


Three-Country Snapshot of Ornithine Transcarbamylase Deficiency

 ,
,  , , , , , , , , ,
, , , , , , , , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
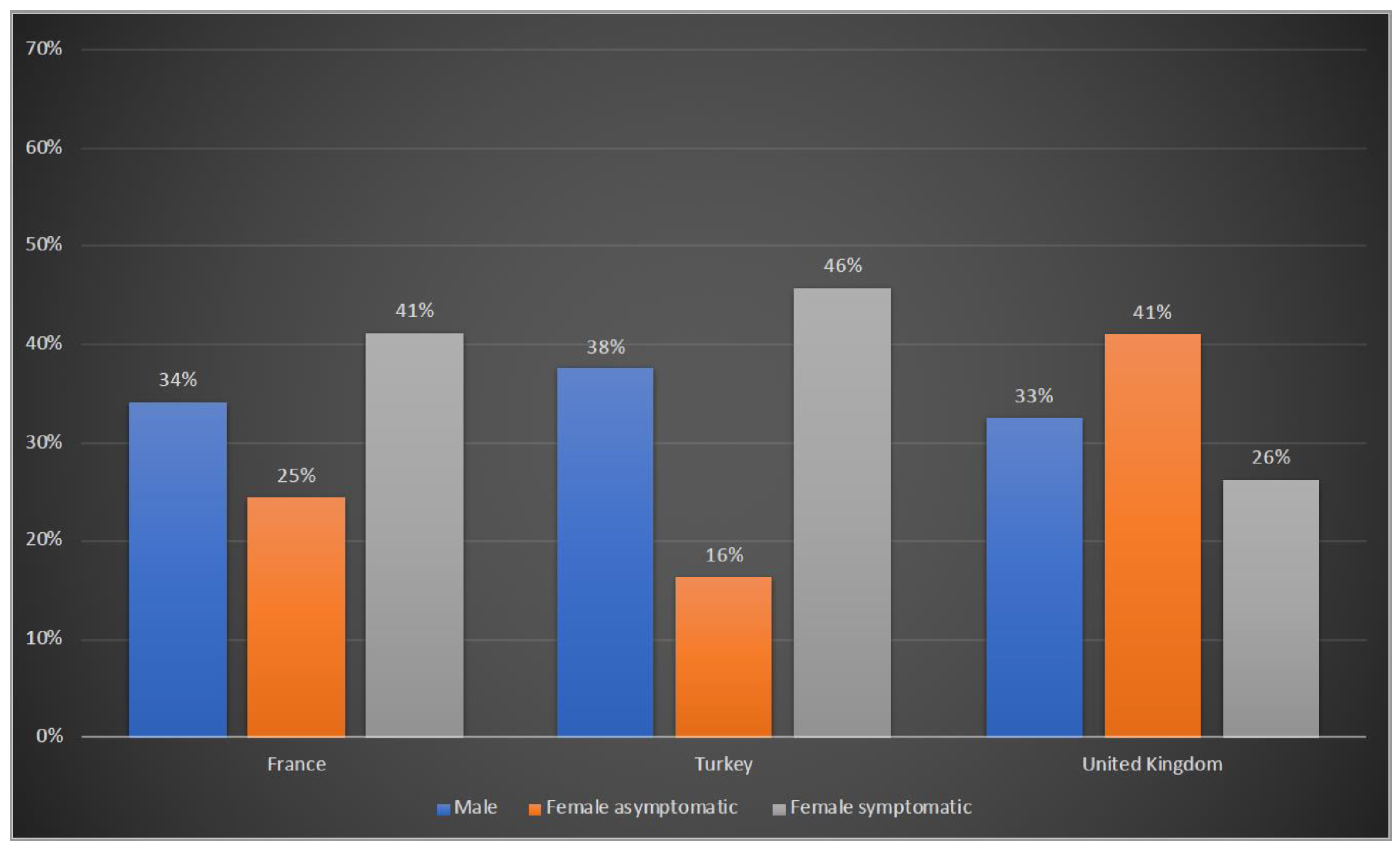
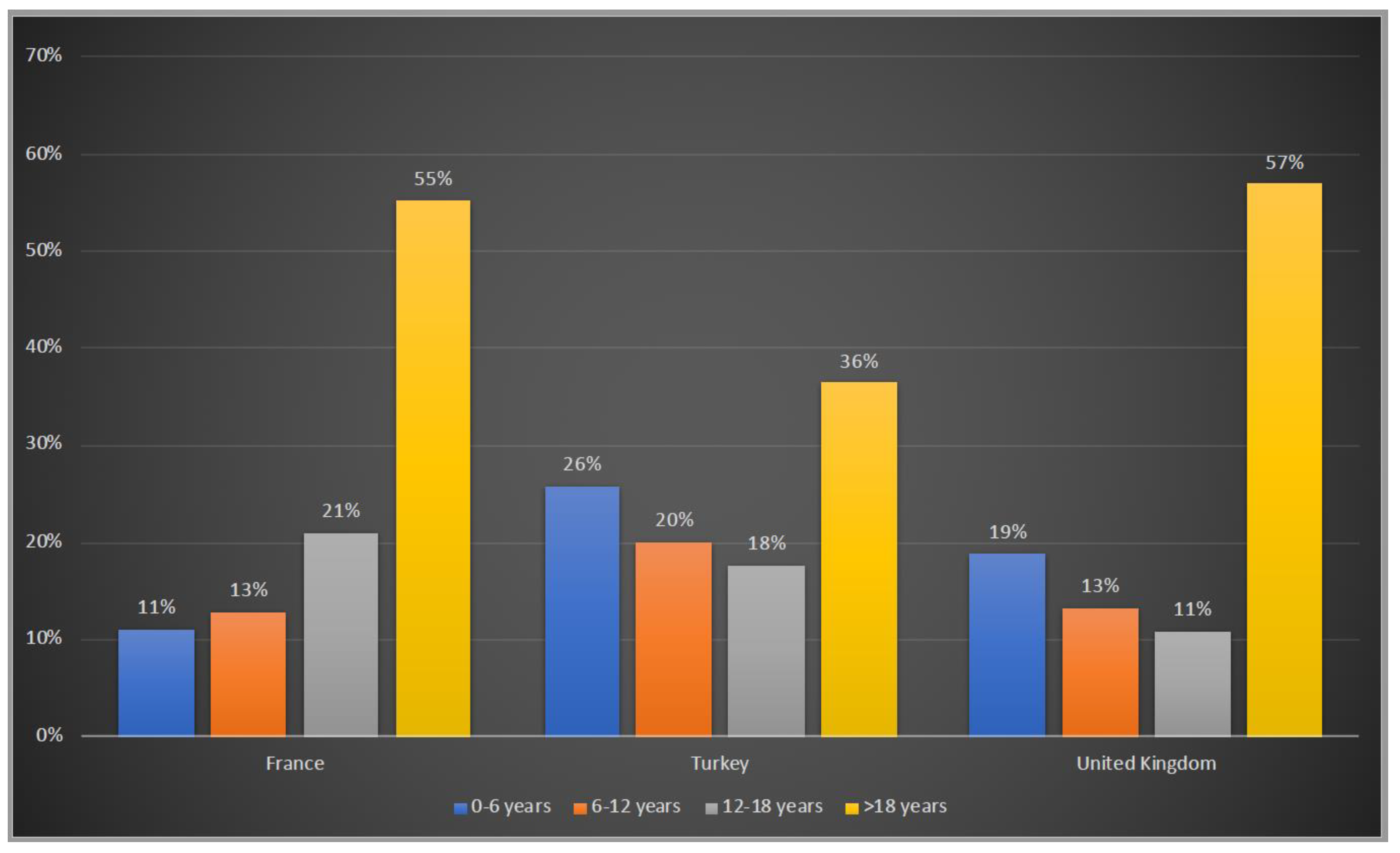
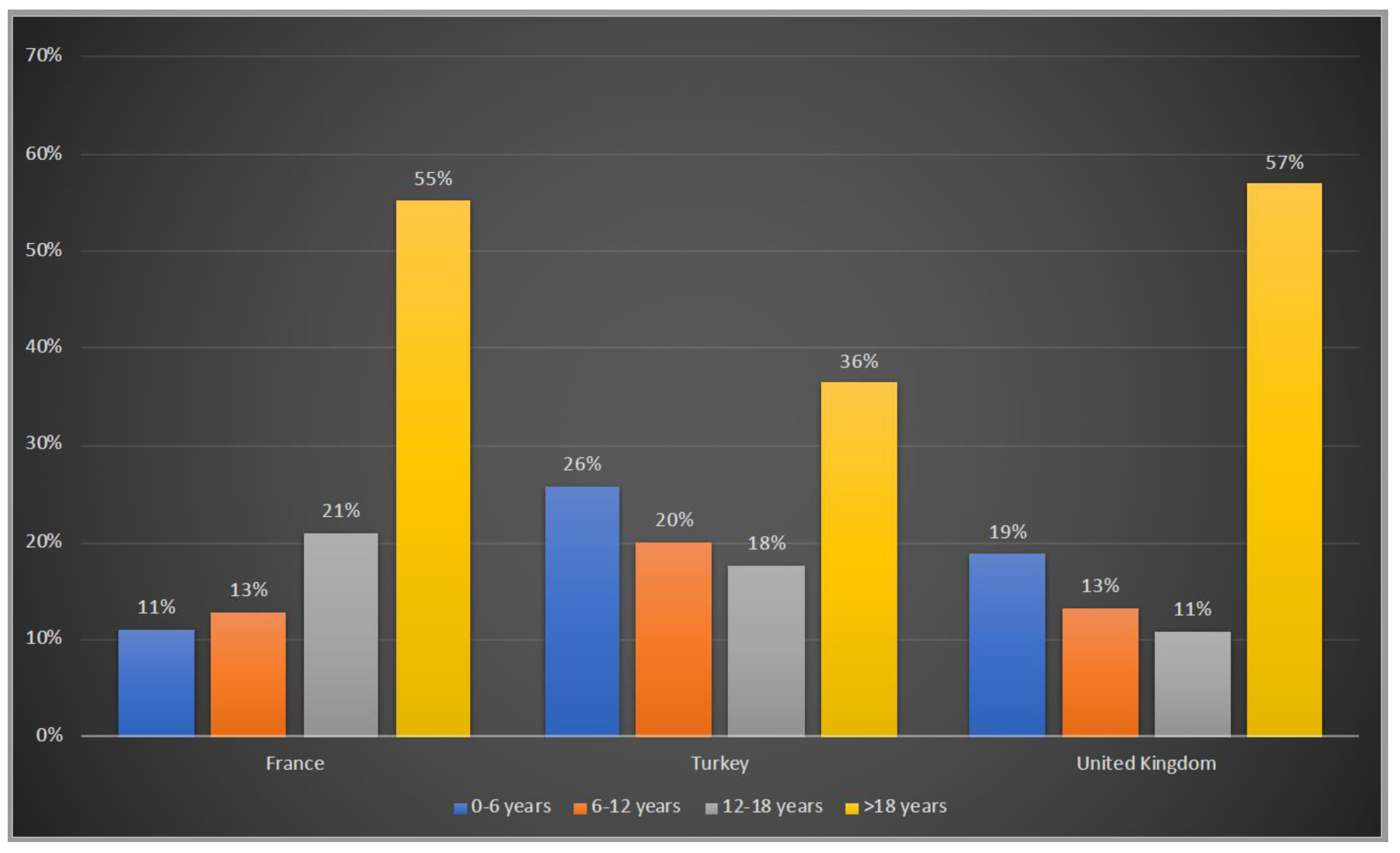
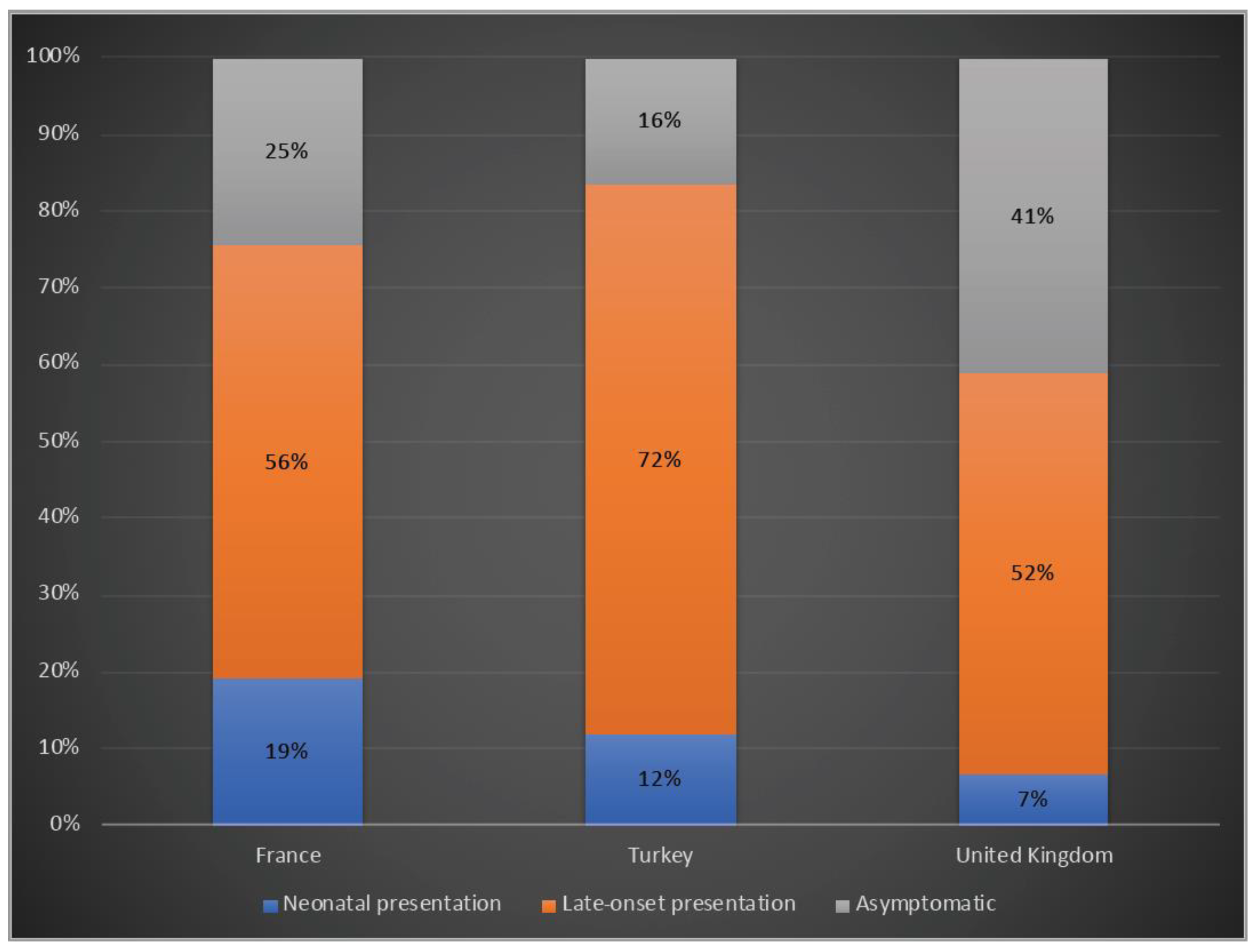
3.1. Demographic Characteristics of Patients
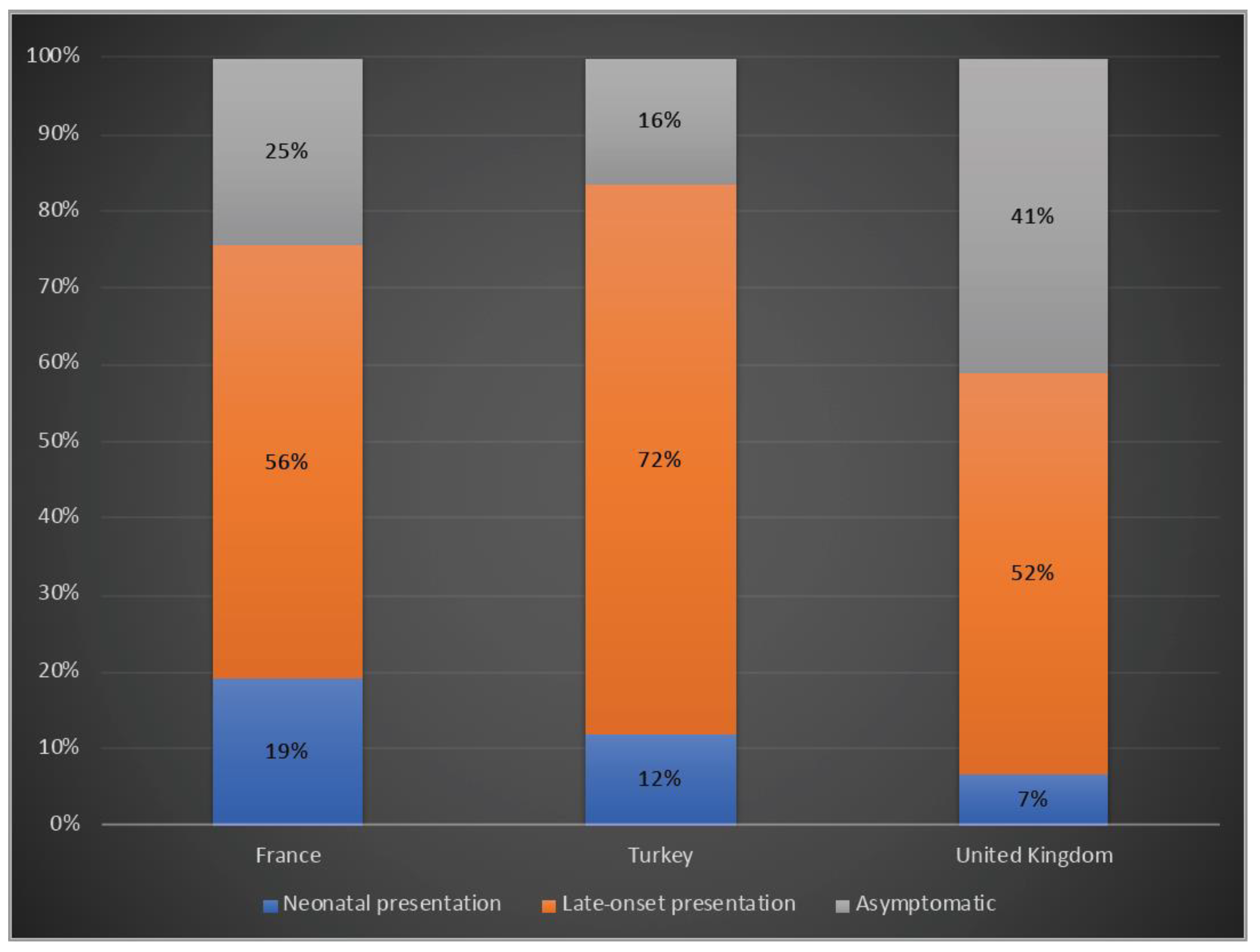
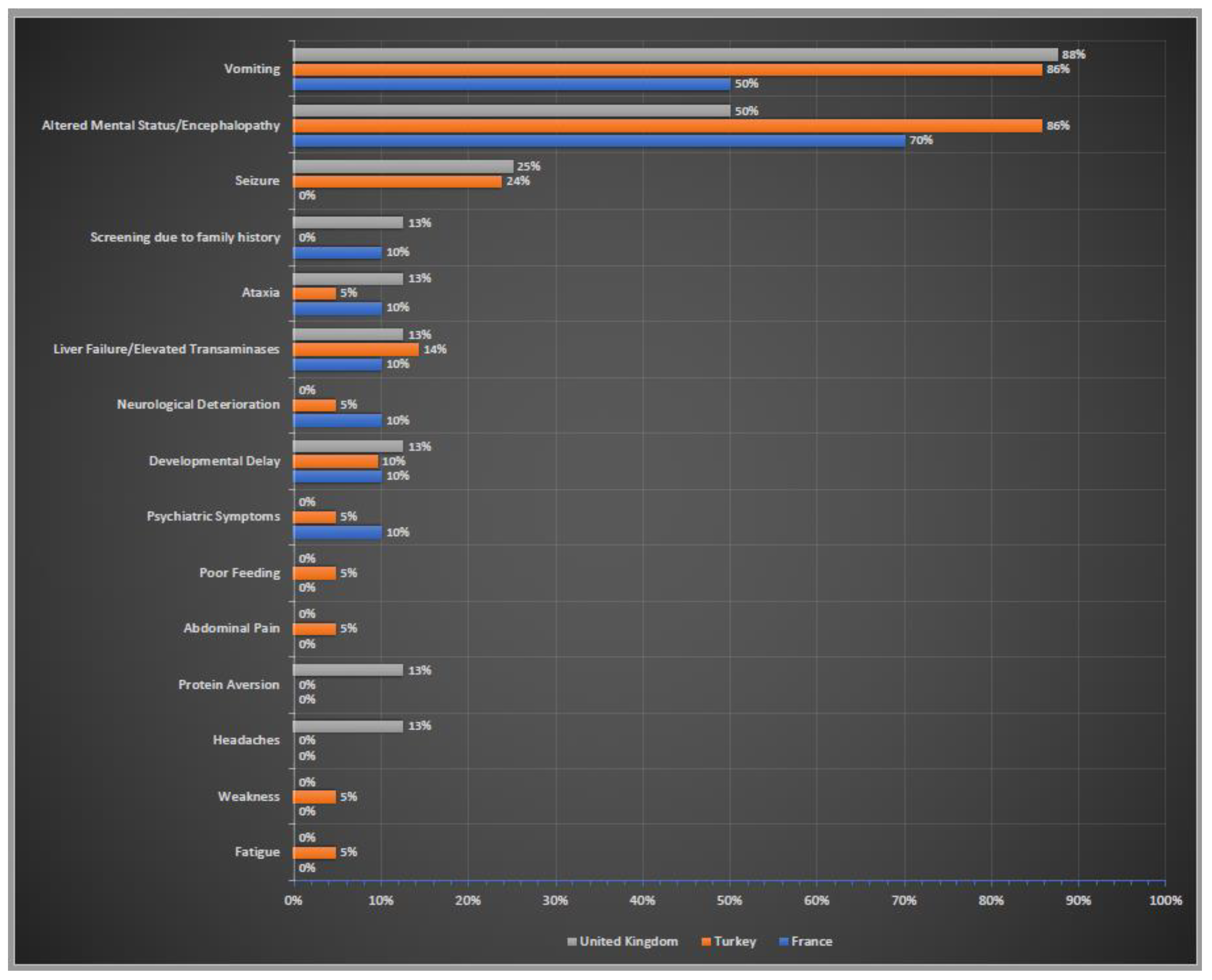
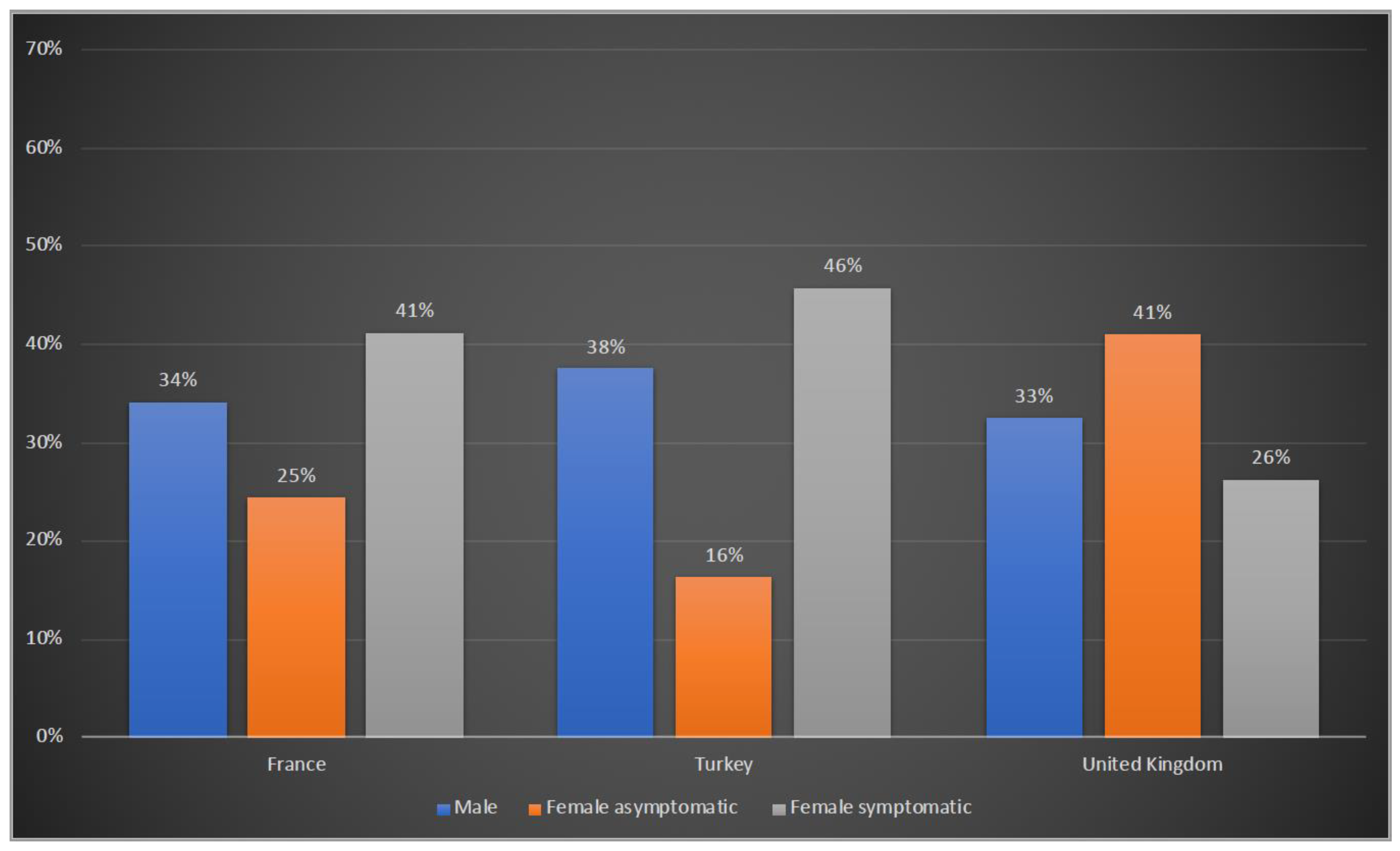
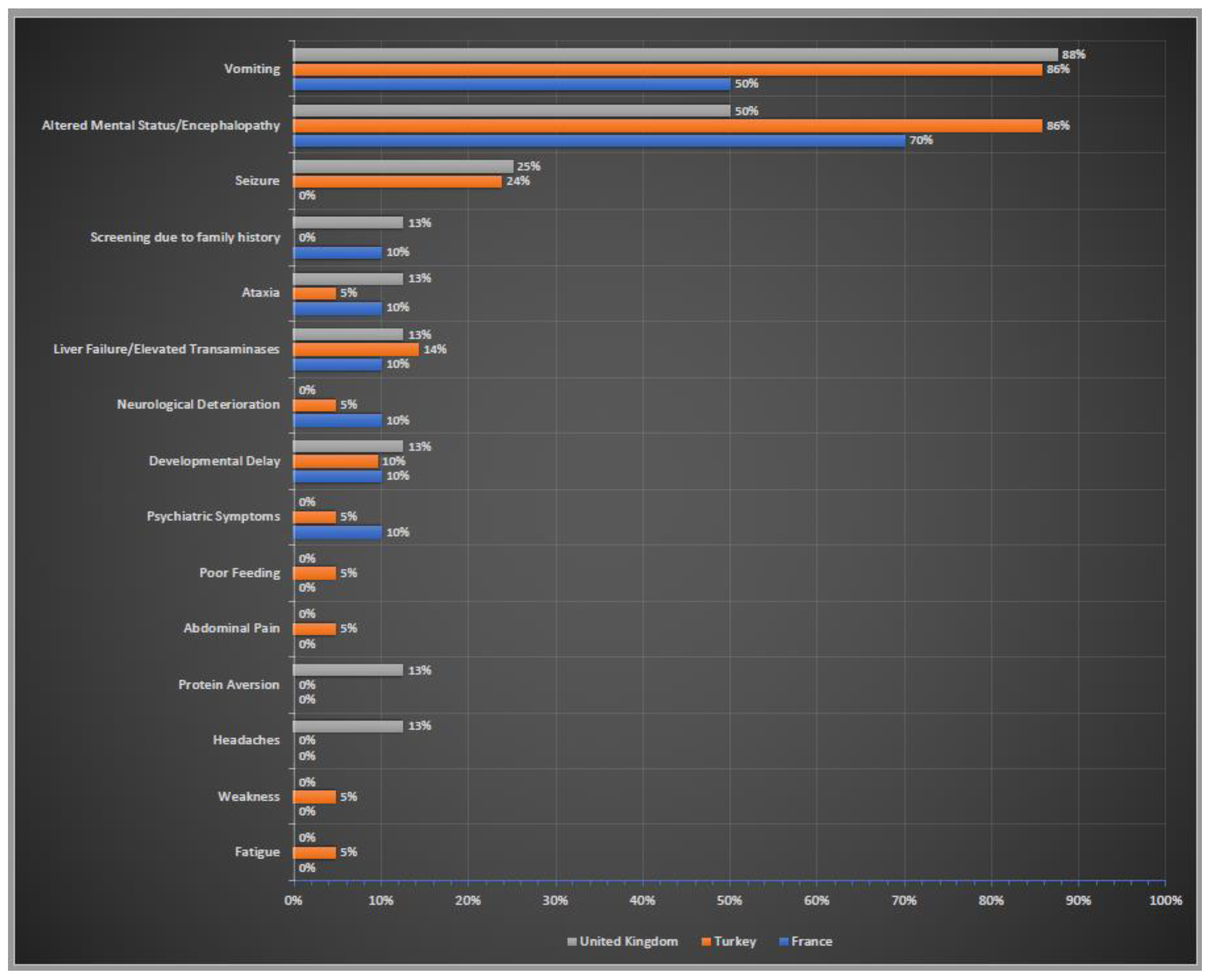
3.2. Clinical Presentation
3.3. Diagnosis of OTCD Patients
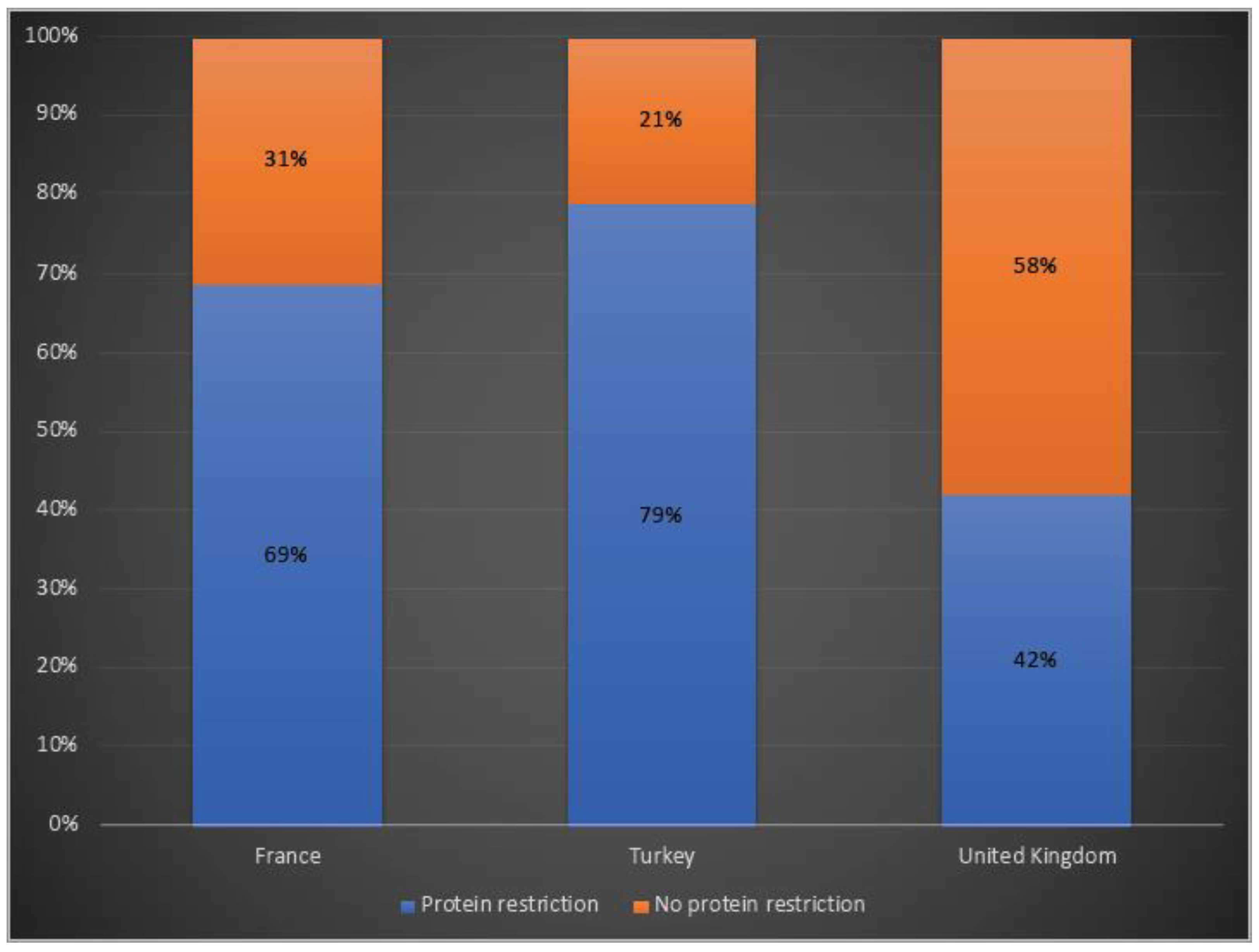
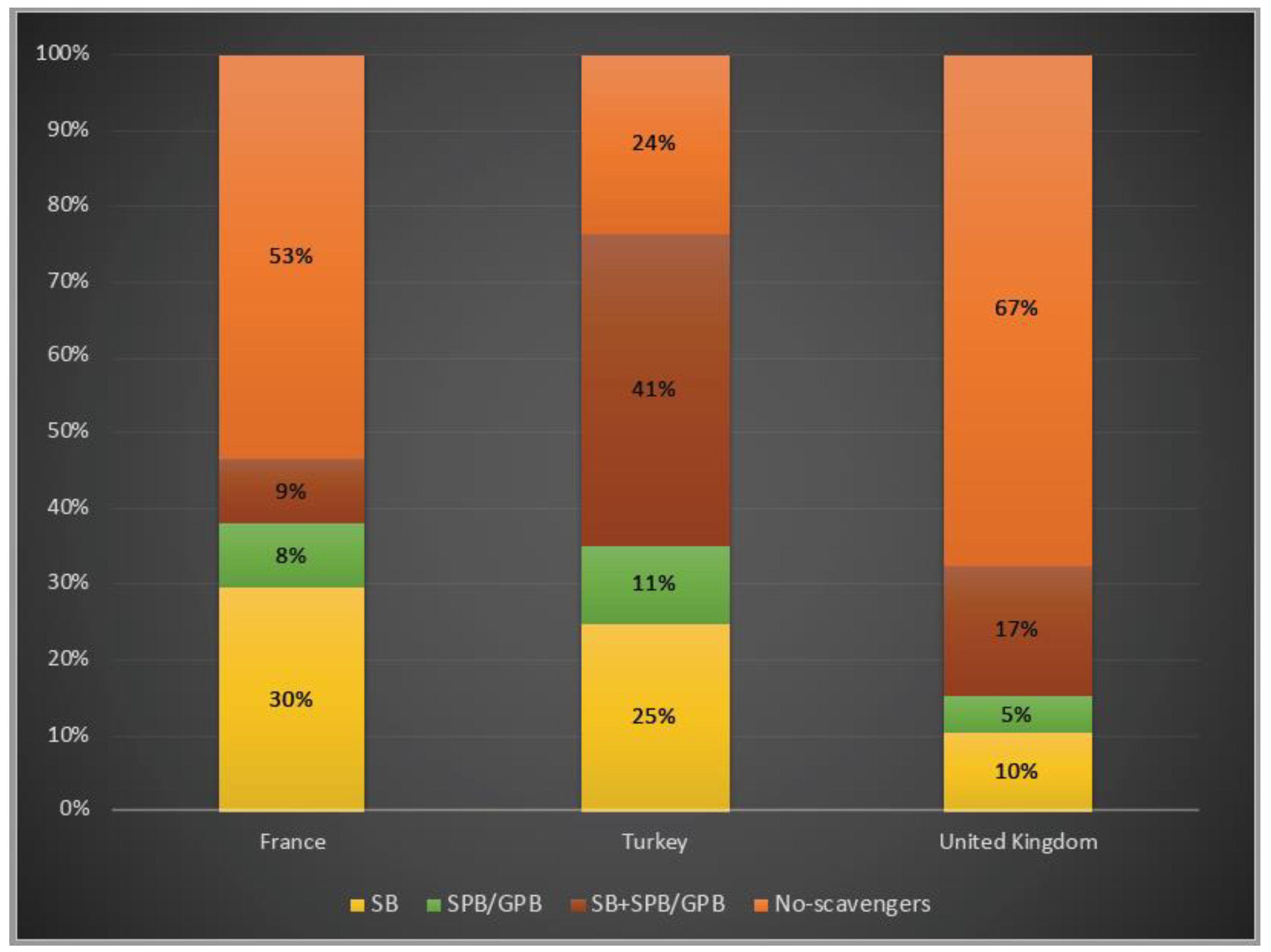
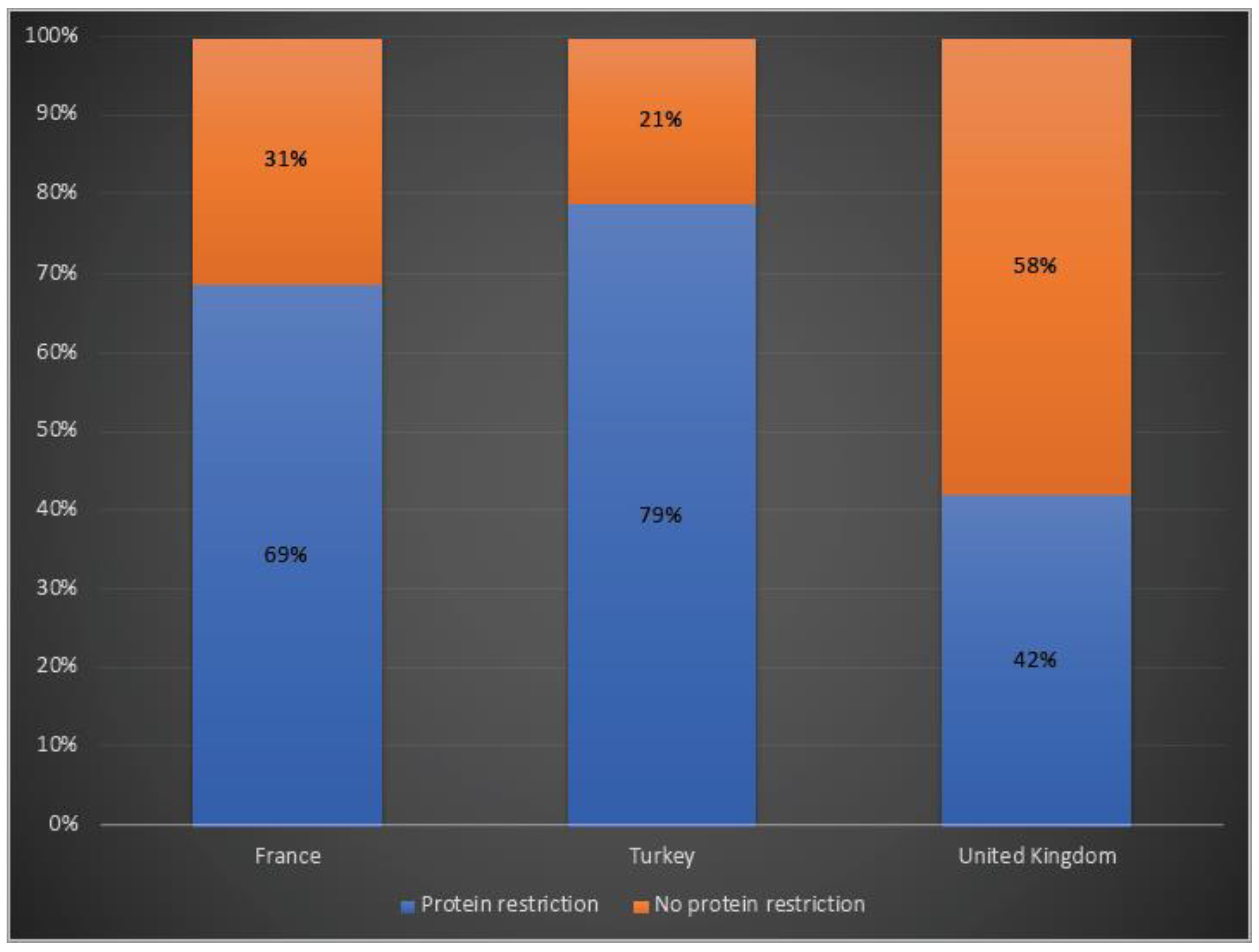
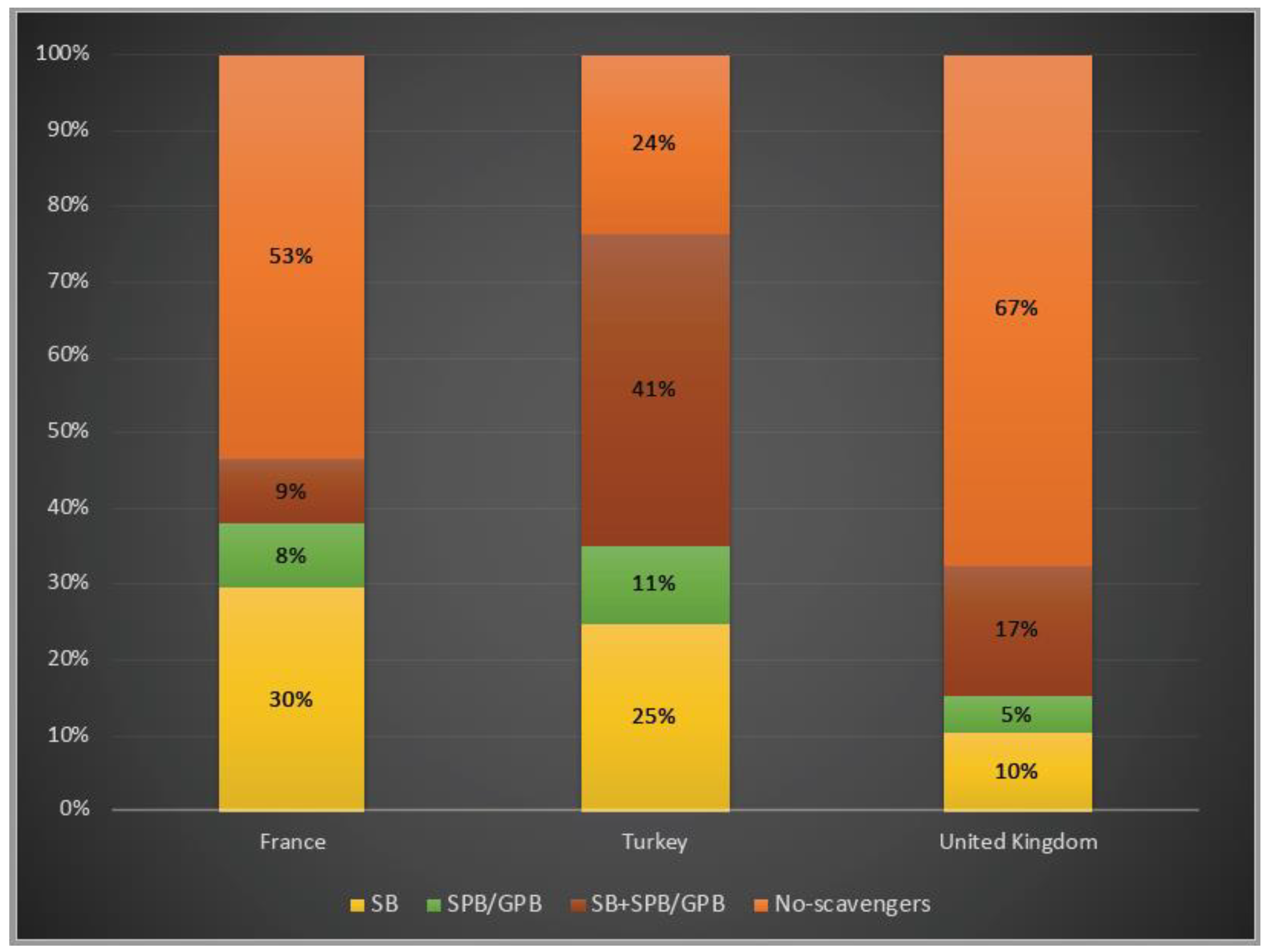
3.4. Disease Management
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seminara, J.; Tuchman, M.; Krivitzky, L.; Krischer, J.; Lee, H.-S.; LeMons, C.; Baumgartner, M.; Cederbaum, S.; Diaz, G.A.; Feigenbaum, A.; et al. Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium. Mol. Genet. Metab. 2010, 100 (Suppl. 1), S97–S105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusilow, S.W.; Maestri, N.E. Urea cycle disorders: Diagnosis, pathophysiology, and therapy. Adv. Pediatr. 1996, 43, 127–170. [Google Scholar] [PubMed]
- Nagata, N.; Matsuda, I.; Oyanagi, K. Estimated frequency of urea cycle enzymopathies in Japan. Am. J. Med. Genet. 1991, 39, 228–229. [Google Scholar] [CrossRef] [PubMed]
- Batshaw, M.L.; Tuchman, M.; Summar, M.; Seminara, J. A longitudinal study of urea cycle disorders. Mol. Genet. Metab. 2014, 113, 127–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionisi-Vici, C.; Rizzo, C.; Burlina, A.B.; Caruso, U.; Sabetta, G.; Uziel, G.; Abeni, D. Inborn errors of metabolism in the Italian pediatric population: A national retrospective survey. J. Pediatr. 2002, 140, 321–329. [Google Scholar] [CrossRef]
- Keskinen, P.; Siitonen, A.; Salo, M. Hereditary urea cycle diseases in Finland. Acta Paediatr. 2008, 97, 1412–1419. [Google Scholar] [CrossRef]
- Couchet, M.; Breuillard, C.; Corne, C.; Rendu, J.; Morio, B.; Schlattner, U.; Moinard, C. Ornithine Transcarbamylase—From Structure to Metabolism: An Update. Front. Physiol. 2021, 12, 748249. [Google Scholar] [CrossRef]
- Gropman, A.L.; Summar, M.; Leonard, J.V. Neurological implications of urea cycle disorders. J. Inherit. Metab. Dis. 2007, 30, 865–879. [Google Scholar] [CrossRef] [Green Version]
- Bigot, A.; Tchan, M.C.; Thoreau, B.; Blasco, H.; Maillot, F. Liver involvement in urea cycle disorders: A review of the literature. J. Inherit. Metab. Dis. 2017, 40, 757–769. [Google Scholar] [CrossRef]
- Caldovic, L.; Abdikarim, I.; Narain, S.; Tuchman, M.; Morizono, H. Genotype–Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J. Genet. Genom. 2015, 42, 181–194. [Google Scholar] [CrossRef]
- Yorifuji, T.; Muroi, J.; Uematsu, A.; Tanaka, K.; Kiwaki, K.; Endo, F.; Matsuda, I.; Nagasaka, H.; Furusho, K. X-inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clin. Genet. 1998, 54, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders—Update. J. Hum. Genet. 2019, 64, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Gobin-Limballe, S.; Ottolenghi, C.; Reyal, F.; Arnoux, J.; Magen, M.; Simon, M.; Brassier, A.; Jabot-Hanin, F.; De Lonlay, P.; Pontoizeau, C.; et al. OTC deficiency in females: Phenotype–genotype correlation based on a 130-family cohort. J. Inherit. Metab. Dis. 2021, 44, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Brassier, A.; Gobin, S.; Arnoux, J.B.; Valayannopoulos, V.; Habarou, F.; Kossorotoff, M.; Servais, A.; Barbier, V.; Dubois, S.; Touati, G.; et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: A series of 90 patients. Orphanet J. Rare Dis. 2015, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, R.C.; Lam, C.; Wong, D.; Cederbaum, S.; Sokol, R.J. Significant Hepatic Involvement in Patients with Ornithine Transcarbamylase Deficiency. J. Pediatr. 2014, 164, 720–725.e6. [Google Scholar] [CrossRef] [Green Version]
- Häberle, J.; Burlina, A.; Chakrapani, A.; Dixon, M.; Karall, D.; Lindner, M.; Mandel, H.; Martinelli, D.; Pintos-Morell, G.; Santer, R.; et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J. Inherit. Metab. Dis. 2019, 42, 1192–1230. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Nuoffer, J.-M.; Mühlhausen, C.; Klaus, V.; Largiadèr, C.R.; Tsiakas, K.; Santer, R.; Wermuth, B.; Häberle, J. Analysis of mRNA transcripts improves the success rate of molecular genetic testing in OTC deficiency. Mol. Genet. Metab. 2008, 94, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Shchelochkov, O.A.; Li, F.-Y.; Geraghty, M.T.; Gallagher, R.C.; Van Hove, J.L.; Lichter-Konecki, U.; Fernhoff, P.M.; Copeland, S.; Reimschisel, T.; Cederbaum, S.; et al. High-frequency detection of deletions and variable rearrangements at the ornithine transcarbamylase (OTC) locus by oligonucleotide array CGH. Mol. Genet. Metab. 2009, 96, 97–105. [Google Scholar] [CrossRef]
- Kido, J.; Matsumoto, S.; Häberle, J.; Inomata, Y.; Kasahara, M.; Sakamoto, S.; Horikawa, R.; Tanemura, A.; Okajima, H.; Suzuki, T.; et al. Role of liver transplantation in urea cycle disorders: Report from a nationwide study in Japan. J. Inherit. Metab. Dis. 2021, 44, 1311–1322. [Google Scholar] [CrossRef]
- Leonard, J.V.; McKiernan, P.J. The role of liver transplantation in urea cycle disorders. Mol. Genet. Metab. 2004, 81, 74–78. [Google Scholar] [CrossRef]
- Meyburg, J.; Opladen, T.; Spiekerkötter, U.; Schlune, A.; Schenk, J.-P.; Schmidt, J.; Weitz, J.; Okun, J.; Bürger, F.; Ben Omran, T.; et al. Human heterologous liver cells transiently improve hyperammonemia and ureagenesis in individuals with severe urea cycle disorders. J. Inherit. Metab. Dis. 2018, 41, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Cunningham, S.C.; Yilmaz, B.S.; Perocheau, D.P.; Eaglestone, S.; Burke, D.; Thrasher, A.J.; Waddington, S.N.; Lisowski, L.; Alexander, I.E.; et al. Safety and efficacy of an engineered hepatotropic AAV gene therapy for ornithine transcarbamylase deficiency in cynomolgus monkeys. Mol. Ther. Methods Clin. Dev. 2021, 23, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baruteau, J.; Waddington, S.E.; Alexander, I.; Gissen, P. Gene therapy for monogenic liver diseases: Clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef] [Green Version]
- Raper, S.E.; Chirmule, N.; Lee, F.S.; Wivel, N.A.; Bagg, A.; Gao, G.-P.; Wilson, J.M.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Summar, M.L.; Dobbelaere, D.; Brusilow, S.; Lee, B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008, 97, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Stepien, K.M.; Geberhiwot, T.; Hendriksz, C.J.; Treacy, E.P. Challenges in diagnosing and managing adult patients with urea cycle disorders. J. Inherit. Metab. Dis. 2019, 42, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Garbade, S.F.; Alber, F.D.; Waisbren, S.E.; McCarter, R.; Kölker, S.; Burgard, P.; Mew, N.A.; Batshaw, M.L.; Baumgartner, M.R.; et al. Impairment of cognitive function in ornithine transcarbamylase deficiency is global rather than domain-specific and is associated with disease onset, sex, maximum ammonium, and number of hyperammonemic events. J. Inherit. Metab. Dis. 2019, 42, 243–253. [Google Scholar] [CrossRef]
- Posset, R.; Garbade, S.F.; Boy, N.; Burlina, A.B.; Dionisi-Vici, C.; Dobbelaere, D.; Garcia-Cazorla, A.; De Lonlay, P.; Teles, E.L.; Vara, R.; et al. Transatlantic combined and comparative data analysis of 1095 patients with urea cycle disorders—A successful strategy for clinical research of rare diseases. J. Inherit. Metab. Dis. 2019, 42, 93–106. [Google Scholar] [CrossRef]
- Sprouse, C.; King, J.; Helman, G.; Pacheco-Colón, I.; Shattuck, K.; Breeden, A.; Seltzer, R.; VanMeter, J.W.; Gropman, A.L. Investigating neurological deficits in carriers and affected patients with ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2014, 113, 136–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, A.; Gropman, A.; Le Mons, C.; Stratakis, C.; Gandjbakhche, A. Evaluation of neurocognitive function of prefrontal cortex in ornithine transcarbamylase deficiency. Mol. Genet. Metab. 2020, 129, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Nakip, S.; Yıldız, Y.; Tokatlı, A. Retrospective evaluation of 85 patients with urea cycle disorders: One center experience, three new mutations. J. Pediatr. Endocrinol. Metab. 2020, 33, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Han, F.; Qiu, W.; Zhang, H.; Ye, J.; Liang, L.; Wang, Y.; Ji, W.; Zhan, X.; Gu, X.; et al. Clinical and molecular characteristics of 69 Chinese patients with ornithine transcarbamylase deficiency. Orphanet J. Rare Dis. 2020, 15, 340. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, A.; Gallagher, R.C.; Keogh, A.; Stricker, T.; Gautschi, M.; Nuoffer, J.-M.; Baumgartner, M.R.; Häberle, J. Frequency and Pathophysiology of Acute Liver Failure in Ornithine Transcarbamylase Deficiency (OTCD). PLoS ONE 2016, 11, e0153358. [Google Scholar] [CrossRef] [Green Version]
- Enns, G.M.; Porter, M.H.; Francis-Sedlak, M.; Burdett, A.; Vockley, J. Perspectives on urea cycle disorder management: Results of a clinician survey. Mol. Genet. Metab. 2019, 128, 102–108. [Google Scholar] [CrossRef]
- Adam, S.; Almeida, M.; Assoun, M.; Baruteau, J.; Bernabei, S.; Bigot, S.; Champion, H.; Daly, A.; Dassy, M.; Dawson, S.; et al. Dietary management of urea cycle disorders: European practice. Mol. Genet. Metab. 2013, 110, 439–445. [Google Scholar] [CrossRef]
- Enns, G.M.; Berry, S.A.; Berry, G.T.; Rhead, W.J.; Brusilow, S.W.; Hamosh, A. Survival after Treatment with Phenylacetate and Benzoate for Urea-Cycle Disorders. N. Engl. J. Med. 2007, 356, 2282–2292. [Google Scholar] [CrossRef]
- Longo, N.; Diaz, G.A.; Lichter-Konecki, U.; Schulze, A.; Inbar-Feigenberg, M.; Conway, R.L.; Bannick, A.A.; McCandless, S.E.; Zori, R.; Hainline, B.; et al. Glycerol phenylbutyrate efficacy and safety from an open label study in pediatric patients under 2 months of age with urea cycle disorders. Mol. Genet. Metab. 2021, 132, 19–26. [Google Scholar] [CrossRef]
- Yeo, M.; Rehsi, P.; Dorman, M.; Grunewald, S.; Baruteau, J.; Chakrapani, A.; Footitt, E.; Prunty, H.; McSweeney, M. Direct replacement of oral sodium benzoate with glycerol phenylbutyrate in children with urea cycle disorders. JIMD Rep. 2022, 63, 137–145. [Google Scholar] [CrossRef]
- Crowe, L.; Anderson, V.; Hardikar, W.; Boneh, A. Cognitive and Behavioural Outcomes of Paediatric Liver Transplantation for Ornithine Transcarbamylase Deficiency. JIMD Rep. 2019, 43, 19–25. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seker Yilmaz, B.; Baruteau, J.; Arslan, N.; Aydin, H.I.; Barth, M.; Bozaci, A.E.; Brassier, A.; Canda, E.; Cano, A.; Chronopoulou, E.; et al. Three-Country Snapshot of Ornithine Transcarbamylase Deficiency. Life 2022, 12, 1721. https://doi.org/10.3390/life12111721
Seker Yilmaz B, Baruteau J, Arslan N, Aydin HI, Barth M, Bozaci AE, Brassier A, Canda E, Cano A, Chronopoulou E, et al. Three-Country Snapshot of Ornithine Transcarbamylase Deficiency. Life. 2022; 12(11):1721. https://doi.org/10.3390/life12111721
Chicago/Turabian StyleSeker Yilmaz, Berna, Julien Baruteau, Nur Arslan, Halil Ibrahim Aydin, Magalie Barth, Ayse Ergul Bozaci, Anais Brassier, Ebru Canda, Aline Cano, Efstathia Chronopoulou, and et al. 2022. "Three-Country Snapshot of Ornithine Transcarbamylase Deficiency" Life 12, no. 11: 1721. https://doi.org/10.3390/life12111721
APA StyleSeker Yilmaz, B., Baruteau, J., Arslan, N., Aydin, H. I., Barth, M., Bozaci, A. E., Brassier, A., Canda, E., Cano, A., Chronopoulou, E., Connolly, G. M., Damaj, L., Dawson, C., Dobbelaere, D., Douillard, C., Eminoglu, F. T., Erdol, S., Ersoy, M., Fang, S., ... Gissen, P. (2022). Three-Country Snapshot of Ornithine Transcarbamylase Deficiency. Life, 12(11), 1721. https://doi.org/10.3390/life12111721