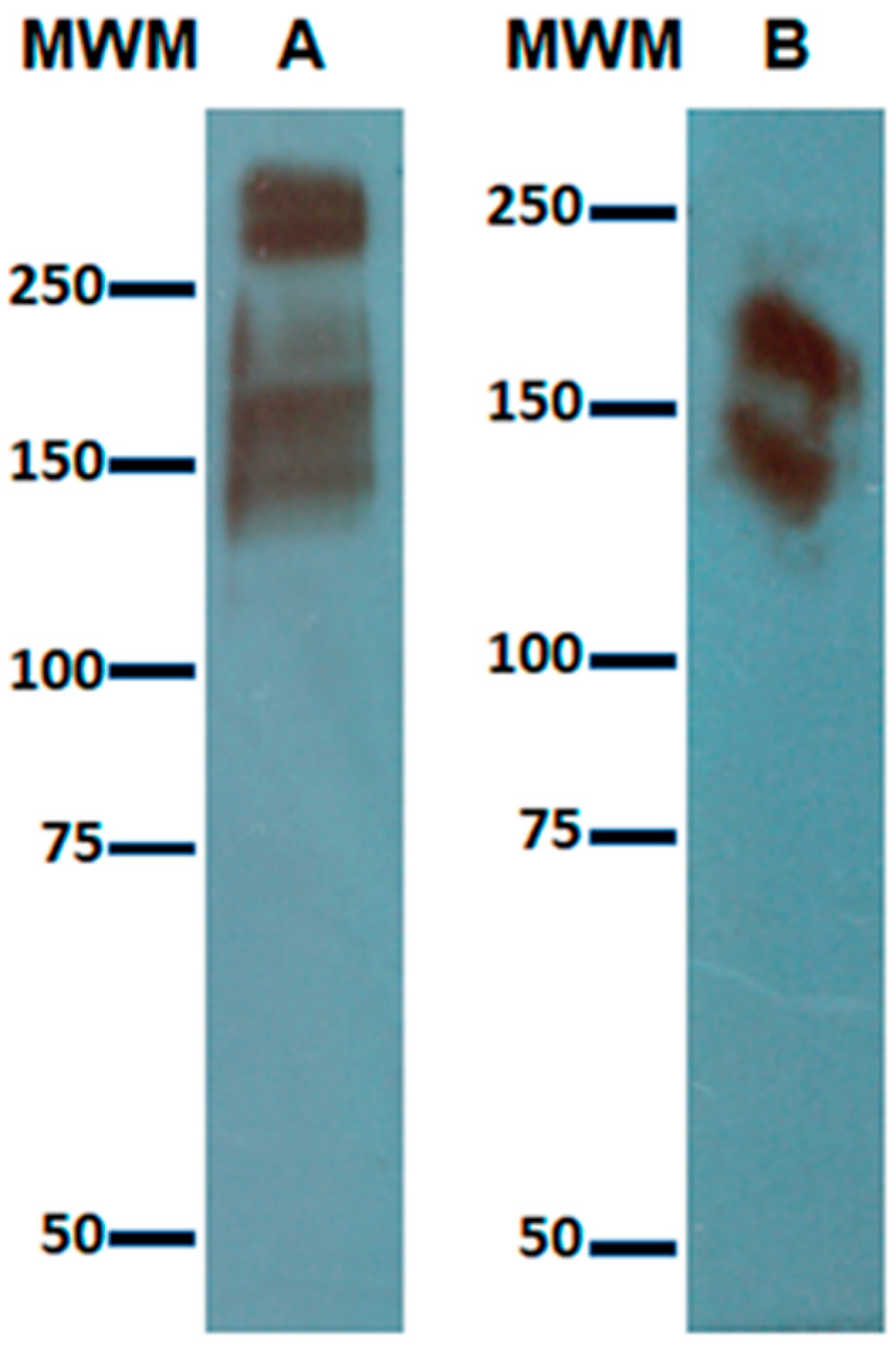
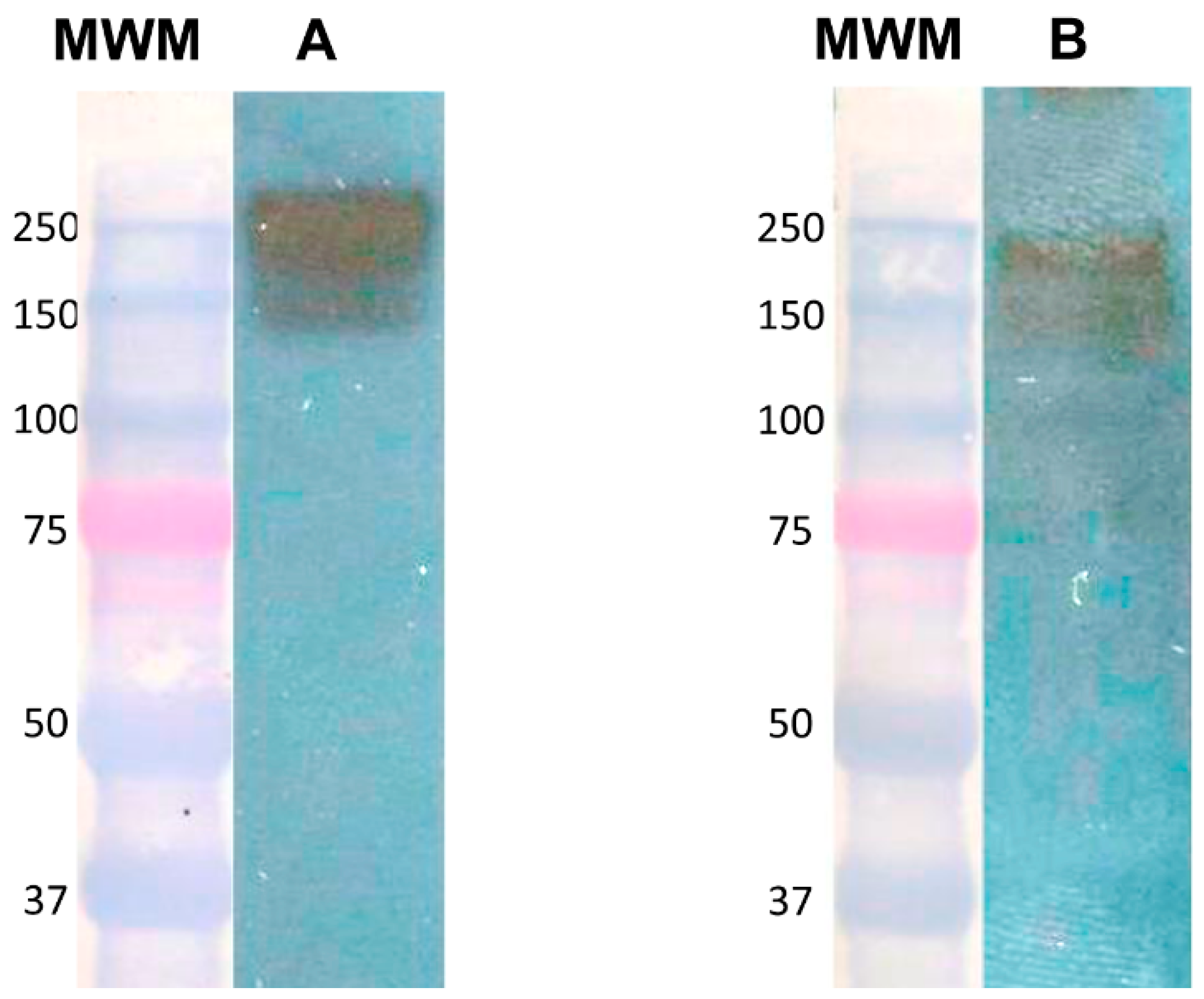
7]. Total cell lysates from cultures, cocultures, or immunoprecipitated samples were resolved by 7.5% SDS–PAGE. Molecular weight markers were precision plus protein dual-color standards, BIO-RAD catalog #161-0374. Proteins were transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in TBS-T for 1 h at room temperature and then incubated overnight at 4 °C with the corresponding antibodies: rabbit anti-Gal lectin (0.4 μg/mL made in our laboratory) [
6], goat anti-human c-Met receptor (0.2 μg/mL SIGMA H9786), or goat anti-human HGF (0.2 μg/mL SIGMA H7157). Membranes were washed with TBS-T and incubated with goat anti-rabbit IgG (H + L) (1/60,000 Cell signaling 7074) or bovine anti-goat IgG (H + L) (1:10,000 Santa Cruz Biotechnology sc-2350), all conjugated to horseradish peroxidase for 90 min at room temperature. After washing with TBS-T, antibody-reactive proteins were detected by chemiluminescence.
2.5. Hematoxylin and Eosin Staining
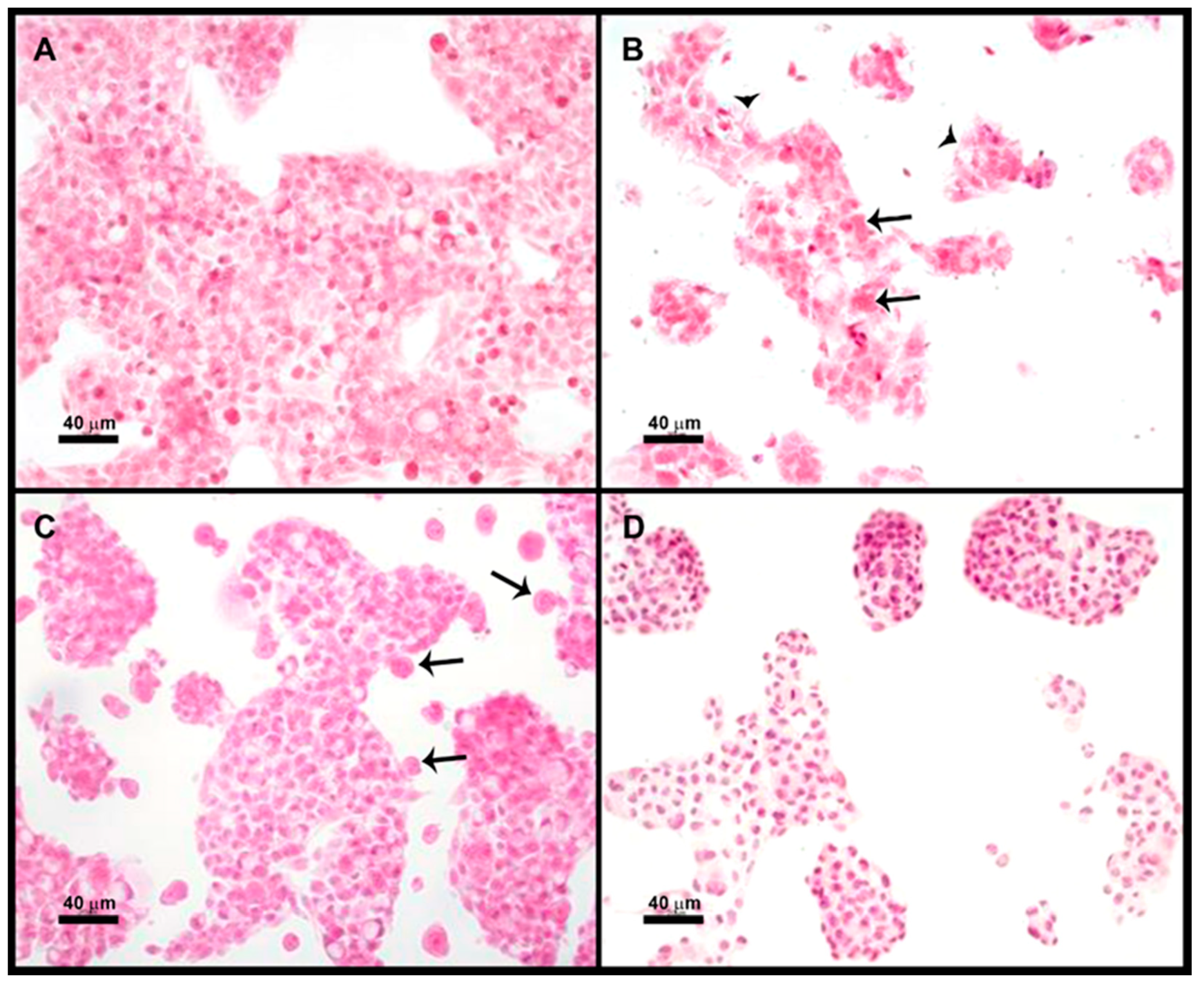
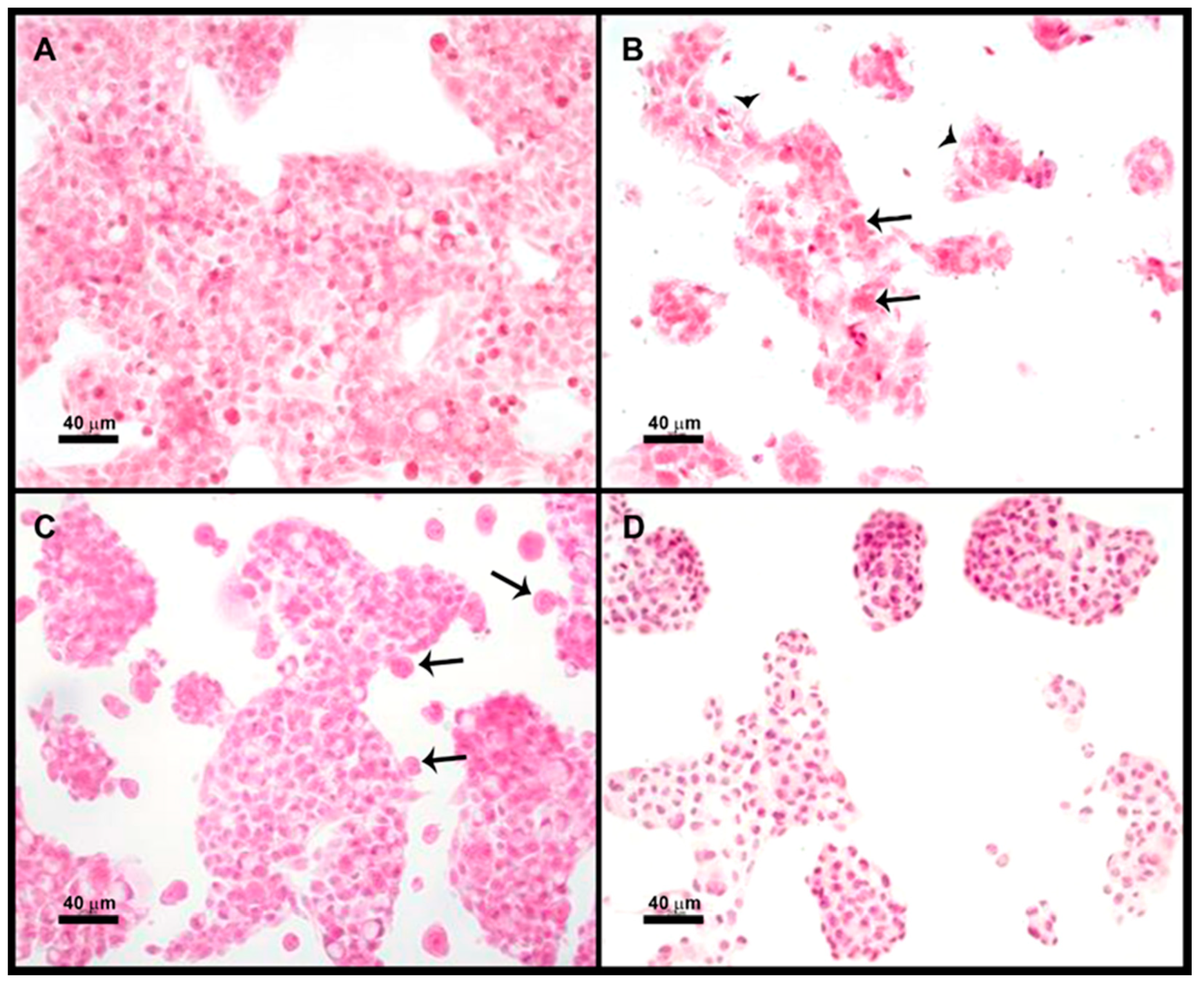
Four groups were used in duplicate: (1) HepG2 cells, (2) HepG2 cells + E. histolytica, (3) HepG2 cells pretreated with 200 ng/mL HGF (1 h) + E. histolytica, and (4) HepG2 + E. histolytica pretreated (30 min) with 180 mM Galactose. The incubation with the trophozoites was performed in a 3:1 ratio (HepG2:amoebas) in 0.5 mL of TYI medium at 37 °C for 15 min.
After completion of the different treatments, the coverslips were washed once with PBS, fixed for 30 min in 4% PFA, and postfixed in 0.4% PFA; finally, they were stained with hematoxylin and eosin. Micrographs were taken with Nikon microscope DMX1200 and processed with Nikon ACT-1 software (Version 2.63, Nikon, Tokyo, Japan)
2.6. Immunofluorescence Assay
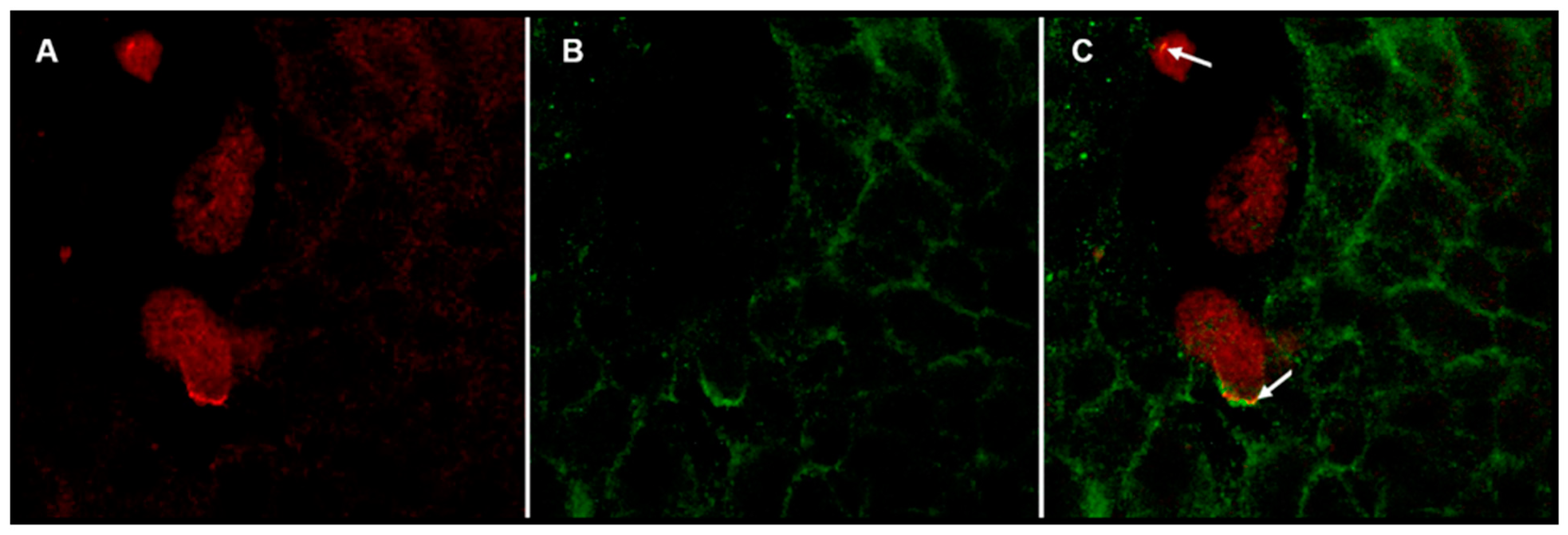
After coculture, the coverslips were washed once with PBS and fixed for 10 min in 4% PFA. Samples were washed thrice with PBS at room temperature and then were incubated in 1% BSA for 1 hr; after that time, samples were incubated whit a dilution (2 μg/mL) of goat anti-human c-Met receptor antibody (SIGMA H9786) overnight at 4 °C. The next day samples were washed three times with PBS and by last were incubated with bovine anti-goat antibody coupled to CF 488A fluorophore (1:1000 SIGMA SAB4600233-2) for 90 min; the preparations were washed thrice with PBS and then incubated with a dilution of rabbit anti-Gal lectin antibody (1 μg/mL, the one made in our laboratory) overnight at 4 °C. The next day samples were washed three times with PBS and then were incubated with goat anti-rabbit antibody coupled to rhodamine fluorophore (1:2000 Jackson Immuno Research 111-295-144) for 90 min. Subsequently, three washes were carried with PBS, and coverslips were mounted on slides with fluorescence mounting medium. The next day sealant was placed around the coverslips. The confocal micrographs were obtained by fluorescence confocal microscopy Olympus FV1000 Upright BX61WI at the National Laboratory for Advanced Microscopy, Biotechnology Institute from UNAM and were processed with “FV Viewer 4.2b software (Copyright © 2003-2016 OLYMPUS CORPORATION, Allentown, PA, USA).
4. Discussion
Entamoeba histolytica has a potent cytotoxic activity on diverse cell types. Even though the mechanism by which the parasite induces killing has not been completely elucidated, one thing is for certain: adherence of the parasite is required, and adherence is achieved by the Gal lectin, as the presence of an excess of galactose prevents adherence and target killing [
12,
13]. In addition to mediating adherence, Gal lectin may also participate in the killing. Maybe the best proof that such is the case resulted from the fact that a monoclonal anti-Gal lectin antibody blocked cytotoxicity without blocking adherence [
14]. On the other hand, the reported identity of the sequence of the heavy subunit of Gal lectin with the HGF [
2], which could be fortuitous, was suggestive for a parasite with a special tropism for the liver. The aim of this work was to analyze the possible interaction of the amoebic trophozoite’s Gal lectin with the c-Met receptor in the surface of cells derived from the human liver.
The recognition of the heavy subunit of the Gal lectin by the anti-HGF antibodies from all the proteins present in the lysate of a pellet from E. histolytica trophozoites indicated that although the reported sequence identity was slight, it was enough to be recognized. The fact that our anti-Gal lectin did not recognize recombinant HGF may be explained because these antibodies are polyclonal and were not purified with an affinity method as were those anti-HGF antibodies.
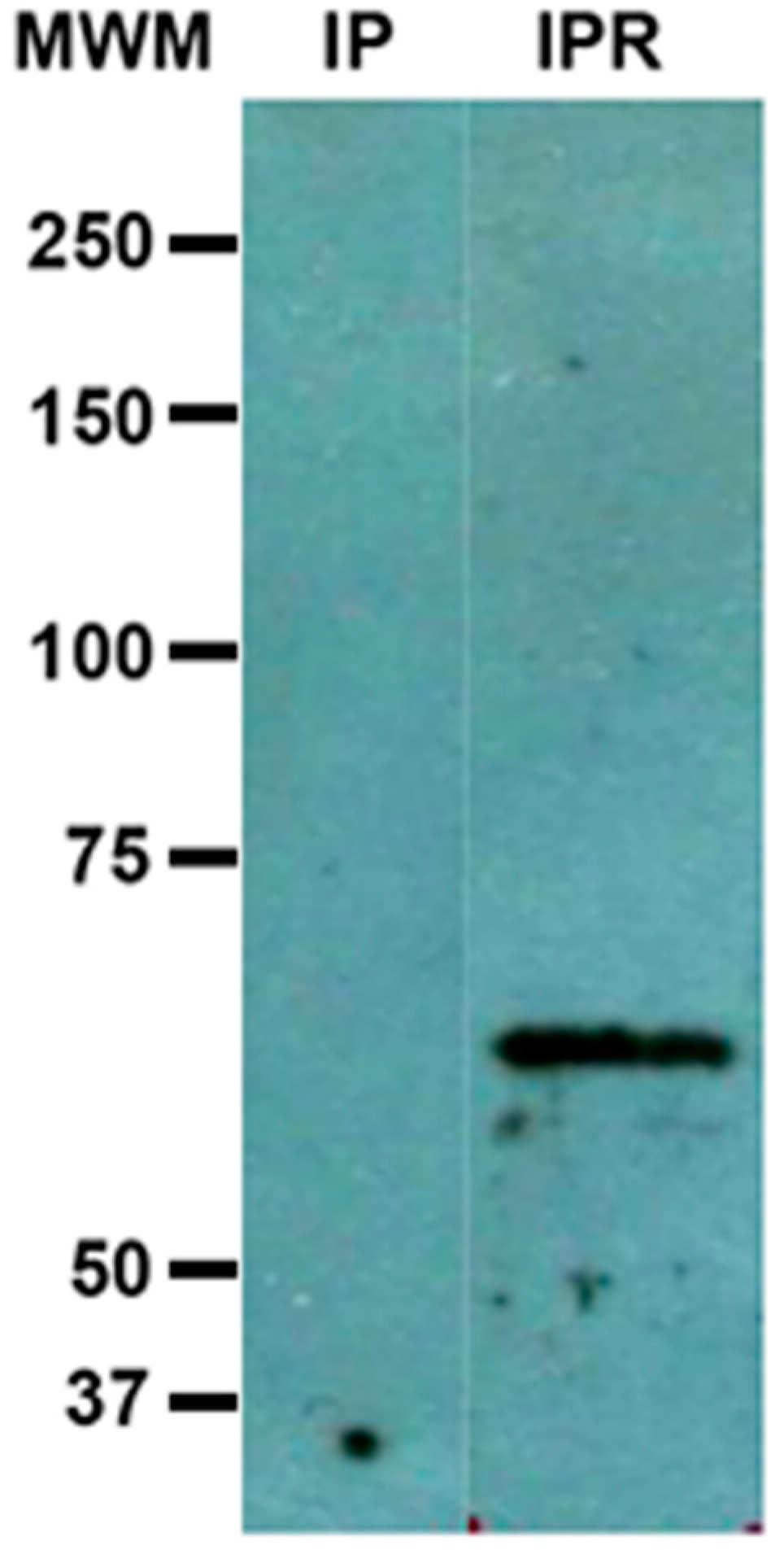
Results from immunoprecipitation with anti-c-Met antibodies revealed under re-duced conditions a 60 kDa peptide identified by anti-Gal lectin antibodies, which may be actually part of the Gal lectin. We do not consider that this result is a cross-reaction of any of the antibodies used in the test. As we mentioned above in
Section 3.3, it has happened to us, when working with pure samples of Gal lectin (obtained as described in [
7]), that the band of 260 kDa, originally present and recognized by anti-Gal antibody, is no longer apparent when the sample is treated with a reducing agent, but a new 60 kDa band ap-pears that is recognized by the same antibody. In those cases, the initial sample comes ex-clusively from a total amoebic lysate, and therefore cannot be attributed to a cross-recognition of a protein from another cell type, for example from HepG2 cells. how-ever, it would be necessary to analyze our “pure” sample by means of two-dimensional electrophoresis, to have a more complete image of the sample components, we are currently working on it. A plausible explanation to this result would be once bound to its ligand (probably c-Met) the action of the rhomboid protease [
15] on the Gal lectin generates a peptide with a smaller molecular weight.
As previously mentioned, it was difficult to discern if the Gal lectin participates directly in the cytotoxicity of the parasite or is the first step in target cell killing. We assayed a microscopic study of cocultures of trophozoites + HepG2 cells; as expected, both adherence and cytotoxicity of the parasite on HepG2 cells can be prevented by pretreatment of amoebas with galactose; however, pretreatment of HepG2 cells with HGF inhibited cytotoxicity but not adherence of amoebas. These results seem to indicate that amoebas need contact c-Met for cytotoxicity. Therefore, amoebic Gal lectin could be participating in adherence and cytotoxicity.
Finally, by means of confocal fluorescence microscopy, we demonstrated that in cocultures of E. histolytica trophozoites and HepG2, Gal lectin colocalizes with c-Met, supporting the idea that this pair of molecules interact with each other.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}