
Altered Serum Uric Acid Levels in Kidney Disorders



Abstract
:1. Introduction

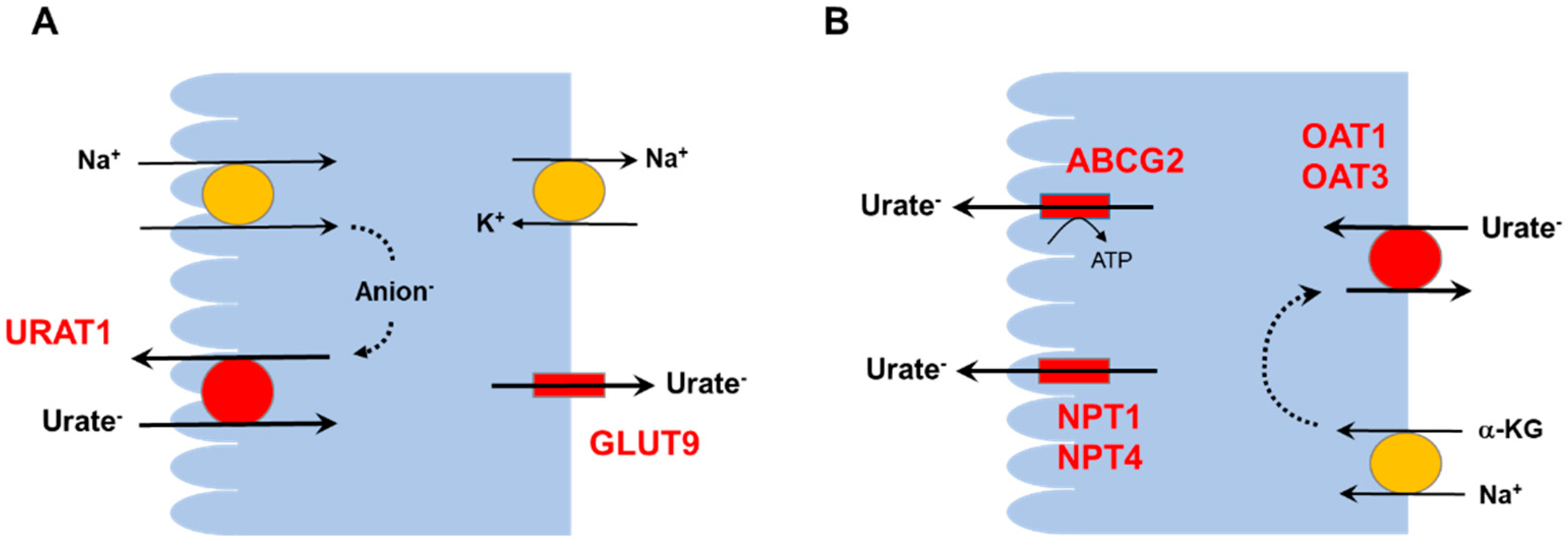
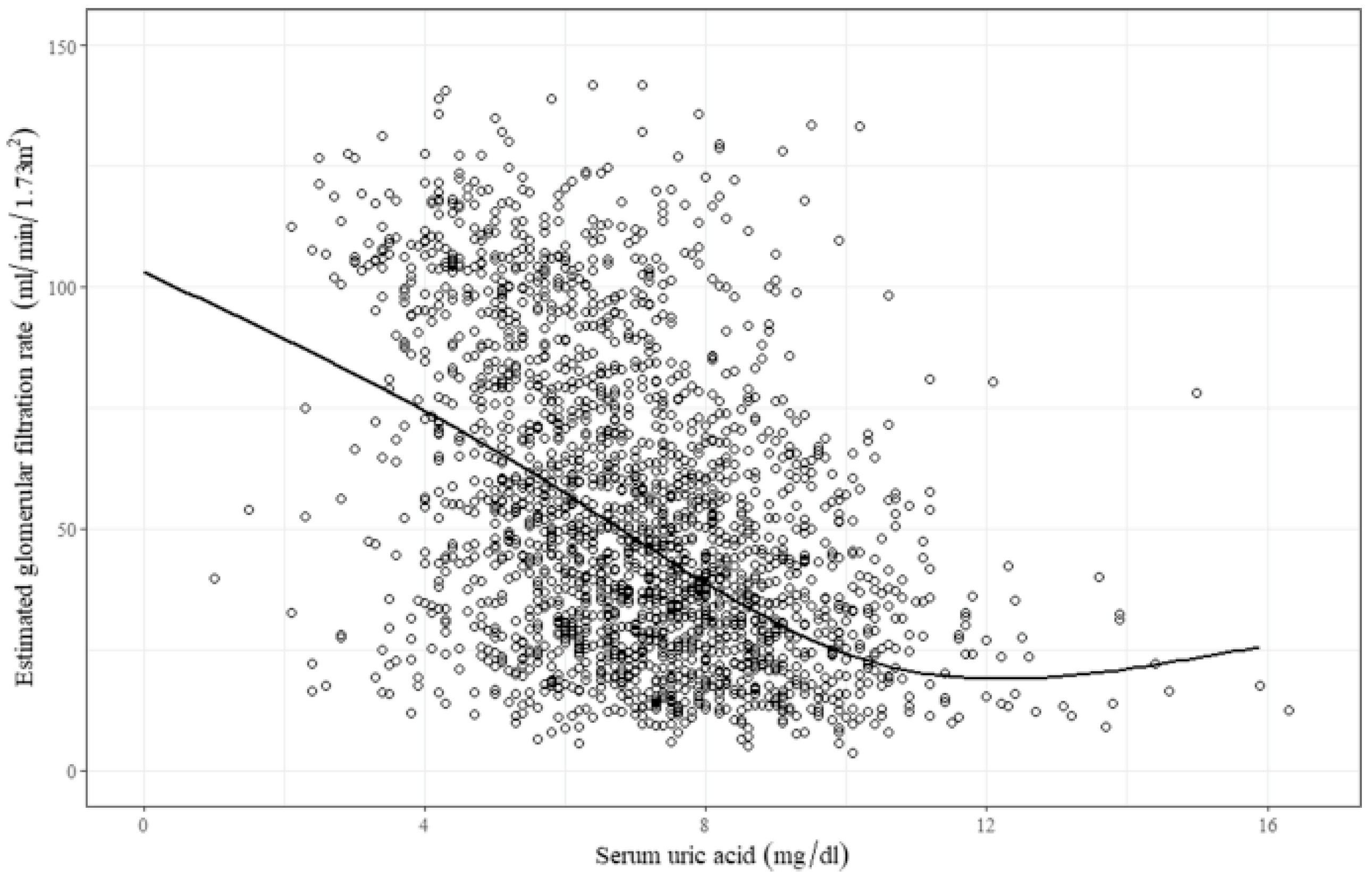
2. Hyperuricemia and the Kidneys

2.1. Asymptomatic Hyperuricemia in CKD
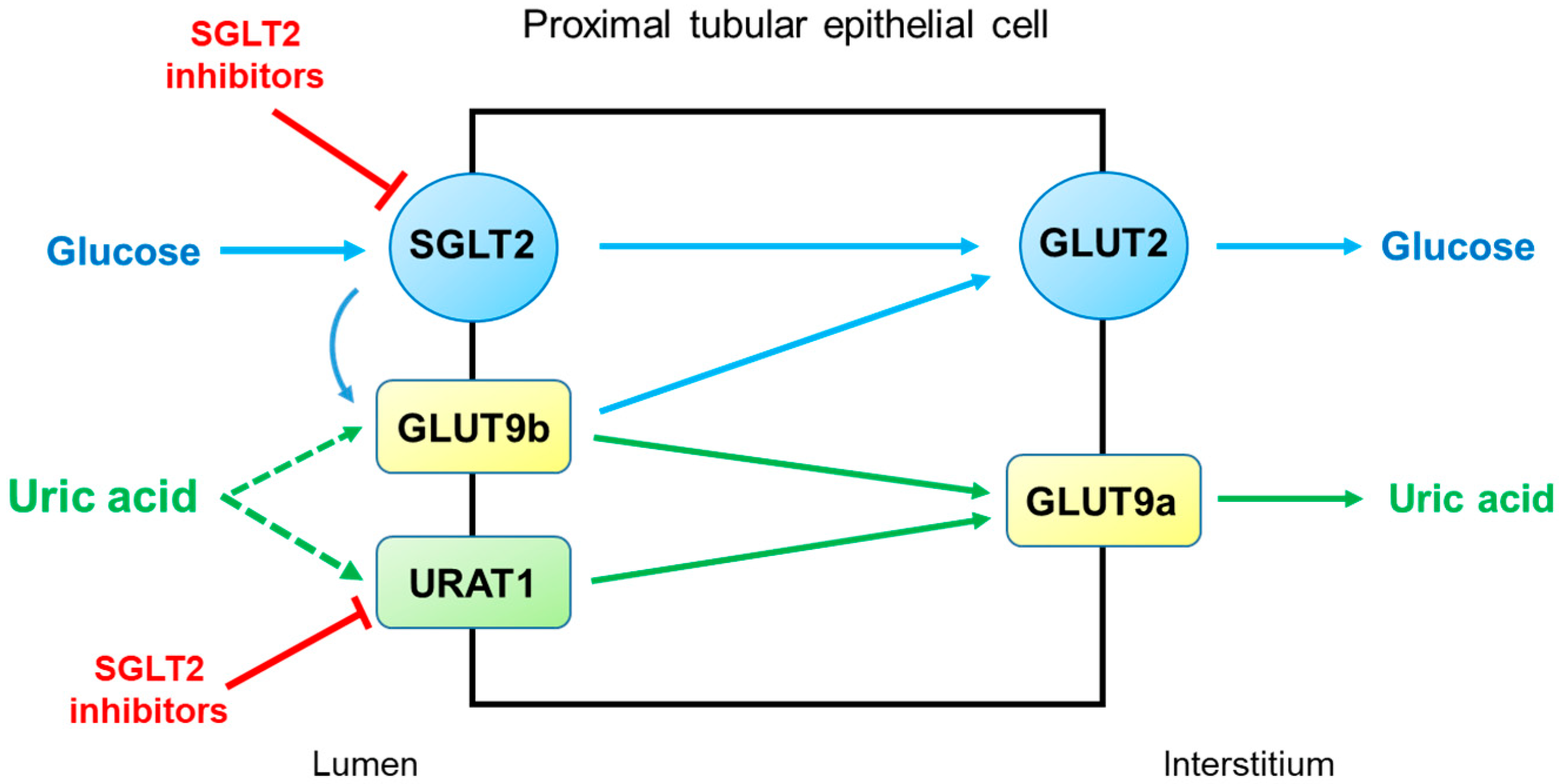
2.2. Diabetic Kidney Disease (DKD)
2.3. ADTKD
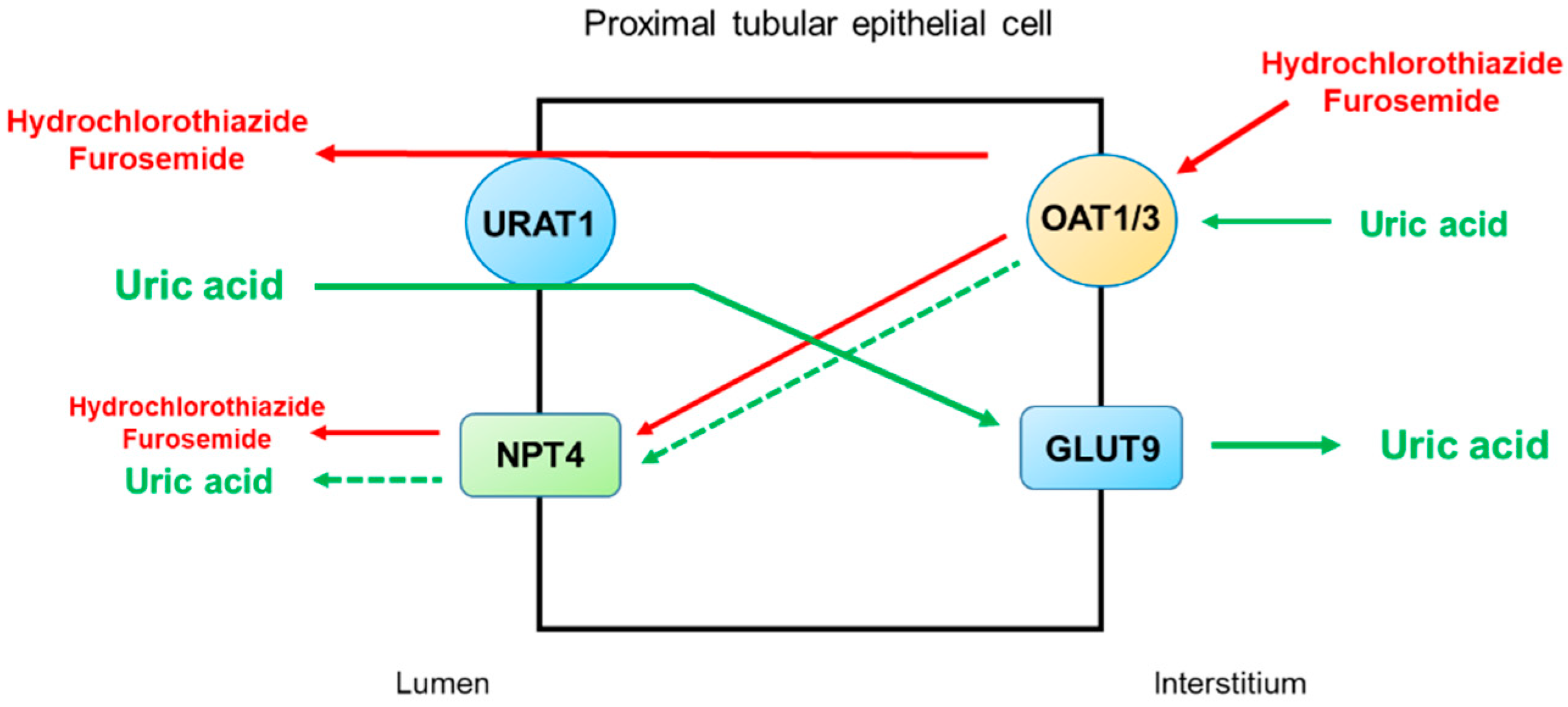
2.4. Thiazide and Loop Diuretics
3. Hypouricemia and the Kidneys
3.1. Renal Hypouricemia
3.2. Fanconi Syndrome
3.3. Coronavirus Disease 2019 (COVID-19)
3.4. Hyponatremic Disorders: SIAD, RSW, and Thiazide-Induced Hyponatremia
3.4.1. SIAD
3.4.2. CSW/RSW
3.4.3. Thiazide-Induced Hyponatremia
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Álvarez-Lario, B.; Macarrón-Vicente, J. Uric acid and evolution. Rheumatology 2010, 49, 2010–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.S.; Jeong, H.J.; Son, C.N.; Kim, S.H.; Kim, H.J.; Kim, G.H.; Jun, J.B. Distribution of serum uric acid levels and prevalence of hyper- and hypouricemia in a Korean general population of 172,970. Korean J. Intern. Med. 2021, 36, S264–S272. [Google Scholar] [CrossRef] [PubMed]
- Halperin Kuhns, V.L.; Woodward, O.M. Sex differences in urate handling. Int. J. Mol. Sci. 2020, 21, 4269. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Chang, Y.; Kim, I.; Ryu, S. U-shaped association between serum uric acid level and risk of mortality: A cohort study. Arthritis Rheumatol. 2018, 70, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular biological and clinical understanding of the pathophysiology and treatments of hyperuricemia and its association with metabolic syndrome, cardiovascular diseases and chronic kidney disease. Int. J. Mol. Sci. 2021, 22, 9221. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Kim, G.H. Urate transporters in the kidney: What clinicians need to know. Electrolyte Blood Press. 2021, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.K.; Ostertag, T.M.; Miner, J.N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci. Rep. 2016, 6, 34995. [Google Scholar]
- Hamada, T.; Ichida, K.; Hosoyamada, M.; Mizuta, E.; Yanagihara, K.; Sonoyama, K.; Sugihara, S.; Igawa, O.; Hosoya, T.; Ohtahara, A.; et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am. J. Hypertens. 2008, 21, 1157–1162. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Qu, Q.; Qu, J.; Lou, X.Y.; Peng, Y.; Zeng, Y.; Wang, G. URAT1 gene polymorphisms influence uricosuric action of losartan in hypertensive patients with hyperuricemia. Pharmacogenomics 2015, 16, 855–863. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Fan, Y.; Wang, Y.; Li, Z.; Mao, D.; Zhuang, W. The impact of an URAT1 polymorphism on the losartan treatment of hypertension and hyperuricemia. J. Clin. Lab. Anal. 2021, 35, e23949. [Google Scholar] [CrossRef]
- Otani, N.; Ouchi, M.; Misawa, K.; Hisatome, I.; Anzai, N. Hypouricemia and urate transporters. Biomedicines 2022, 10, 652. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.R.; Choi, H.S.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Sung, S.A.; Kim, Y.S.; Oh, K.H.; Ahn, C.; Kim, S.W. Hyperuricemia has increased the risk of progression of chronic kidney disease: Propensity score matching analysis from the KNOW-CKD study. Sci. Rep. 2019, 9, 6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzali, M.; Hughes, J.; Kim, Y.G.; Jefferson, J.A.; Kang, D.H.; Gordon, K.L.; Lan, H.Y.; Kivlighn, S.; Johnson, R.J. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension 2001, 38, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eräranta, A.; Kurra, V.; Tahvanainen, A.M.; Vehmas, T.I.; Kööbi, P.; Lakkisto, P.; Tikkanen, I.; Niemelä, O.J.; Mustonen, J.T.; Pörsti, I.H. Oxonic acid-induced hyperuricemia elevates plasma aldosterone in experimental renal insufficiency. J. Hypertens. 2008, 26, 1661–1668. [Google Scholar] [CrossRef]
- Mulè, G.; Castiglia, A.; Morreale, M.; Geraci, G.; Cusumano, C.; Guarino, L.; Altieri, D.; Panzica, M.; Vaccaro, F.; Cottone, S. Serum uric acid is not independently associated with plasma renin activity and plasma aldosterone in hypertensive adults. Nutr Metab. Cardiovasc. Dis. 2017, 27, 350–359. [Google Scholar] [CrossRef]
- Perlstein, T.S.; Gumieniak, O.; Hopkins, P.N.; Murphey, L.J.; Brown, N.J.; Williams, G.H.; Hollenberg, N.K.; Fisher, N.D. Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney Int. 2004, 66, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, D.L.N.; Moreira, T.R.; da Silva, L.S. A systematic review and meta-analysis of the association between uric acid levels and chronic kidney disease. Sci. Rep. 2022, 12, 6251. [Google Scholar] [CrossRef]
- Floege, J.; Johnson, R.J. Hyperuricemia and progression of chronic kidney disease: To treat or not to treat? Kidney Int. 2021, 99, 14–16. [Google Scholar] [CrossRef]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of allopurinol on the progression of chronic kidney disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef]
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N. Engl. J. Med. 2020, 382, 2493–2503. [Google Scholar] [CrossRef]
- Jordan, D.M.; Choi, H.K.; Verbanck, M.; Topless, R.; Won, H.H.; Nadkarni, G.; Merriman, T.R.; Do, R. No causal effects of serum urate levels on the risk of chronic kidney disease: A Mendelian randomization study. PLoS Med. 2019, 16, e1002725. [Google Scholar] [CrossRef] [PubMed]
- Sircar, D.; Chatterjee, S.; Waikhom, R.; Golay, V.; Raychaudhury, A.; Chatterjee, S.; Pandey, R. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: A 6-month, double-blind, randomized, placebo-controlled trial. Am. J. Kidney Dis. 2015, 66, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezai, A.; Soma, M.; Nakata, K.; Osaka, S.; Ishii, Y.; Yaoita, H.; Hata, H.; Shiono, M. Comparison of febuxostat and allopurinol for hyperuricemia in cardiac surgery patients with chronic kidney disease (NU-FLASH trial for CKD). J. Cardiol. 2015, 66, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.J.; Nakagawa, T.; Jalal, D.; Sánchez-Lozada, L.G.; Kang, D.H.; Ritz, E. Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transpl. 2013, 28, 2221–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, W.; Shrestha, P.; Sumida, K.; Thomas, F.; Sweeney, P.L.; Potukuchi, P.K.; Rhee, C.M.; Streja, E.; Kalantar-Zadeh, K.; Kovesdy, C.P. Association of uric acid-lowering therapy with incident chronic kidney disease. JAMA Netw. Open. 2022, 5, e2215878. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.; Chen, Y.D.; Hollenbeck, C.B.; Reaven, G.M. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991, 266, 3008–3011. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Zhu, J.; Feng, J.; Li, H.; Yu, Q.; Qin, H.; Wei, L.; Zhang, J. Serum uric acid levels and diabetic kidney disease in patients with type 2 diabetes mellitus: A dose-response meta-analysis. Prim. Care Diabetes 2022, 16, 457–465. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef]
- Bailey, C.J. Uric acid and the cardio-renal effects of SGLT2 inhibitors. Diabetes Obes. Metab. 2019, 21, 1291–1298. [Google Scholar] [CrossRef] [Green Version]
- Chino, Y.; Samukawa, Y.; Sakai, S.; Nakai, Y.; Yamaguchi, J.; Nakanishi, T.; Tamai, I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm. Drug Dispos. 2014, 35, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Ren. Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef] [PubMed]
- Suijk, D.L.S.; van Baar, M.J.B.; van Bommel, E.J.M.; Iqbal, Z.; Krebber, M.M.; Vallon, V.; Touw, D.; Hoorn, E.J.; Nieuwdorp, M.; Kramer, M.M.H.; et al. SGLT2 Inhibition and uric acid excretion in patients with type 2 diabetes and normal kidney function. Clin. J. Am. Soc. Nephrol. 2022, 17, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Mabillard, H.; Sayer, J.A.; Olinger, E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol. Dial. Transpl. 2021, 98, gfab268. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Prim. 2019, 5, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleyer, A.J.; Kmoch, S. Autosomal dominant tubulointerstitial kidney disease: Of names and genes. Kidney Int. 2014, 86, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Bleyer, A.J.; Kidd, K.; Živná, M.; Kmoch, S. Autosomal dominant tubulointerstitial kidney disease. Adv. Chronic Kidney Dis. 2017, 24, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Scolari, F.; Caridi, G.; Rampoldi, L.; Tardanico, R.; Izzi, C.; Pirulli, D.; Amoroso, A.; Casari, G.; Ghiggeri, G.M. Uromodulin storage diseases: Clinical aspects and mechanisms. Am. J. Kidney Dis. 2004, 44, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Goldfarb, D.S.; El-Achkar, T.M.; Lieske, J.C.; Wu, X.R. Tamm-Horsfall protein/uromodulin deficiency elicits tubular compensatory responses leading to hypertension and hyperuricemia. Am. J. Physiol. Renal. Physiol. 2018, 314, F1062–F1076. [Google Scholar] [CrossRef] [Green Version]
- Stavrou, C.; Koptides, M.; Tombazos, C.; Psara, E.; Patsias, C.; Zouvani, I.; Kyriacou, K.; Hildebrandt, F.; Christofides, T.; Pierides, A.; et al. Autosomal-dominant medullary cystic kidney disease type 1: Clinical and molecular findings in six large Cypriot families. Kidney Int. 2002, 62, 1385–1394. [Google Scholar] [CrossRef]
- Okorn, C.; Goertz, A.; Vester, U.; Beck, B.B.; Bergmann, C.; Habbig, S.; König, J.; Konrad, M.; Müller, D.; Oh, J.; et al. HNF1B nephropathy has a slow-progressive phenotype in childhood-with the exception of very early onset cases: Results of the German Multicenter HNF1B Childhood Registry. Pediatr. Nephrol. 2019, 34, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Bingham, C.; Hattersley, A.T. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol. Dial. Transpl. 2004, 19, 2703–2708. [Google Scholar] [CrossRef] [PubMed]
- Ferrè, S.; Veenstra, G.J.; Bouwmeester, R.; Hoenderop, J.G.; Bindels, R.J. HNF-1B specifically regulates the transcription of the γa-subunit of the Na+/K+-ATPase. Biochem. Biophys. Res. Commun. 2011, 404, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Ben Salem, C.; Slim, R.; Fathallah, N.; Hmouda, H. Drug-induced hyperuricaemia and gout. Rheumatology 2017, 56, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.F. Metabolic complications associated with use of diuretics. Semin. Nephrol. 2011, 31, 542–552. [Google Scholar] [CrossRef]
- Pascual, E.; Perdiguero, M. Gout, diuretics and the kidney. Ann. Rheum. Dis. 2006, 65, 981–982. [Google Scholar] [CrossRef]
- Jutabha, P.; Anzai, N.; Wempe, M.F.; Wakui, S.; Endou, H.; Sakurai, H. Apical voltage-driven urate efflux transporter NPT4 in renal proximal tubule. Nucleosides Nucleotides Nucleic Acids. 2011, 30, 1302–1311. [Google Scholar] [CrossRef]
- Hamada, T.; Kuwabara, M.; Watanabe, A.; Mizuta, E.; Ohtahara, A.; Omodani, H.; Watanabe, M.; Nakamura, H.; Hirota, Y.; Miyazaki, S.; et al. A comparative study on the effectiveness of losartan/hydrochlorothiazide and telmisartan/hydrochlorothiazide in patients with hypertension. Clin. Exp. Hypertens. 2014, 36, 251–257. [Google Scholar] [CrossRef]
- Mancikova, A.; Krylov, V.; Hurba, O.; Sebesta, I.; Nakamura, M.; Ichida, K.; Stiburkova, B. Functional analysis of novel allelic variants in URAT1 and GLUT9 causing renal hypouricemia type 1 and 2. Clin. Exp. Nephrol. 2016, 20, 578–584. [Google Scholar] [CrossRef]
- Ichida, K.; Hosoyamada, M.; Hisatome, I.; Enomoto, A.; Hikita, M.; Endou, H.; Hosoya, T. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J. Am. Soc. Nephrol. 2004, 15, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.I.; Kang, J.H.; Lee, J.H.; Ha, I.S.; Kim, S.; Komoda, F.; Sekine, T.; Igarashi, T.; Choi, Y. Mutational analysis of idiopathic renal hypouricemia in Korea. Pediatr. Nephrol. 2005, 20, 886–890. [Google Scholar] [CrossRef]
- Shen, H.; Feng, C.; Jin, X.; Mao, J.; Fu, H.; Gu, W.; Liu, A.; Shu, Q.; Du, L. Recurrent exercise-induced acute kidney injury by idiopathic renal hypouricemia with a novel mutation in the SLC2A9 gene and literature review. BMC Pediatr. 2014, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, M.; Kurata, K.; Iwasaki, Y. Acute renal failure with severe loin pain and patchy renal vasoconstriction in a patient with march hemoglobinuria. Clin. Nephrol. 2022, 97, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids. 2008, 27, 608–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Wakabayashi, K.; Totsuka, A.; Hayashi, Y.; Nitta, S.; Hara, K.; Akira, M.; Tomino, Y.; Suzuki, Y. Exercise-induced acute kidney injury in a police officer with hereditary renal hypouricemia. Case Rep. Nephrol. Dial. 2019, 9, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Hosoyamada, M. Hypothetical mechanism of exercise-induced acute kidney injury associated with renal hypouricemia. Biomedicines 2021, 9, 1847. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Uchida, S.; Shibata, S.; Tomioka, N.H.; Matsumoto, K.; Hosoyamada, M. Xanthine oxidoreductase inhibitors suppress the onset of exercise-induced AKI in high HPRT activity Urat1-Uox double knockout mice. J. Am. Soc. Nephrol. 2022, 33, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Klootwijk, E.D.; Reichold, M.; Unwin, R.J.; Kleta, R.; Warth, R.; Bockenhauer, D. Renal Fanconi syndrome: Taking a proximal look at the nephron. Nephrol. Dial. Transpl. 2015, 30, 1456–1460. [Google Scholar]
- Dalmak, S.; Erek, E.; Serdengecti, K.; Okar, I.; Ulku, U.; Basaran, M. A case study of adult-onset hypophosphatemic osteomalacia with idiopathic fanconi syndrome. Nephron 1996, 72, 121–122. [Google Scholar] [CrossRef]
- Hall, A.M.; Bass, P.; Unwin, R.J. Drug-induced renal Fanconi syndrome. QJM 2014, 107, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Perazella, M.A. Tenofovir-induced kidney disease: An acquired renal tubular mitochondriopathy. Kidney Int. 2010, 78, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Park, D.J.; Jang, K.S.; Kim, G.H. Adult idiopathic renal Fanconi syndrome: A case report. Electrolyte Blood Press. 2018, 16, 19–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichter-Konecki, U.; Broman, K.W.; Blau, E.B.; Konecki, D.S. Genetic and physical mapping of the locus for autosomal dominant renal Fanconi syndrome, on chromosome 15q15.3. Am. J. Hum. Genet. 2001, 68, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Magen, D.; Berger, L.; Coady, M.J.; Ilivitzki, A.; Militianu, D.; Tieder, M.; Selig, S.; Lapointe, J.Y.; Zelikovic, I.; Skorecki, K. A loss-of-function mutation in NaPi-IIa and renal Fanconi’s syndrome. N. Engl. J. Med. 2010, 362, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Klootwijk, E.D.; Reichold, M.; Helip-Wooley, A.; Tolaymat, A.; Broeker, C.; Robinette, S.L.; Reinders, J.; Peindl, D.; Renner, K.; Eberhart, K.; et al. Mistargeting of peroxisomal EHHADH and inherited renal Fanconi’s syndrome. N. Engl. J. Med. 2014, 370, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar]
- Sharma, P.; Ng, J.H.; Bijol, V.; Jhaveri, K.D.; Wanchoo, R. Pathology of COVID-19-associated acute kidney injury. Clin. Kidney J. 2021, 14, i30–i39. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar]
- Hassler, L.; Reyes, F.; Sparks, M.A.; Welling, P.; Batlle, D. Evidence for and against direct kidney infection by SARS-CoV-2 in patients with COVID-19. Clin. J. Am. Soc. Nephrol. 2021, 16, 1755–1765. [Google Scholar] [CrossRef]
- Werion, A.; Belkhir, L.; Perrot, M.; Schmit, G.; Aydin, S.; Chen, Z.; Penaloza, A.; De Greef, J.; Yildiz, H.; Pothen, L.; et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020, 98, 1296–1307. [Google Scholar] [CrossRef]
- Dufour, I.; Werion, A.; Belkhir, L.; Wisniewska, A.; Perrot, M.; De Greef, J.; Schmit, G.; Yombi, J.C.; Wittebole, X.; Laterre, P.F.; et al. Serum uric acid, disease severity and outcomes in COVID-19. Crit. Care. 2021, 25, 212. [Google Scholar] [CrossRef]
- Serafinelli, J.; Mastrangelo, A.; Morello, W.; Cerioni, V.F.; Salim, A.; Nebuloni, M.; Montini, G. Kidney involvement and histological findings in two pediatric COVID-19 patients. Pediatr. Nephrol. 2021, 36, 3789–3793. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lu, C.; Gu, H.Q.; Li, Y.; Zhang, G.; Lio, J.; Luo, X.; Zhang, L.; Hu, Y.; Lan, X.; et al. Serum uric acid concentrations and risk of adverse outcomes in patients with COVID-19. Front. Endocrinol. 2021, 12, 633767. [Google Scholar]
- Ellison, D.H.; Ber, T. Clinical practice. The syndrome of inappropriate antidiuresis. N. Engl. J. Med. 2007, 356, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Dorhout Mees, E.J.; Blom van Assendelft, P.; Nieuwenhuis, M.G. Elevation of uric aicd clearance caused by inappropriate antidiuretic hormone secretion. Acta Med. Scand. 1971, 189, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.H. Hypouricemia in the syndrome of inappropriate secretion of antidiuretic hormone. N. Engl. J. Med. 1979, 301, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Decaux, G.; Namias, B.; Gulbis, B.; Soupart, A. Evidence in hyponatremia related to inappropriate secretion of ADH that V1 receptor stimulation contributes to the increase in renal uric acid clearance. J. Am. Soc. Nephrol. 1996, 7, 805–810. [Google Scholar] [CrossRef]
- Taniguchi, K.; Tamura, Y.; Kumagai, T.; Shibata, S.; Uchida, S. Stimulation of V1a receptor increases renal uric acid clearance via urate transporters: Insight into pathogenesis of hypouricemia in SIADH. Clin. Exp. Nephrol. 2016, 20, 845–852. [Google Scholar] [CrossRef]
- Peters, J.P.; Welt, L.G.; Sims, E.A.; Orloff, J.; Needham, J. A salt-wasting syndrome associated with cerebral disease. Trans. Assoc. Am. Physicians 1950, 63, 57–64. [Google Scholar]
- Palmer, B.; Clegg, D. Cerebral salt wasting is a real cause of hyponatremia: Commentary. Kidney360 2022, 3. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Verbalis, J.G. The curious story of cerebral salt wasting: Fact or fiction? Clin. J. Am. Soc. Nephrol. 2020, 15, 1666–1668. [Google Scholar] [CrossRef]
- Sterns, R.; Rondon-Berrios, H. Cerebral salt wasting is a real cause of hyponatremia: CON. Kidney360 2022, 3. Publish Ahead of Print. [Google Scholar] [CrossRef]
- Maesaka, J.K.; Imbriano, L.J.; Miyawaki, N. High prevalence of renal salt wasting without cerebral disease as cause of hyponatremia in general medical wards. Am. J. Med. Sci. 2018, 356, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Maesaka, J.K.; Imbriano, L.J.; Ali, N.M.; Ilamathi, E. Is it cerebral or renal salt wasting? Kidney Int. 2009, 76, 934–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maesaka, J.K.; Imbriano, L.J.; Miyawaki, N. Evolution and evolving resolution of controversy over existence and prevalence of cerebral/renal salt wasting. Curr. Opin. Nephrol. Hypertens 2020, 29, 213–220. [Google Scholar] [PubMed]
- Bitew, S.; Imbriano, L.; Miyawaki, N.; Fishbane, S.; Maesaka, J.K. More on renal salt wasting without cerebral disease: Response to saline infusion. Clin. J. Am. Soc. Nephrol. 2009, 4, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.S.; Kim, G.H. Thiazide-induced hyponatremia. Electrolyte Blood Press. 2010, 8, 51–57. [Google Scholar] [CrossRef]
- Kim, S.; Jo, C.H.; Kim, G.H. The role of vasopressin V2 receptor in drug-Induced hyponatremia. Front. Physiol. 2021, 12, 797039. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperuricemia |
| Chronic kidney disease (CKD) |
| Diabetes mellitus and diabetic kidney disease (DKD) |
| Autosomal dominant tubulointerstitial kidney disease (ADTKD) |
| Thiazide and loop diuretics |
| Hypouricemia |
| Renal hypouricemia type 1 and type 2 |
| Fanconi syndrome: inherited or acquired |
| Coronavirus disease 2019 (COVID-19) |
| Hyponatremic disorders |
| Syndrome of inappropriate antidiuresis (SIAD) |
| Cerebral/renal salt wasting |
| Thiazide-induced hyponatremia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.-H.; Jun, J.-B. Altered Serum Uric Acid Levels in Kidney Disorders. Life 2022, 12, 1891. https://doi.org/10.3390/life12111891
Kim G-H, Jun J-B. Altered Serum Uric Acid Levels in Kidney Disorders. Life. 2022; 12(11):1891. https://doi.org/10.3390/life12111891
Chicago/Turabian StyleKim, Gheun-Ho, and Jae-Bum Jun. 2022. "Altered Serum Uric Acid Levels in Kidney Disorders" Life 12, no. 11: 1891. https://doi.org/10.3390/life12111891
APA StyleKim, G.-H., & Jun, J.-B. (2022). Altered Serum Uric Acid Levels in Kidney Disorders. Life, 12(11), 1891. https://doi.org/10.3390/life12111891