
Loss of Corticostriatal Mu-Opioid Receptors in α-Synuclein Transgenic Mouse Brains



Abstract
:1. Introduction
2. Materials and Methods
3. Results
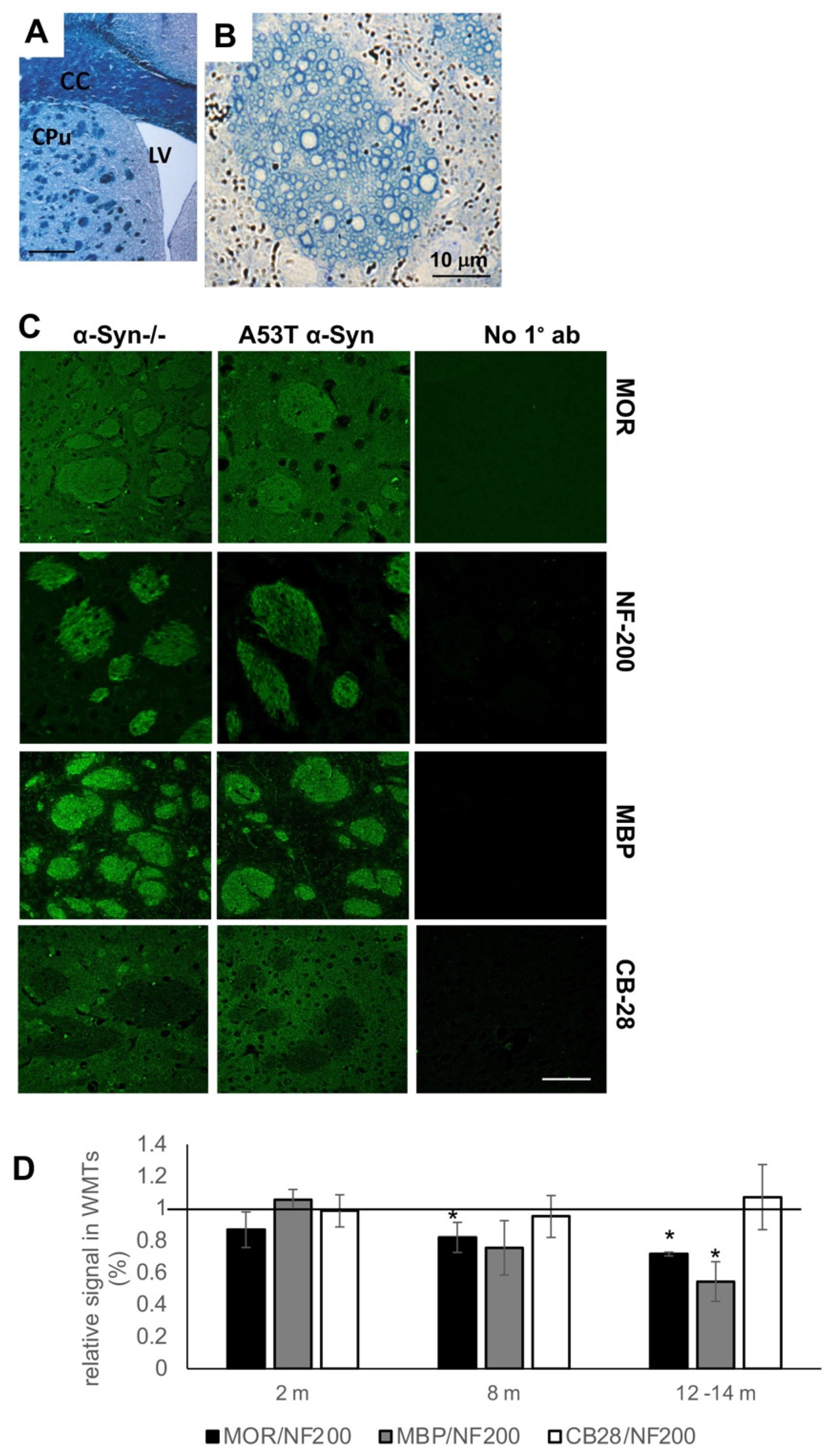
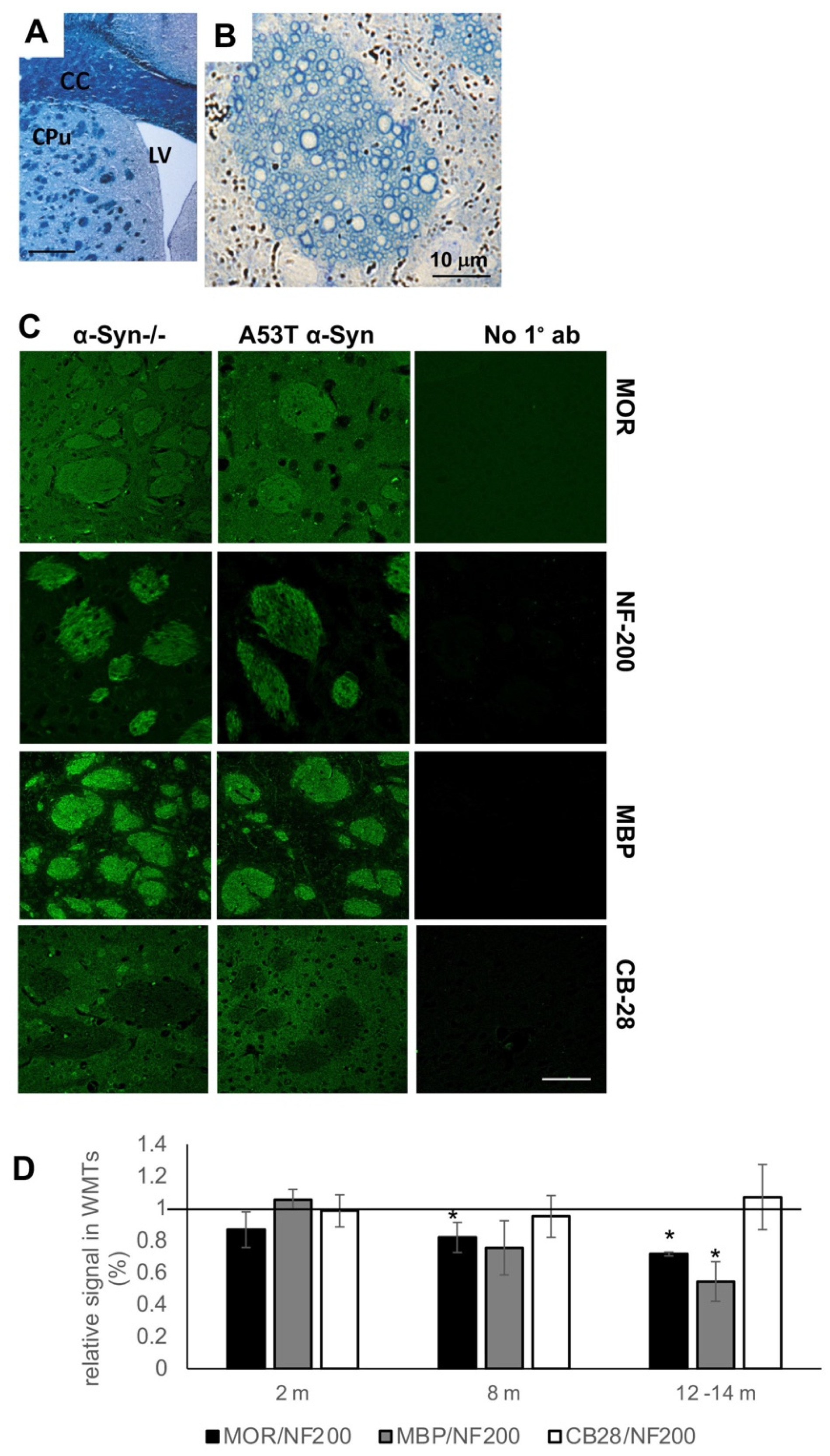
3.1. Loss of MOR Immunoreactivity within Corticostriatal WMTs of A53T α-Syn tg Mouse Brains
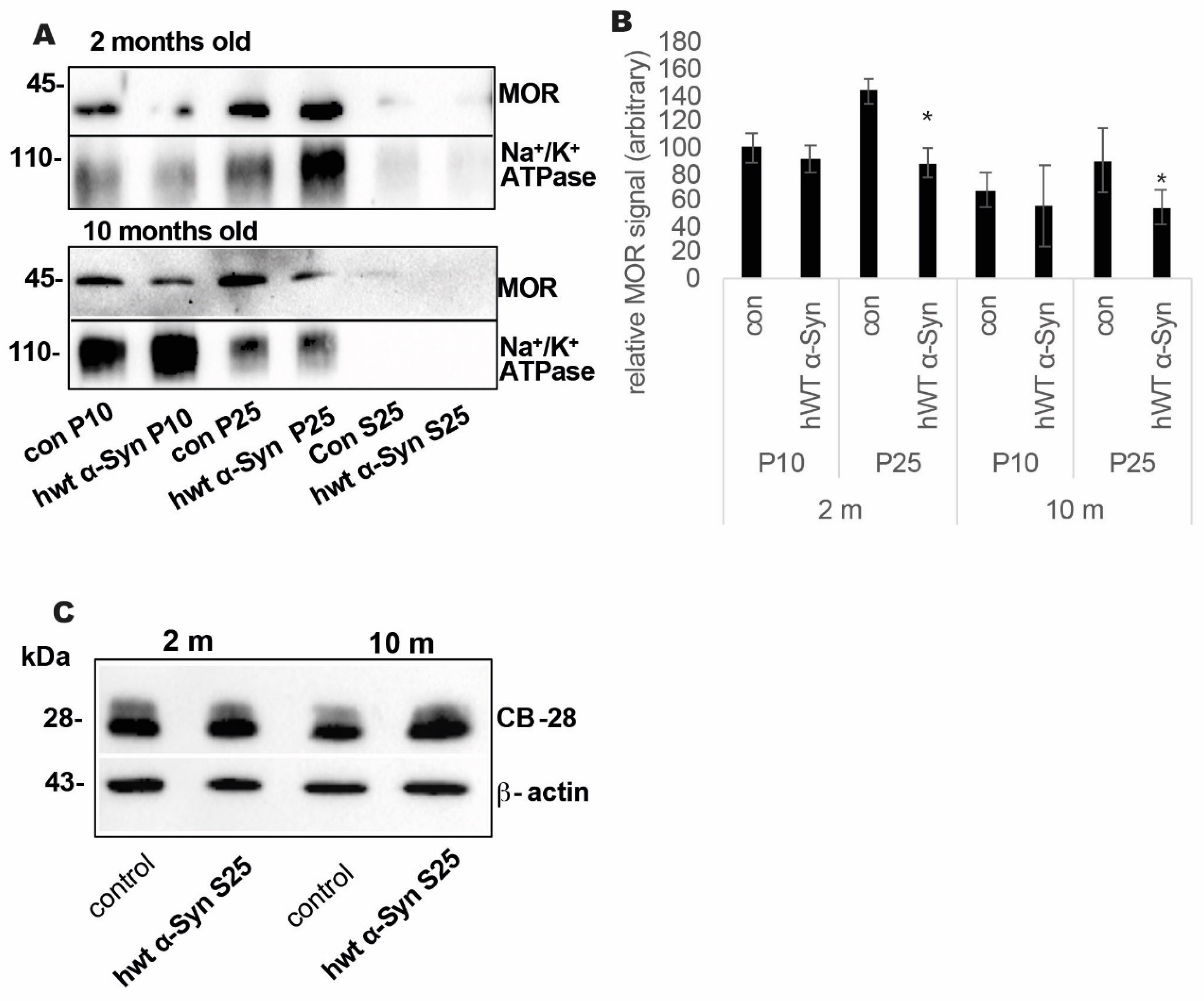
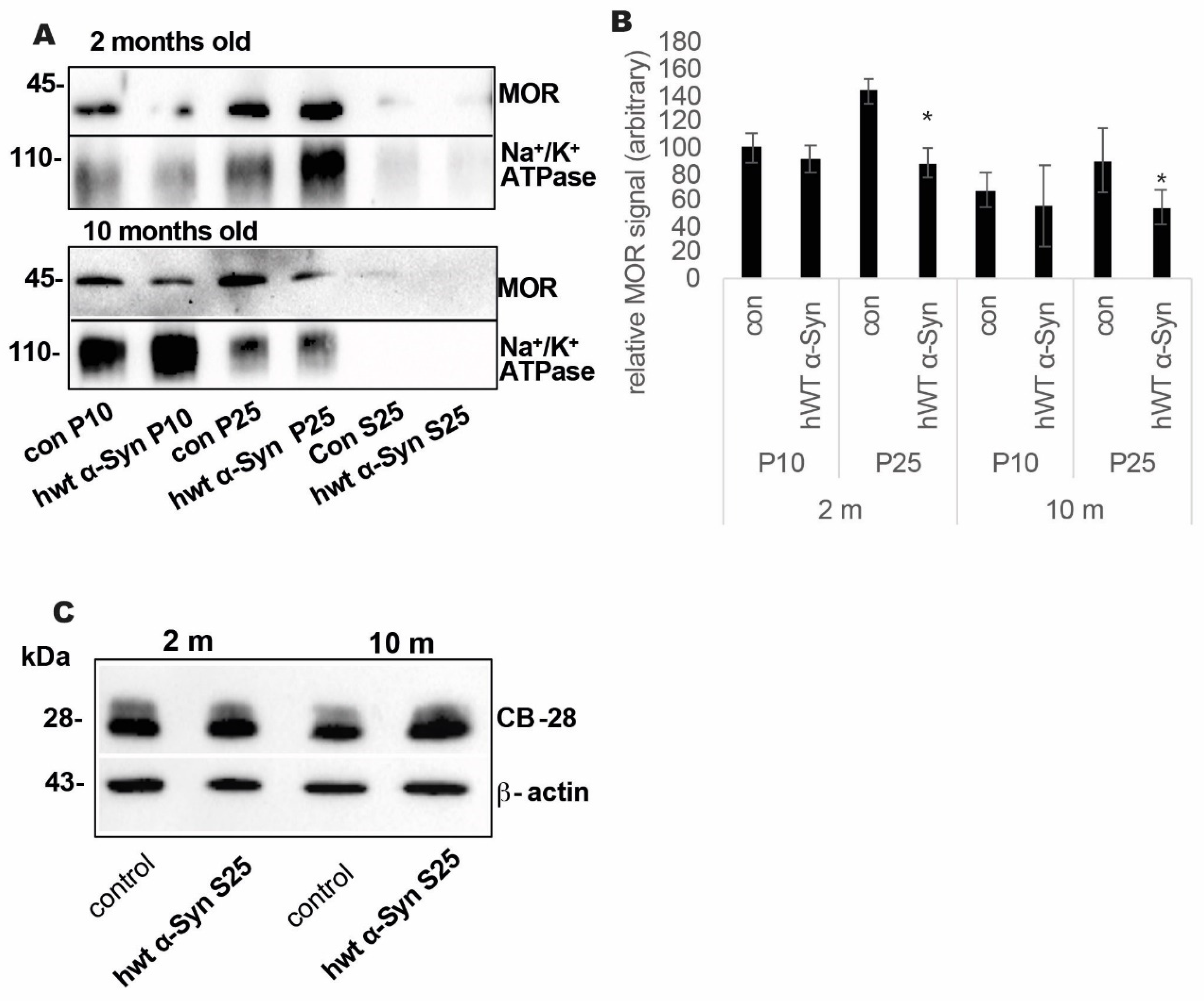
3.2. Loss of MOR Protein Expression Levels in the Striatum of Thy-1 hWT α-Syn Mouse Brains
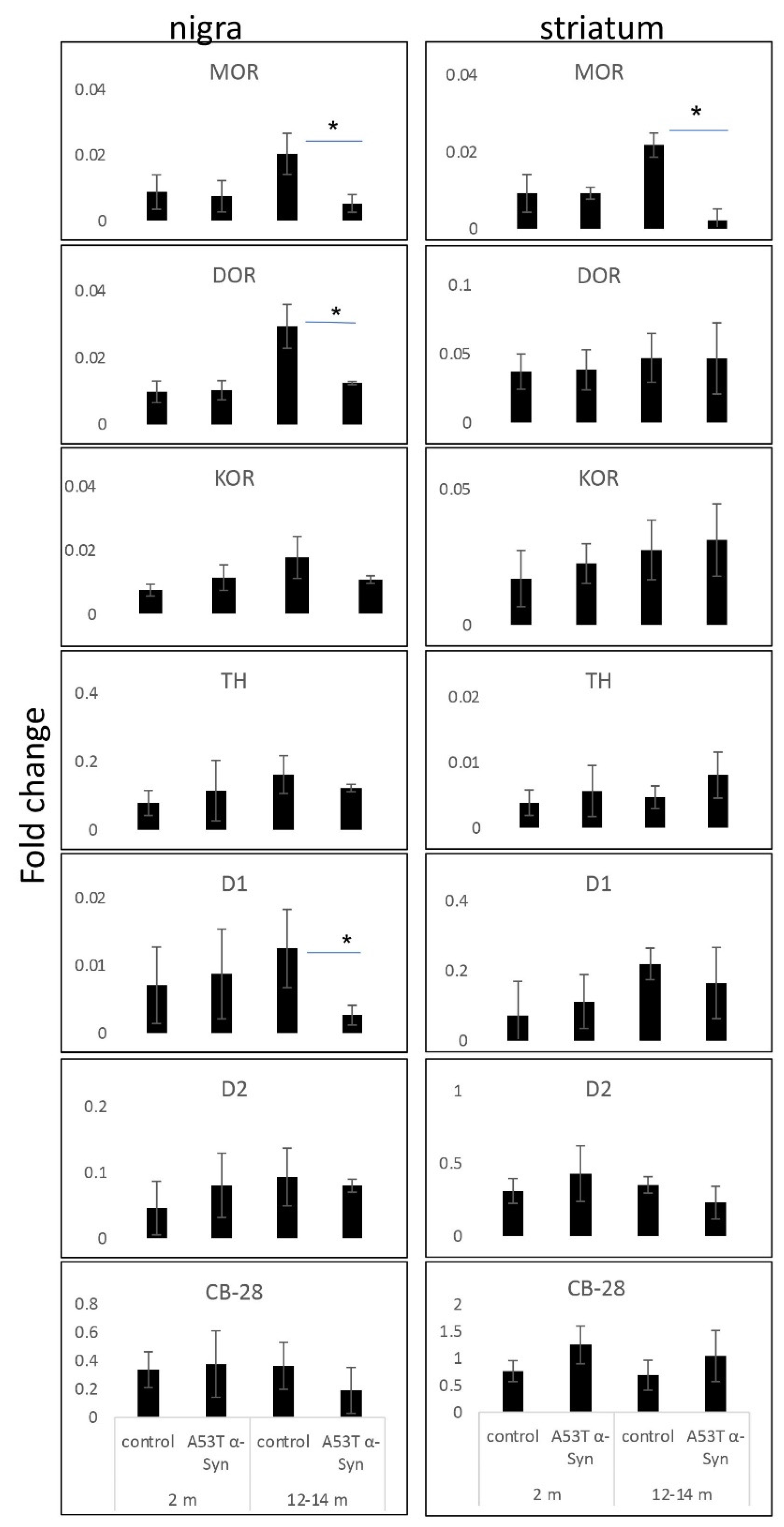
3.3. Loss of MOR Signals Occur at the mRNA Level
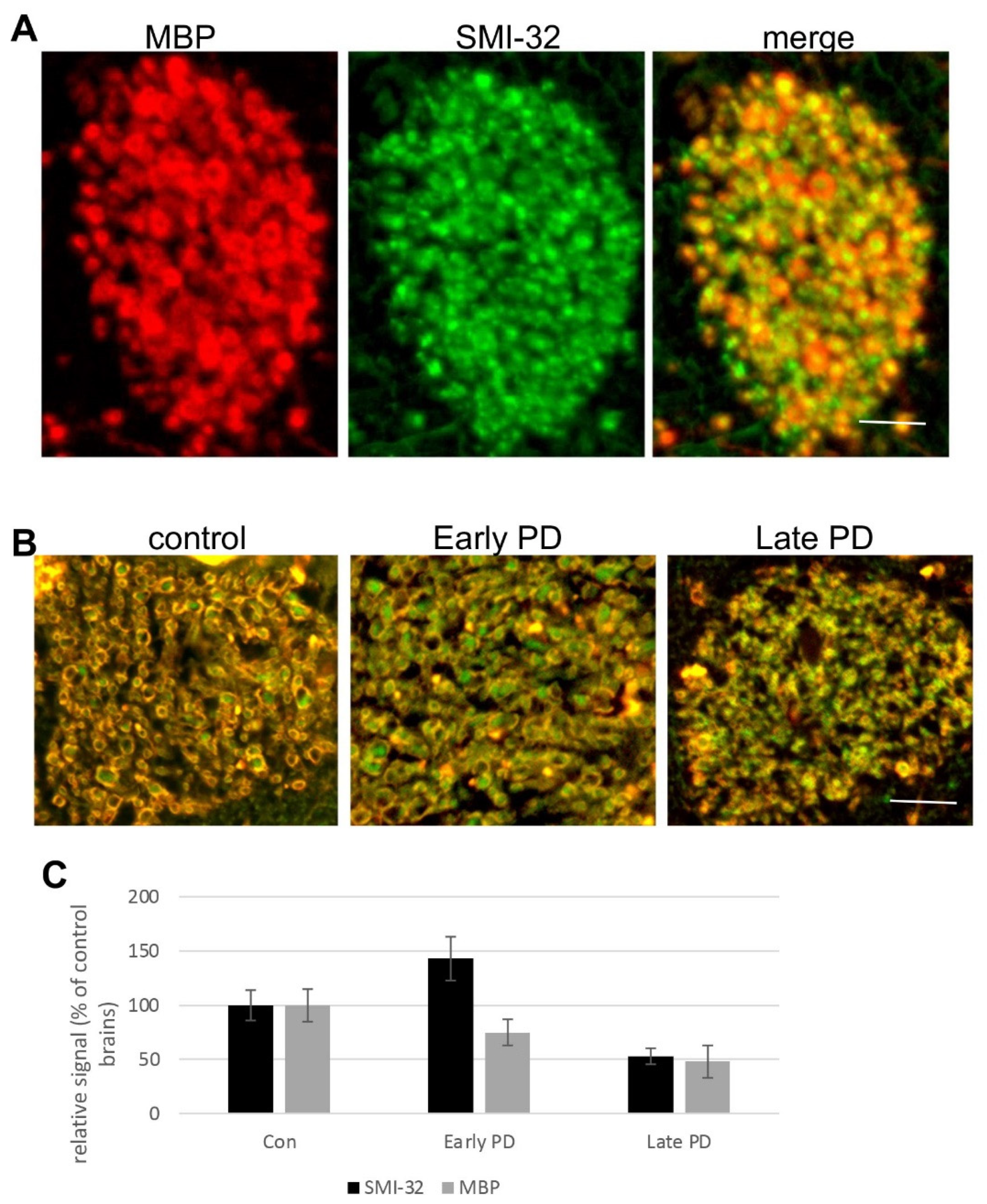
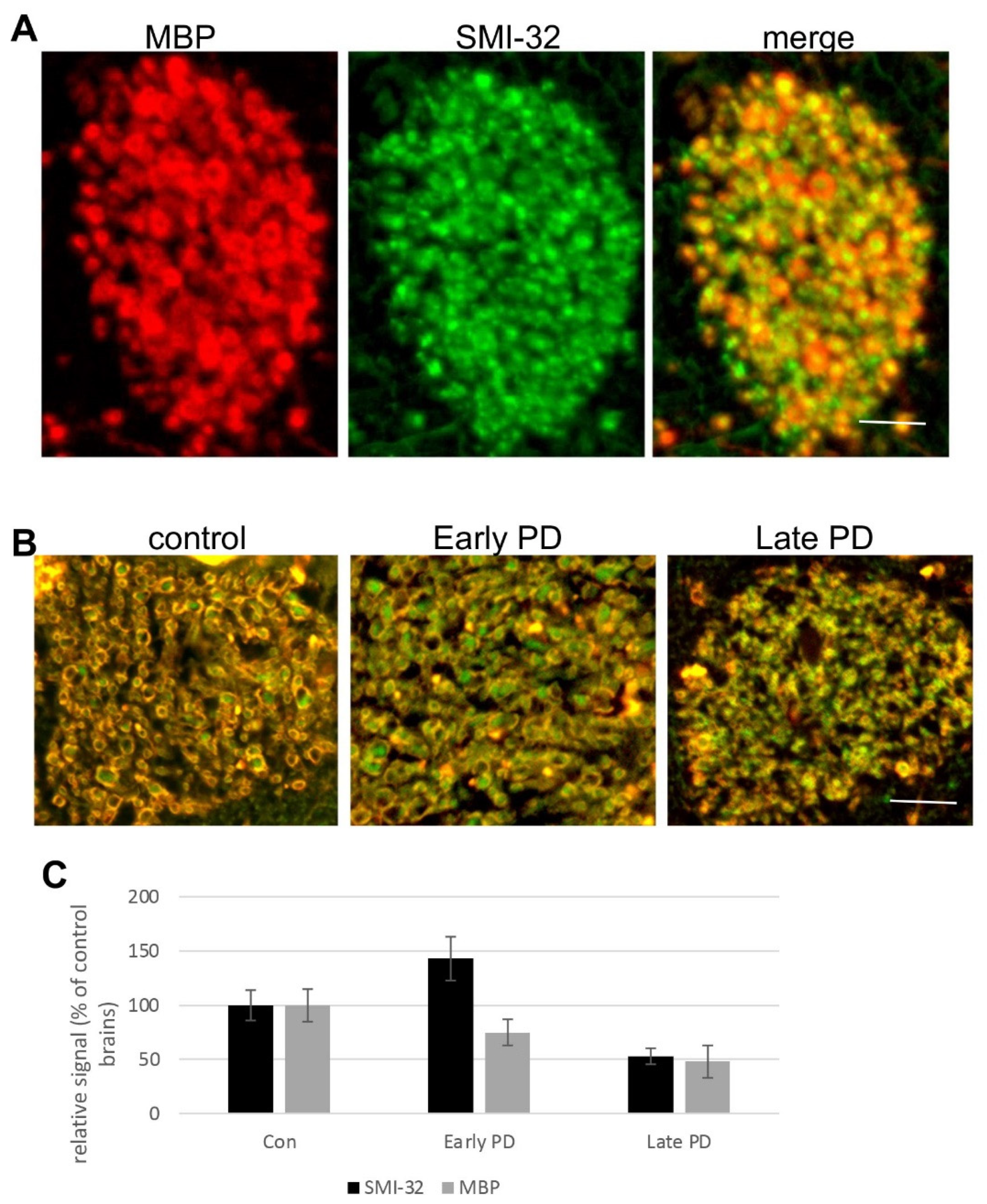
3.4. Degeneration of Corticostriatal WMT in Human Brains with PD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huston, J.P. Hand Book of Basal Ganglia Structure and Function, 2nd ed.; Steiner, H., Tseng, K.Y., Eds.; Elsevier: Cambridge, MA, USA, 2016; Volume 24, pp. 3–32. [Google Scholar] [CrossRef]
- Haber, S.N. Corticostriatal circuitry. Dialogues Clin. Neurosci. 2016, 18, 7–21. [Google Scholar]
- Arkadir, D.; Bergman, H.; Fahn, S. Redundant dopaminergic activity may enable compensatory axonal sprouting in Parkinson disease. Neurology 2014, 82, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Villalba, R.M.; Smith, Y. Loss and remodeling of striatal dendritic spines in Parkinson’s disease: From homeostasis to maladaptive plasticity? J. Neural Transm. 2018, 125, 431–447. [Google Scholar] [CrossRef]
- Sgroi, S.; Tonini, R. Opioidergic Modulation of Striatal Circuits, Implications in Parkinson’s Disease and Levodopa Induced Dyskinesia. Front Neurol. 2018, 9, 524. [Google Scholar] [CrossRef]
- Puryear, C.B.; Brooks, J.; Tan, L.; Smith, K.; Li, Y.; Cunningham, J.; Todtenkopf, M.S.; Dean, R.L.; Sanchez, C. Opioid receptor modulation of neural circuits in depression: What can be learned from preclinical data? Neurosci. Biobehav. Rev. 2020, 108, 658–678. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Bezard, E.; Eleopra, R.; Zaveri, N.T.; Morari, M. Managing Parkinson’s disease: Moving ON with NOP. Br. J. Pharm. 2020, 177, 28–47. [Google Scholar] [CrossRef] [Green Version]
- Bezard, E.; Gross, C.E.; Brotchie, J.M. Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci. 2003, 26, 215–221. [Google Scholar] [CrossRef]
- Samadi, P.; Bedard, P.J.; Rouillard, C. Opioids and motor complications in Parkinson’s disease. Trends Pharm. Sci. 2006, 27, 512–517. [Google Scholar] [CrossRef]
- Toll, L.; Bruchas, M.R.; Calo, G.; Cox, B.M.; Zaveri, N.T. Nociceptin/Orphanin FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharm. Rev. 2016, 68, 419–457. [Google Scholar] [CrossRef]
- Hsu, D.T.; Sanford, B.J.; Meyers, K.K.; Love, T.M.; Hazlett, K.E.; Walker, S.J.; Mickey, B.J.; Koeppe, R.A.; Langenecker, S.A.; Zubieta, J.K. It still hurts: Altered endogenous opioid activity in the brain during social rejection and acceptance in major depressive disorder. Mol. Psychiatry 2015, 20, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.H.; Myers, J.; Reis Marques, T.; Rabiner, E.A.; Howes, O.D. Reduced mu opioid receptor availability in schizophrenia revealed with [(11)C]-carfentanil positron emission tomographic Imaging. Nat. Commun. 2019, 10, 4493. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Ostlund, S.B.; James, A.S.; Park, C.S.; Ge, W.; Roberts, K.W.; Mittal, N.; Murphy, N.P.; Cepeda, C.; Kieffer, B.L.; et al. Targeted expression of μ-opioid receptors in a subset of striatal direct-pathway neurons restores opiate reward. Nat. Neurosci. 2014, 17, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Brimblecombe, K.R.; Cragg, S.J. The Striosome and Matrix Compartments of the Striatum: A Path through the Labyrinth from Neurochemistry toward Function. ACS Chem. Neurosci. 2017, 8, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Moriwaki, A.; Wang, J.B.; Uhl, G.R.; Pickel, V.M. Ultrastructural immunocytochemical localization of mu opioid receptors and Leu5-enkephalin in the patch compartment of the rat caudate-putamen nucleus. J. Comp. Neurol. 1996, 375, 659–674. [Google Scholar] [CrossRef]
- Wang, H.; Moriwaki, A.; Wang, J.B.; Uhl, G.R.; Pickel, V.M. Ultrastructural immunocytochemical localization of mu-opioid receptors in dendritic targets of dopaminergic terminals in the rat caudate-putamen nucleus. Neuroscience 1997, 81, 757–771. [Google Scholar] [CrossRef]
- Wang, H.; Pickel, V.M. Dendritic spines containing mu-opioid receptors in rat striatal patches receive asymmetric synapses from prefrontal corticostriatal afferents. J. Comp. Neurol. 1998, 396, 223–237. [Google Scholar] [CrossRef]
- Miura, M.; Masuda, M.; Aosaki, T. Roles of micro-opioid receptors in GABAergic synaptic transmission in the striosome and matrix compartments of the striatum. Mol. Neurobiol. 2008, 37, 104–115. [Google Scholar] [CrossRef]
- Wang, H.; Pickel, V.M. Preferential cytoplasmic localization of delta-opioid receptors in rat striatal patches: Comparison with plasmalemmal mu-opioid receptors. J. Neurosci. 2001, 21, 3242–3250. [Google Scholar] [CrossRef] [Green Version]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Crittenden, J.R.; Graybiel, A.M. Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat. 2011, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Eblen, F.; Graybiel, A.M. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J. Neurosci. 1995, 15, 5999–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, A.E.; Wilson, C.J. Corticostriatal innervation of the patch and matrix in the rat neostriatum. J. Comp. Neurol. 1996, 374, 578–592. [Google Scholar] [CrossRef]
- Kupferschmidt, D.A.; Cody, P.A.; Lovinger, D.M.; Davis, M.I. Brain BLAQ: Post-hoc thick-section histochemistry for localizing optogenetic constructs in neurons and their distal terminals. Front Neuroanat. 2015, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging. 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef] [Green Version]
- Schechter, M.; Grigoletto, J.; Abd-Elhadi, S.; Glickstein, H.; Friedman, A.; Serrano, G.E.; Beach, T.G.; Sharon, R. A role for alpha-Synuclein in axon growth and its implications in corticostriatal glutamatergic plasticity in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Grigoletto, J.; Pukass, K.; Gamliel, A.; Davidi, D.; Katz-Brull, R.; Richter-Landsberg, C.; Sharon, R. Higher levels of myelin phospholipids in brains of neuronal alpha-Synuclein transgenic mice precede myelin loss. Acta Neuropathol. Commun. 2017, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Specht, C.G.; Schoepfer, R. Deletion of the alpha-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci. 2001, 2, 11. [Google Scholar] [CrossRef]
- Yakunin, E.; Loeb, V.; Kisos, H.; Biala, Y.; Yehuda, S.; Yaari, Y.; Selkoe, D.J.; Sharon, R. Alpha-synuclein neuropathology is controlled by nuclear hormone receptors and enhanced by docosahexaenoic acid in a mouse model for Parkinson’s disease. Brain Pathol. 2012, 22, 280–294. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Mallory, M.; Hashimoto, M.; Song, D.; Shults, C.W.; Lang, I.; Masliah, E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002, 68, 568–578. [Google Scholar] [CrossRef]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Fleming, S.M.; Salcedo, J.; Fernagut, P.O.; Rockenstein, E.; Masliah, E.; Levine, M.S.; Chesselet, M.F. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J. Neurosci. 2004, 24, 9434–9440. [Google Scholar] [CrossRef] [Green Version]
- Chesselet, M.F.; Richter, F.; Zhu, C.; Magen, I.; Watson, M.B.; Subramaniam, S.R. A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 2012, 9, 297–314. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Standaert, D.G. Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J. Neurosci. 2001, 21, 5546–5558. [Google Scholar] [CrossRef] [Green Version]
- Voelker, C.C.; Garin, N.; Taylor, J.S.; Gahwiler, B.H.; Hornung, J.P.; Molnar, Z. Selective neurofilament (SMI-32, FNP-7 and N200) expression in subpopulations of layer V pyramidal neurons in vivo and in vitro. Cereb. Cortex 2004, 14, 1276–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, A.; Homma, D.; Bloem, B.; Gibb, L.G.; Amemori, K.I.; Hu, D.; Delcasso, S.; Truong, T.F.; Yang, J.; Hood, A.S.; et al. Chronic Stress Alters Striosome-Circuit Dynamics, Leading to Aberrant Decision-Making. Cell 2017, 171, 1191–1205.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, A.; Hueske, E.; Drammis, S.M.; Toro Arana, S.E.; Nelson, E.D.; Carter, C.W.; Delcasso, S.; Rodriguez, R.X.; Lutwak, H.; DiMarco, K.S.; et al. Striosomes Mediate Value-Based Learning Vulnerable in Age and a Huntington’s Disease Model. Cell 2020, 183, 918–934.e49. [Google Scholar] [CrossRef] [PubMed]
- Piccini, P.; Weeks, R.A.; Brooks, D.J. Alterations in opioid receptor binding in Parkinson’s disease patients with levodopa-induced dyskinesias. Ann. Neurol. 1997, 42, 720–726. [Google Scholar] [CrossRef]
- Fernandez, A.; de Ceballos, M.L.; Jenner, P.; Marsden, C.D. Neurotensin, substance P, delta and mu opioid receptors are decreased in basal ganglia of Parkinson’s disease patients. Neuroscience 1994, 61, 73–79. [Google Scholar] [CrossRef]
- Gertler, T.S.; Chan, C.S.; Surmeier, D.J. Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 2008, 28, 10814–10824. [Google Scholar] [CrossRef] [PubMed]
- Seizinger, B.R.; Grimm, C.; Hollt, V.; Herz, A. Evidence for a selective processing of proenkephalin B into different opioid peptide forms in particular regions of rat brain and pituitary. J. Neurochem. 1984, 42, 447–457. [Google Scholar] [CrossRef]
- Breslin, M.B.; Lindberg, I.; Benjannet, S.; Mathis, J.P.; Lazure, C.; Seidah, N.G. Differential processing of proenkephalin by prohormone convertases 1(3) and 2 and furin. J. Biol. Chem. 1993, 268, 27084–27093. [Google Scholar] [CrossRef]
- Atwood, B.K.; Kupferschmidt, D.A.; Lovinger, D.M. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat. Neurosci. 2014, 17, 540–548. [Google Scholar] [CrossRef]
- Koprich, J.B.; Fox, S.H.; Johnston, T.H.; Goodman, A.; Le Bourdonnec, B.; Dolle, R.E.; DeHaven, R.N.; DeHaven-Hudkins, D.L.; Little, P.J.; Brotchie, J.M. The selective mu-opioid receptor antagonist ADL5510 reduces levodopa-induced dyskinesia without affecting antiparkinsonian action in MPTP-lesioned macaque model of Parkinson’s disease. Mov. Disord. 2011, 26, 1225–1233. [Google Scholar] [CrossRef]
- Hanrieder, J.; Ljungdahl, A.; Falth, M.; Mammo, S.E.; Bergquist, J.; Andersson, M. L-DOPA-induced dyskinesia is associated with regional increase of striatal dynorphin peptides as elucidated by imaging mass spectrometry. Mol. Cell Proteom. 2011, 10, M111.009308. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Cai, H. Opioid system in L-DOPA-induced dyskinesia. Transl. Neurodegener. 2017, 6, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosnell, B.A.; Levine, A.S. Reward systems and food intake: Role of opioids. Int. J. Obes. 2009, 33 (Suppl. 2), S54–S58. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, C.; Robbins, T.W.; Nathan, P.J.; Bullmore, E.T.; Everitt, B.J. Inhibition of opioid transmission at the μ-opioid receptor prevents both food seeking and binge-like eating. Neuropsychopharmacology 2012, 37, 2643–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peciña, S.; Smith, K.S. Hedonic and motivational roles of opioids in food reward: Implications for overeating disorders. Pharm. Biochem. Behav. 2010, 97, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Tuominen, L.; Tuulari, J.J.; Hirvonen, J.; Parkkola, R.; Helin, S.; Salminen, P.; Nuutila, P.; Nummenmaa, L. Obesity is associated with decreased μ-opioid but unaltered dopamine D2 receptor availability in the brain. J. Neurosci. 2015, 35, 3959–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, T.; Höllt, V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol. Ther. 2008, 117, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Whistler, J.L. Examining the role of mu opioid receptor endocytosis in the beneficial and side-effects of prolonged opioid use: From a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol. Depend 2012, 121, 189–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, A.; Reimold, M.; Wrase, J.; Hermann, D.; Croissant, B.; Mundle, G.; Dohmen, B.M.; Braus, D.F.; Schumann, G.; Machulla, H.J.; et al. Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: A positron emission tomography study using carbon 11-labeled carfentanil. Arch. Gen. Psychiatry 2005, 62, 57–64. [Google Scholar] [CrossRef]
- Weerts, E.M.; Wand, G.S.; Kuwabara, H.; Munro, C.A.; Dannals, R.F.; Hilton, J.; Frost, J.J.; McCaul, M.E. Positron emission tomography imaging of mu- and delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcohol. Clin. Exp. Res. 2011, 35, 2162–2173. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, D.A.; Kim, Y.K.; Bencherif, B.; Boyd, S.J.; Nelson, R.; Copersino, M.; Endres, C.J.; Dannals, R.F.; Frost, J.J. Imaging brain mu-opioid receptors in abstinent cocaine users: Time course and relation to cocaine craving. Biol. Psychiatry 2005, 57, 1573–1582. [Google Scholar] [CrossRef]
- Chen, T.; Wang, Q.; Chao, D.; Xia, T.C.; Sheng, S.; Li, Z.R.; Zhao, J.N.; Wen, G.Q.; Ding, G.; Xia, Y. δ-Opioid Receptor Activation Attenuates the Oligomer Formation Induced by Hypoxia and/or α-Synuclein Overexpression/Mutation Through Dual Signaling Pathways. Mol. Neurobiol. 2019, 56, 3463–3475. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Chao, D.; Sandhu, H.K.; Liao, X.; Zhao, J.; Wen, G.; Xia, Y. δ-Opioid receptor activation reduces α-synuclein overexpression and oligomer formation induced by MPP(+) and/or hypoxia. Exp. Neurol. 2014, 255, 127–136. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Prime Sequence (5′->3′) | Pre-Treatment Condition |
|---|---|---|
| 18S | Forward: 5′-GCCAGAACCTGGCTGTACTT-3′ Reverse: 5′-GAGCGAGTGATCACCATCAT-3′ | |
| MOR | Forward: 5′-GCCTTAGCCACTAGCACG-3′ Reverse: 5′-AACATTACGGGCAGACCA-3′ | DNase * |
| DOR | Forward: 5′-ATCGTCCGGTACACCAAATTGA-3′ Reverse: 5′-GTACTTGGCGCTCTGGAAGG-3′ | |
| KOR | Forward: 5′-GTTTGTCATCATCCGATACACGAA-3′ Reverse: 5′-GCATAGTGGTAGTAACCAAAGCATCT-3′ | |
| CD-28K | Forward: 5′-CGCTGACGGAAGTGGTTACC -3′ Reverse: 5′-TTCCGGTGATAGCTCCAATCC-3′ | |
| D1 | Forward: 5′-AACTGTATGGTGCCCTTCTGTGG -3′ Reverse: 5′-CAGCCCCGTTGTTGTTGATG-3′ | DNase * |
| D2 | Forward: 5′CATCAGCATCGACAGGTACACA-3′ Reverse: 5′-CAGTAACTCGGCGCTTGGA-3′ | |
| TH | Forward: 5′-AAATGCTGTTCTCAACCTG-3′ Reverse: 5′-GCTTCAAATGTCTCAAACAC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoletto, J.; Schechter, M.; Sharon, R. Loss of Corticostriatal Mu-Opioid Receptors in α-Synuclein Transgenic Mouse Brains. Life 2022, 12, 63. https://doi.org/10.3390/life12010063
Grigoletto J, Schechter M, Sharon R. Loss of Corticostriatal Mu-Opioid Receptors in α-Synuclein Transgenic Mouse Brains. Life. 2022; 12(1):63. https://doi.org/10.3390/life12010063
Chicago/Turabian StyleGrigoletto, Jessica, Meir Schechter, and Ronit Sharon. 2022. "Loss of Corticostriatal Mu-Opioid Receptors in α-Synuclein Transgenic Mouse Brains" Life 12, no. 1: 63. https://doi.org/10.3390/life12010063
APA StyleGrigoletto, J., Schechter, M., & Sharon, R. (2022). Loss of Corticostriatal Mu-Opioid Receptors in α-Synuclein Transgenic Mouse Brains. Life, 12(1), 63. https://doi.org/10.3390/life12010063