
Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency


Abstract
:1. Introduction
2. Materials and Methods
3. Results
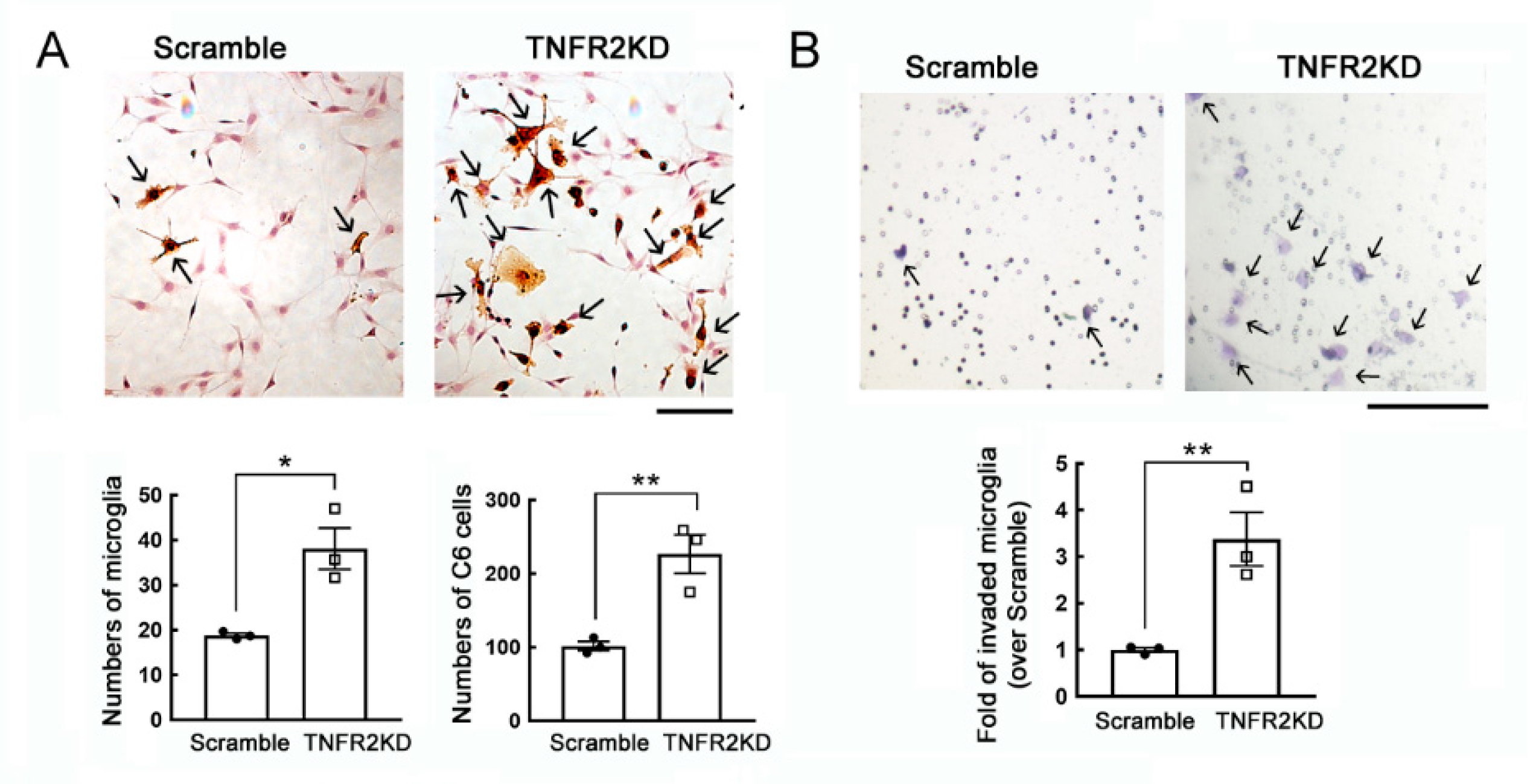
3.1. Growth and Migration of Microglia Increased by TNFR2 Gene Knockdown
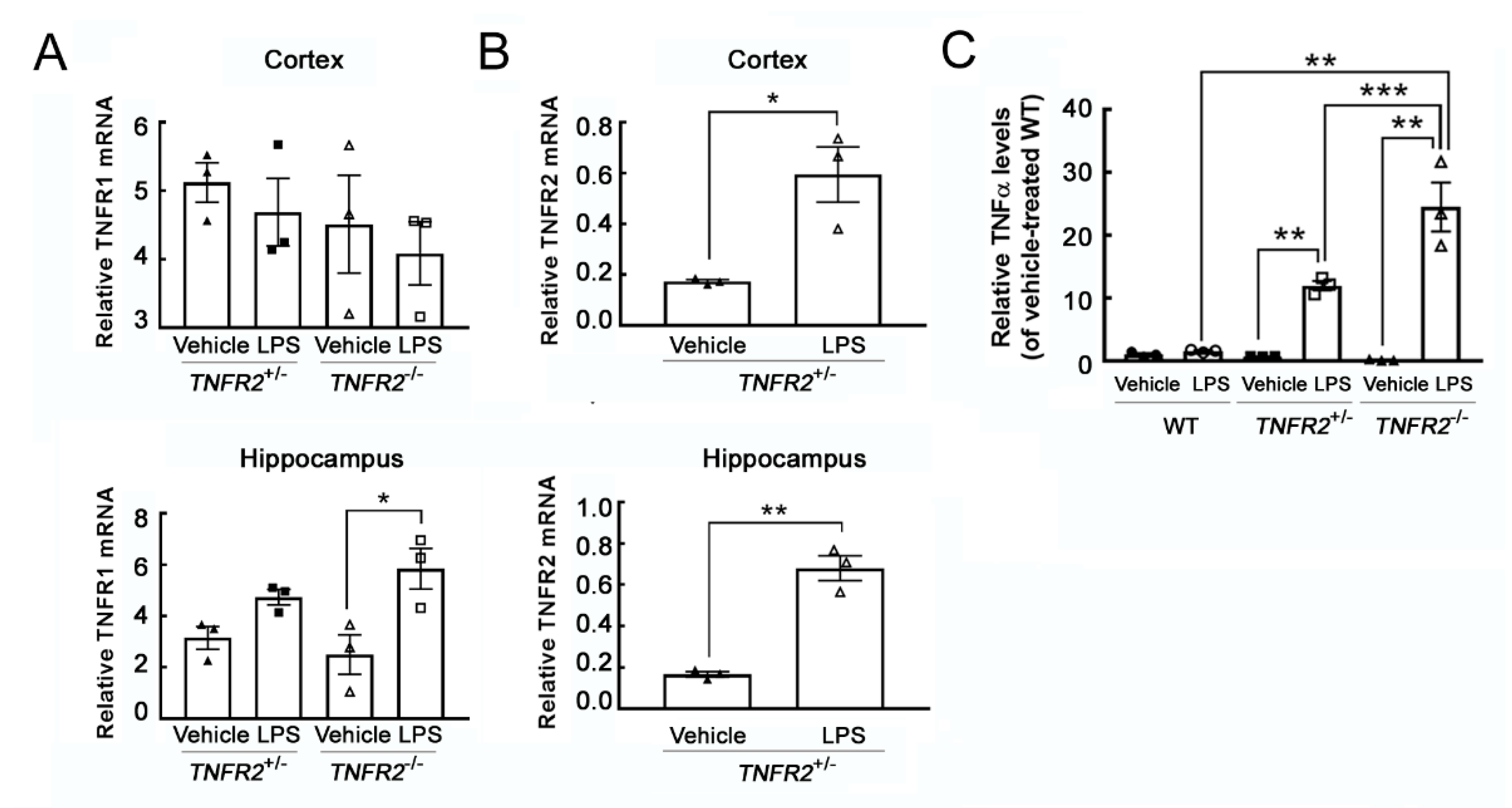
3.2. Enhanced Microglial Activation in TNFR2-Deficient Mouse Brain by Peripheral LPS Administration
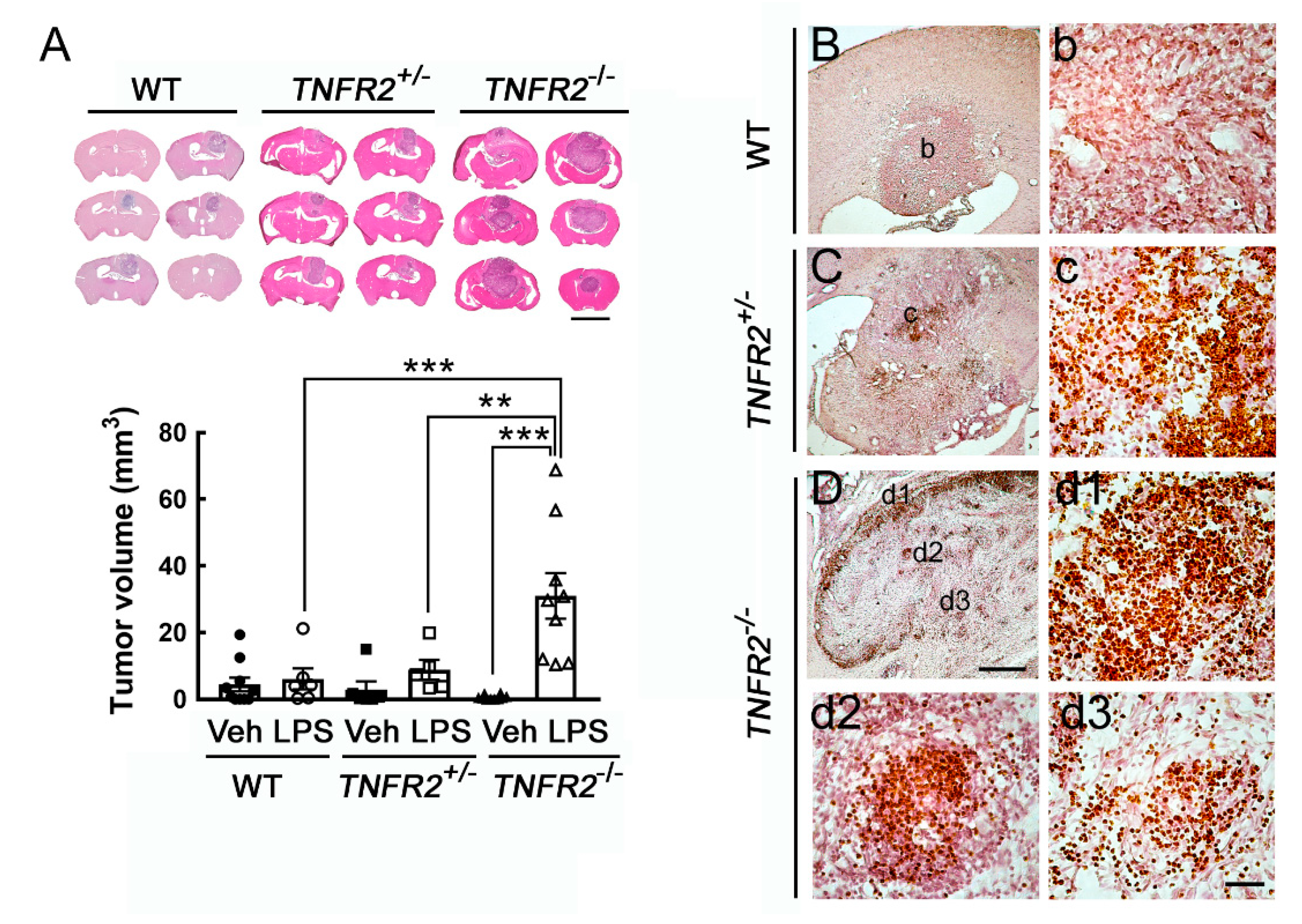
3.3. Progressive Growth of Rodent Glioma Cells in TNFR2 Deficient Mice Receiving Peripheral LPS Administration
3.4. Progressive Growth of Mouse GL261 Glioma Cells in TNFR2 Deficient Mice Receiving LPS Pretreatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watters, J.J.; Schartner, J.M.; Badie, B. Microglia function in brain tumors. J. Neurosci. Res. 2005, 81, 447–455. [Google Scholar] [CrossRef]
- Walker, D.; Hamilton, W.; Walter, F.M.; Watts, C. Strategies to accelerate diagnosis of primary brain tumors at the primary-secondary care interface in children and adults. CNS Oncol. 2013, 2, 447–462. [Google Scholar] [CrossRef]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225, discussion 226–219. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Wei, J.; Chen, P.; Gupta, P.; Ott, M.; Zamler, D.; Kassab, C.; Bhat, K.P.; Curran, M.A.; de Groot, J.F.; Heimberger, A.B. Immune biology of glioma-associated macrophages and microglia: Functional and therapeutic implications. Neuro-Oncology 2020, 22, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011, 59, 472–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwa, M.; Markovic, D.; Gabrusiewicz, K.; Synowitz, M.; Glass, R.; Zawadzka, M.; Wesolowska, A.; Kettenmann, H.; Kaminska, B. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain 2007, 130, 476–489. [Google Scholar] [CrossRef]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellert-Miklaszewska, A.; Dabrowski, M.; Lipko, M.; Sliwa, M.; Maleszewska, M.; Kaminska, B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia 2013, 61, 1178–1190. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Ogden, A.T.; Horgan, D.; Waziri, A.; Anderson, D.; Louca, J.; McKhann, G.M.; Sisti, M.B.; Parsa, A.T.; Bruce, J.N. Defective receptor expression and dendritic cell differentiation of monocytes in glioblastomas. Neurosurgery 2006, 59, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.M.; Yang, C.S.; Lin, T.C.; Chan, T.C.; Tzeng, S.F. Induced interleukin-33 expression enhances the tumorigenic activity of rat glioma cells. Neuro-Oncology 2014, 16, 552–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Hsieh, Y.T.; Fang, K.M.; Yang, C.S.; Tzeng, S.F. Reduction of CD200 expression in glioma cells enhances microglia activation and tumor growth. J. Neurosci. Res. 2016, 94, 1460–1471. [Google Scholar] [CrossRef]
- Wang, L.W.; Chang, Y.C.; Chen, S.J.; Tseng, C.H.; Tu, Y.F.; Liao, N.S.; Huang, C.C.; Ho, C.J. TNFR1-JNK signaling is the shared pathway of neuroinflammation and neurovascular damage after LPS-sensitized hypoxic-ischemic injury in the immature brain. J. Neuroinflamm. 2014, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Fang, K.M.; Wang, Y.L.; Huang, M.C.; Sun, S.H.; Cheng, H.; Tzeng, S.F. Expression of macrophage inflammatory protein-1alpha and monocyte chemoattractant protein-1 in glioma-infiltrating microglia: Involvement of ATP and P2X receptor. J. Neurosci. Res. 2011, 89, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.M.; Yang, C.S.; Sun, S.H.; Tzeng, S.F. Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action. J. Neurochem. 2009, 111, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Morrone, F.B.; Oliveira, D.L.; Gamermann, P.; Stella, J.; Wofchuk, S.; Wink, M.R.; Meurer, L.; Edelweiss, M.I.; Lenz, G.; Battastini, A.M. In vivo glioblastoma growth is reduced by apyrase activity in a rat glioma model. BMC Cancer 2006, 6, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.T.; Lin, C.; Hsu, C.T.; Wang, T.F.; Ke, F.Y.; Kuo, Y.M. Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Struct. Funct. 2012, 218, 1051–1060. [Google Scholar] [CrossRef]
- Nadeau, S.; Rivest, S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: A view from the blood-brain barrier. Neuroscience 1999, 93, 1449–1464. [Google Scholar] [CrossRef]
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2089–2102. [Google Scholar] [CrossRef] [Green Version]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Hasler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Aderka, D.; Sorkine, P.; Abu-Abid, S.; Lev, D.; Setton, A.; Cope, A.P.; Wallach, D.; Klausner, J. Shedding kinetics of soluble tumor necrosis factor (TNF) receptors after systemic TNF leaking during isolated limb perfusion. Relevance to the pathophysiology of septic shock. J. Clin. Investig. 1998, 101, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, S.L.; de Sauvage, F.J.; Kikly, K.; Carver-Moore, K.; Pitts-Meek, S.; Gillett, N.; Sheehan, K.C.; Schreiber, R.D.; Goeddel, D.V.; Moore, M.W. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994, 372, 560–563. [Google Scholar] [CrossRef]
- van Mierlo, G.J.; Scherer, H.U.; Hameetman, M.; Morgan, M.E.; Flierman, R.; Huizinga, T.W.; Toes, R.E. Cutting edge: TNFR-shedding by CD4+CD25+ regulatory T cells inhibits the induction of inflammatory mediators. J. Immunol. 2008, 180, 2747–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aderka, D.; Engelmann, H.; Maor, Y.; Brakebusch, C.; Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 1992, 175, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Raymond, D.P.; Crabtree, T.D.; Pelletier, S.J.; Houlgrave, C.W.; Pruett, T.L.; Sawyer, R.G. Enhanced murine macrophage TNF receptor shedding by cytosine-guanine sequences in oligodeoxynucleotides. J. Immunol. 2000, 165, 5153–5160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, A.; Evans, T.J.; Buurman, W.A.; Bemelmans, M.H.; Moyes, D.; Cohen, J. Differences in the shedding of soluble TNF receptors between endotoxin-sensitive and endotoxin-resistant mice in response to lipopolysaccharide or live bacterial challenge. J. Immunol. Immunol. 1995, 155, 2005–2012. [Google Scholar]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Arico, E.; Aloisi, F.; Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roggendorf, W.; Strupp, S.; Paulus, W. Distribution and characterization of microglia/macrophages in human brain tumors. Acta Neuropathol. 1996, 92, 288–293. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Sonabend, A.M.; Han, Y.; Lesniak, M.S. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia 2006, 54, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Ginzkey, C.; Eicker, S.O.; Marget, M.; Krause, J.; Brecht, S.; Westphal, M.; Hugo, H.H.; Mehdorn, H.M.; Steinmann, J.; Hamel, W. Increase in tumor size following intratumoral injection of immunostimulatory CpG-containing oligonucleotides in a rat glioma model. Cancer Immunol. Immunol. Immunother 2010, 59, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.Y.; Wong, M.P.; Chung, L.P.; Chan, A.S.; Yuen, S.T. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol. 1997, 93, 518–527. [Google Scholar] [CrossRef]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef]
- Bechmann, I.; Goldmann, J.; Kovac, A.D.; Kwidzinski, E.; Simburger, E.; Naftolin, F.; Dirnagl, U.; Nitsch, R.; Priller, J. Circulating monocytic cells infiltrate layers of anterograde axonal degeneration where they transform into microglia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 647–649. [Google Scholar] [CrossRef]
- Britschgi, M.; Wyss-Coray, T. Immune cells may fend off Alzheimer disease. Nat. Med. 2007, 13, 408–409. [Google Scholar] [CrossRef]
- Desbaillets, I.; Tada, M.; de Tribolet, N.; Diserens, A.C.; Hamou, M.F.; Van Meir, E.G. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int. J. Cancer. J. Int. Cancer 1994, 58, 240–247. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′->3′) | Reverse (5′->3′) |
|---|---|---|
| TNFR2 allele (t) | AGCTCCAGGCACAAGGGCGGG | CCTCTCATGCTGTCCCGGAA |
| neomycin-resistant gene gene (neoR) | CACGGGTAGCCAACGCTATGTC | GCCCTGAATGAACTGCAGGACG |
| CyPA | CGTCTGCTTCGAGCTGTTTG | GTAAAATGCCCGCAAGTCAA |
| TNFα | ACCGTCAGCCGATTTGCTAT | CCGGACTCCGCAAAGTCTAA |
| TNFR1 | GCTTGTGTCCCCAAGGAAAG | GTCCTGGGGGTTTGTGACAT |
| TNFR2 | GTCGCGCTGGTCTTCGAACTG | GGTATACATGCTTGCCTCACAGTC |
| shRNA | Sequence | |
| sh-ctrl (Scramble) | AAAAGCTACACTATCGAGCAATTTTGGATCCAAAATTGCTCGATAGTGTAGC | |
| sh-mTNFR2-188 | AAAAGCCAGATCTCACAGGAATATTGGATCCAATATTCCTGTGAGATCTGGC | |
| sh-mTNFR2-220 | AAAAGCTCAGATGTGCTGTGCTATTGGATCCAATAGCACAGCACATCTGAGC | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, C.-K.; Fang, K.-M.; Huang, H.-T.; Chang, W.-R.; Chuang, C.-C.; Tzeng, S.-F. Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency. Life 2021, 11, 961. https://doi.org/10.3390/life11090961
Liao C-K, Fang K-M, Huang H-T, Chang W-R, Chuang C-C, Tzeng S-F. Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency. Life. 2021; 11(9):961. https://doi.org/10.3390/life11090961
Chicago/Turabian StyleLiao, Chih-Kai, Kuan-Min Fang, Hui-Ting Huang, Wen-Ruei Chang, Chao-Chi Chuang, and Shun-Fen Tzeng. 2021. "Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency" Life 11, no. 9: 961. https://doi.org/10.3390/life11090961
APA StyleLiao, C.-K., Fang, K.-M., Huang, H.-T., Chang, W.-R., Chuang, C.-C., & Tzeng, S.-F. (2021). Enhanced Microglia Activation and Glioma Tumor Progression by Inflammagen Priming in Mice with Tumor Necrosis Factor Receptor Type 2 Deficiency. Life, 11(9), 961. https://doi.org/10.3390/life11090961