
Novel Circoviruses from Birds Share Common Evolutionary Roots with Fish Origin Circoviruses

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Circovirus Specific PCR and Complete Genome Amplification
2.3. Next-Generation Sequencing
2.4. Software
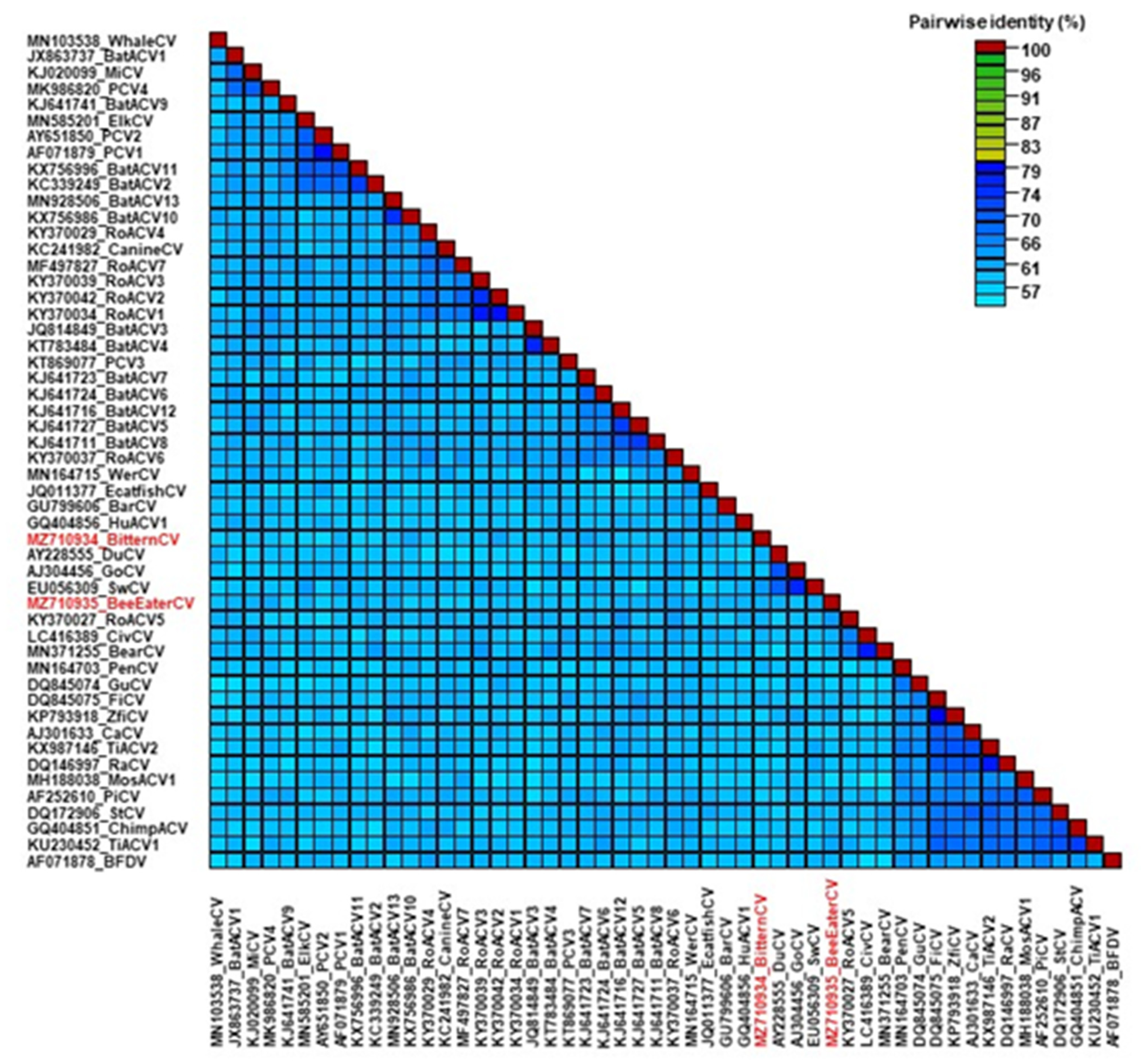
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Ethics approval
References
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A virus phylum unifying seven families of Rep-encoding viruses with single-stranded, circular DNA genomes. J. Virol. 2020, 94, e00582-20. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Circella, E.; Legretto, M.; Pugliese, N.; Caroli, A.; Bozzo, G.; Accogli, G.; Lavazza, A.; Camarda, A. Psittacine beak and feather disease-like illness in Gouldian finches (Chloebia gouldiae). Avian Dis. 2014, 58, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tian, J.; Tan, X.; Yu, H.; Ding, S.; Sun, H.; Yu, X. Pathological observations of an experimental infection of geese with goose circovirus. Avian Pathol. 2011, 40, 55–61. [Google Scholar] [CrossRef]
- Nayar, G.P.; Hamel, A.; Lin, L. Detection and characterization of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. Can. Vet. J. 1997, 38, 385–386. [Google Scholar]
- Raue, R.; Schmidt, V.; Freick, M.; Reinhardt, B.; Johne, R.; Kamphausen, L.; Kaleta, E.; Müller, H.; Krautwald-Junghanns, M.E. A disease complex associated with pigeon circovirus infection, young pigeon disease syndrome. Avian Pathol. 2005, 34, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, B.W.; Niagro, F.D.; Lukert, P.D.; Steffens, W.L.; Latimer, K.S. Characterization of a new virus from cockatoos with psittacine beak and feather disease. Virology 1989, 171, 83–88. [Google Scholar] [CrossRef]
- Shivaprasad, H.L.; Hill, D.; Todd, D.; Smyth, J.A. Circovirus infection in a Gouldian finch (Chloebia gouldiae). Avian Pathol. 2004, 33, 525–529. [Google Scholar] [CrossRef]
- Soike, D.; Kohler, B.; Albrecht, K. A circovirus-like infection of geese related to a runting syndrome. Avian Pathol. 1999, 28, 199–202. [Google Scholar] [CrossRef]
- Soike, D.; Albrecht, K.; Hattermann, K.; Schmitt, C.; Mankertz, A. Novel circovirus in mulard ducks with developmental and feathering disorders. Vet. Rec. 2004, 154, 792–793. [Google Scholar] [CrossRef]
- Todd, D.; Weston, J.; Ball, N.W.; Borghmans, B.J.; Smyth, J.A.; Gelmini, L.; Lavazza, A. Nucleotide sequence-based identification of a novel circovirus of canaries. Avian Pathol. 2001, 30, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Todd, D.; Scott, A.N.; Fringuelli, E.; Shivraprasad, H.L.; Gavier-Widen, D.; Smyth, J.A. Molecular characterization of novel circoviruses from finch and gull. Avian Pathol. 2007, 36, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fehér, E.; Kaszab, E.; Forró, B.; Bali, K.; Marton, S.; Lengyel, G.; Bányai, K. Genome sequence of a mallard duck origin cyclovirus, DuACyV-1. Arch. Virol. 2017, 162, 3925–3929. [Google Scholar] [CrossRef] [PubMed]
- Kaszab, E.; Marton, S.; Forró, B.; Bali, K.; Lengyel, G.; Bányai, K.; Fehér, E. Characterization of the genomic sequence of a novel CRESS DNA virus identified in Eurasian jay (Garrulus glandarius). Arch. Virol. 2018, 163, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef]
- Li, L.; Kapoor, A.; Slikas, B.; Bamidele, O.S.; Wang, C.; Shaukat, S.; Masroor, M.A.; Wilson, M.L.; Ndjango, J.B.; Peeters, M.; et al. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J. Virol. 2010, 84, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Lecis, R.; Mucedda, M.; Pidinchedda, E.; Zobba, R.; Pittau, M.; Alberti, A. Genomic characterization of a novel bat-associated circovirus detected in European Miniopterus schreibersii bats. Virus Genes 2020, 56, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.; Jiang, T.; Hu, T.; Mi, S.; Zhao, Z.; Zhang, F.; Feng, J.; Fan, Q.; He, B.; Tu, C. Molecular characterization of a novel bat-associated circovirus with a poly-T tract in the 3′ intergenic region. Virus Res. 2018, 250, 95–103. [Google Scholar] [CrossRef]
- Moldován, N.; Balázs, Z.S.; Tombácz, D.; Csabai, Z.; Szűcs, A.; Snyder, M.; Boldogkői, Z. Multi-platform analysis reveals a complex transcriptome architecture of a circovirus. Virus Res. 2017, 237, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Graber, J.H. Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 2012, 3, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Fehér, E.; Mihalov-Kovács, E.; Kaszab, E.; Malik, Y.S.; Marton, S.; Bányai, K. Genomic diversity of CRESS DNA viruses in the eukaryotic virome of swine feces. Microorganisms 2021, 9, 1426. [Google Scholar] [CrossRef]
- Morandini, V.; Dugger, K.M.; Ballard, G.; Elrod, M.; Schmidt, A.; Ruoppolo, V.; Lescroël, A.; Jongsomjit, D.; Massaro, M.; Pennycook, J.; et al. Identification of a novel Adelie penguin circovirus at Cape Crozier (Ross Island, Antarctica). Viruses 2019, 11, 1088. [Google Scholar] [CrossRef] [Green Version]
- Nath, B.K.; Das, S.; Roby, J.A.; Sarker, S.; Luque, D.; Raidal, S.R.; Forwood, J.K. Structural perspectives of beak and feather disease virus and porcine circovirus proteins. Viral Immunol. 2021, 34, 49–59. [Google Scholar] [CrossRef]
- Heath, L.; Martin, D.P.; Warburton, L.; Perrin, M.; Horsfield, W.; Kingsley, C.; Rybicki, E.P.; Williamson, A.L. Evidence of unique genotypes of beak and feather disease virus in southern Africa. J. Virol. 2004, 78, 9277–9284. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.M.; Hon, C.C.; Lam, T.Y.; Li, V.Y.; Wong, C.K.; de Oliveira, T.; Leung, F.C. Evidence for recombination in natural populations of porcine circovirus type 2 in Hong Kong and mainland China. J. Gen. Virol. 2007, 88, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Olvera, A.; Cortey, M.; Segalés, J. Molecular evolution of porcine circovirus type 2 genomes: Phylogeny and clonality. Virology 2007, 357, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piewbang, C.; Jo, W.K.; Puff, C.; van der Vries, E.; Kesdangsakonwut, S.; Rungsipipat, A.; Kruppa, J.; Jung, K.; Baumgärtner, W.; Techangamsuwan, S.; et al. Novel canine circovirus strains from Thailand: Evidence for genetic recombination. Sci. Rep. 2018, 8, 7524. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, M.; Cságola, A.; Farkas, S.L.; Székely, C.; Tuboly, T. First detection and analysis of a fish circovirus. J. Gen. Virol. 2011, 92, 1817–1821. [Google Scholar] [CrossRef]
- Lőrincz, M.; Dán, Á.; Láng, M.; Csaba, G.; Tóth, A.G.; Székely, C.; Cságola, A.; Tuboly, T. Novel circovirus in European catfish (Silurus glanis). Arch. Virol. 2012, 157, 1173–1176. [Google Scholar] [CrossRef] [Green Version]
- Patterson, Q.M.; Kraberger, S.; Martin, D.P.; Shero, M.R.; Beltran, R.S.; Kirkham, A.L.; Aleamotu’a, M.; Ainley, D.G.; Kim, S.; Burns, J.M.; et al. Circoviruses and cycloviruses identified in Weddell seal fecal samples from McMurdo Sound, Antarctica. Infect. Genet. Evol. 2021, 95, 105070. [Google Scholar] [CrossRef]
- Todd, D. Circoviruses: Immunosuppressive threats to avian species: A review. Avian Pathol. 2000, 29, 373–394. [Google Scholar] [CrossRef]
- Stenzel, T.; Dziewulska, D.; Tykałowski, B.; Śmiałek, M.; Kowalczyk, J.; Koncicki, A. Immunogenicity of pigeon circovirus recombinant capsid protein in pigeons. Viruses. 2018, 31, 596. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, L.; Shang, H.; Zhou, F.; Wang, C.; Zhang, S.; Gao, P.; Guo, P.; Zhu, R.; Sun, Z.; et al. Effects of duck circovirus on immune function and secondary infection of Avian Pathogenic Escherichia coli. Poult. Sci. 2022, 101799. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Bird Species | Sample | Data about the Rescued Bird |
|---|---|---|
| Common blackbird, Turdus merula | B,K,L,T | Leg injury |
| K,L | Traumatic injuries, internal bleeding | |
| K,L,S | Hemorrhagic fluid in the thoracoabdominal cavity | |
| Common buzzard, Buteo buteo | K,L,S | Caseonecrotic granulomas in lung, liver, and gizzard; mycobacteriosis |
| K,L | Electric shock, necrotizing leg | |
| Common crane, Grus grus | Lesion | Signs of pox virus infection |
| Common house martin, Delichon urbicum | K,L | NA |
| L | Eye lesions, small, pale kidneys | |
| Common kestrel, Falco tinnunculus | K,L | Traumatic injuries |
| K,L | Wing injury | |
| B,K,L | NA | |
| K,L | Electric shock | |
| K,L | Electric shock | |
| Common kingfisher, Alcedo atthis | K,L | NA |
| Common pheasant, Phasianus colchicus | K,L,S | NA |
| Eurasian woodcock, Scolopax rusticola | K,L | Shot injury of the breast |
| European bee-eater, Merops apiaster | K,L | Wing injury, weight loss, degenerated kidneys |
| European Green Woodpecker, Picus viridis | B,K,L,S | Head injury |
| European honey buzzard, Pernis apivorus | K,L | Traumatic injuries |
| Eurasian sparrowhawk, Accipiter nisus | B,K,L | Wing injury |
| K,L | Weight loss, bleeding in the stomach | |
| Great cormorant, Phalacrocorax carbo | K,L | Traumatic injuries, tested positive for polyomavirus |
| Great spotted woodpecker, Dendrocopos major | K,L | Traumatic injuries |
| Grey heron, Ardea cinerea | K,L | Necrotizing wing, visceral gout |
| Little bittern, Ixobrychus minutus | B,K,L | Wing injury, poor body condition, weight loss, enlarged liver with lesions |
| B,K,L | Traumatic injuries of the left body site, kidney injury | |
| B,K,L | Poor body condition, broken lower mandible, visceral gout | |
| Little owl, Athene noctua | K,L | Poor body condition and weight loss |
| Mute swan, Cygnus olor | K,L | Weight loss, diarrhea |
| Tawny owl, Strix aluco | K | NA |
| K,L | Enlarged liver, liver failure, pale kidneys | |
| Water rail, Rallus aquaticus | K,L | Pale kidneys |
| Little Bittern Circovirus | European Bee-Eater Circovirus | |
|---|---|---|
| Genome | 1935 nt | 1960 nt |
| 5′ intergenic region | nt 1859–51 | nt 1894–33 |
| Nonanucleotide | TAGTATTAC | TAGTATTAC |
| Stem-loop inverted repeat | CACAGGCGCCGG | GCCGAGGTGGCCG |
| rep | nt 52–999 (315 aa) | nt 34–945 (303 aa) |
| RCR motif I | MTLNN | FTLNN |
| RCR motif II | PHLQG | PHLQG |
| RCR motif III | YCSK | YCSK |
| Walker-A motif | GPPGCGKT | GPPGCGKS |
| Walker-B motif | VIDDF | IVDDF |
| Motif C | ITSN | ITSN |
| cp | nt 1858–1229 (209 aa) | nt 1893–1171 (240 aa) |
| 3′ intergenic region | nt 1000–1228 | nt 946–1170 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, E.; Kaszab, E.; Bali, K.; Hoitsy, M.; Sós, E.; Bányai, K. Novel Circoviruses from Birds Share Common Evolutionary Roots with Fish Origin Circoviruses. Life 2022, 12, 368. https://doi.org/10.3390/life12030368
Fehér E, Kaszab E, Bali K, Hoitsy M, Sós E, Bányai K. Novel Circoviruses from Birds Share Common Evolutionary Roots with Fish Origin Circoviruses. Life. 2022; 12(3):368. https://doi.org/10.3390/life12030368
Chicago/Turabian StyleFehér, Enikő, Eszter Kaszab, Krisztina Bali, Márton Hoitsy, Endre Sós, and Krisztián Bányai. 2022. "Novel Circoviruses from Birds Share Common Evolutionary Roots with Fish Origin Circoviruses" Life 12, no. 3: 368. https://doi.org/10.3390/life12030368