
Identification of Cell Markers and Their Expression Patterns in Skin Based on Single-Cell RNA-Sequencing Profiles

 ,
,  ,
, 
Abstract
:1. Introduction
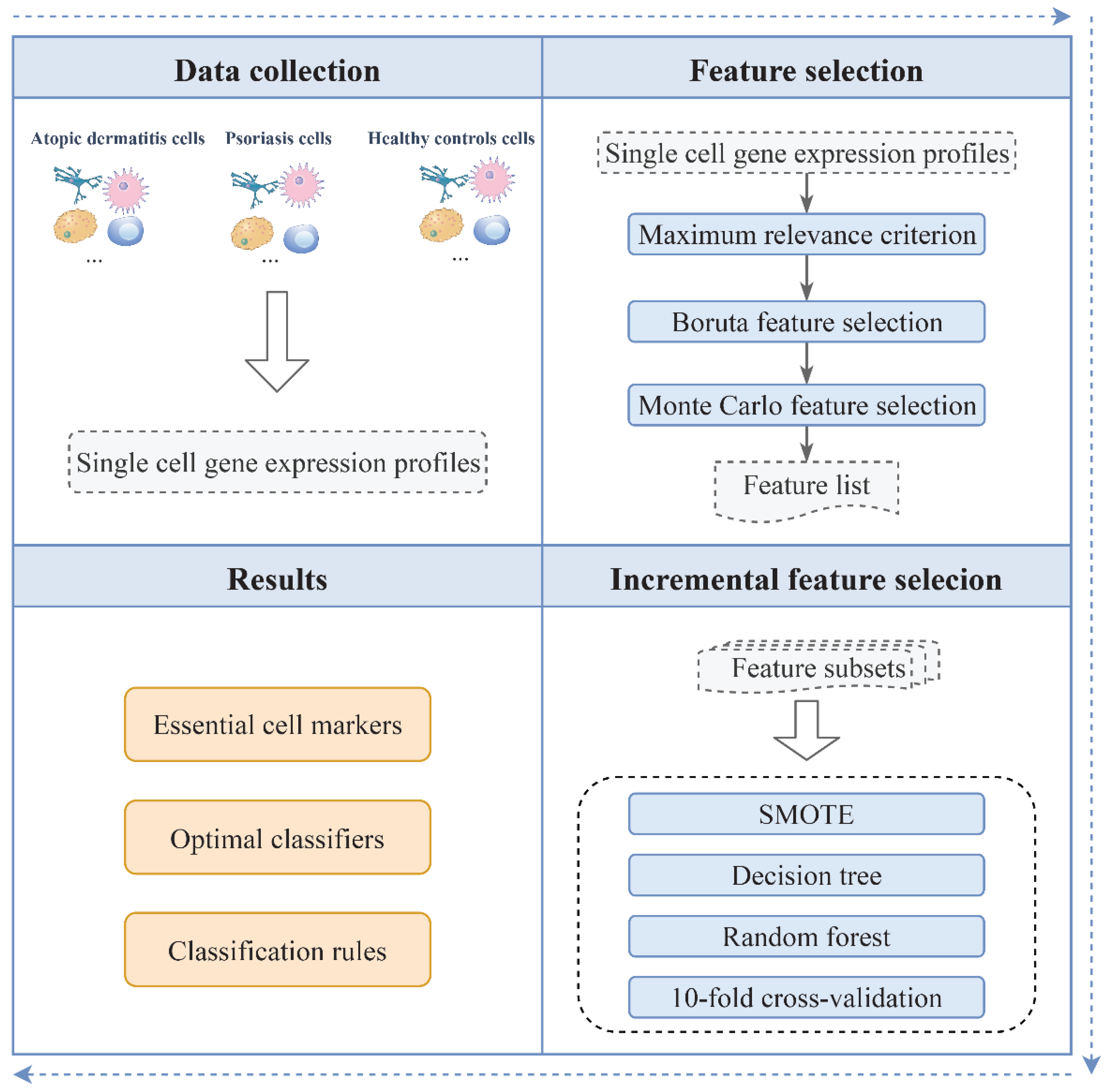
2. Materials and Methods
2.1. Single-Cell RNA-Sequencing Profiles of Skin Samples
2.2. Feature Selection
2.2.1. Feature Exclusion Based on the Maximum Relevance Criterion
2.2.2. Boruta Feature Filtering
2.2.3. Monte Carlo Feature Selection
2.3. Incremental Feature Selection
2.4. Synthetic Minority Oversampling Technique
2.5. Classification Algorithms
2.5.1. RF
2.5.2. DT
2.6. Performance Measurement
3. Results and Discussion
3.1. Features Selected by Using the Maximum Relevance Criterion, Boruta, and MCFS Methods
3.2. Determination of the Optimal Features by the IFS Method
3.3. Classification Rules Extracted by the Optimal DT Classifier
3.4. Computation Time vs. MCC
3.5. Analysis of Features
3.6. Analysis of the Rules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. Eur. J. Dermatol. 2002, 12, 390–401. [Google Scholar]
- McGrath, J.; Eady, R.; Pope, F. Anatomy and organization of human skin. Rook’s Textb. Dermatol. 2004, 1, 3.2–3.8. [Google Scholar]
- Maibach, H.; Honari, G. Applied Dermatotoxicology: Clinical Aspects; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Carlson, B.M. Human Embryology and Developmental Biology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Madison, K.C. Barrier function of the skin:“La raison d’etre” of the epidermis. J. Investig. Dermatol. 2003, 121, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Berke, R.; Singh, A.; Guralnick, M. Atopic dermatitis: An overview. Am. Fam. Physician 2012, 86, 35–42. [Google Scholar] [PubMed]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, ige autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Bieber, T.; Leung, D.Y. Immune mechanisms leading to atopic dermatitis. J. Allergy Clin. Immunol. 2003, 112, S128–S139. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Elder, J.T. Psoriasis: Epidemiology. Clin. Dermatol. 2007, 25, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Davidovici, B.; Krueger, J.G. New insights in the immunologic basis of psoriasis. In Seminars in Cutaneous Medicine and Surgery; NIH Public Access: Washington, DC, USA, 2010; p. 3. [Google Scholar]
- Lowes, M.A.; Suarez-Farinas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Suryawanshi, H.; Morozov, P.; Gay-Mimbrera, J.; Duca, E.D.; Kim, H.J.; Kameyama, N.; Estrada, Y.; Der, E.; Krueger, J.G. Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J. Allergy Clin. Immunol. 2020, 145, 1615–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, H.; Angsana, J.; Bissonnette, R.; Muñoz-Elías, E.J.; Sarfati, M. Inflammatory skin disorders: Monocyte-derived cells take center stage. Front. Immunol. 2021, 12, 691806. [Google Scholar] [CrossRef]
- Penkava, F.; Velasco-Herrera, M.D.C.; Young, M.D.; Yager, N.; Nwosu, L.N.; Pratt, A.G.; Lara, A.L.; Guzzo, C.; Maroof, A.; Mamanova, L. Single-cell sequencing reveals clonal expansions of pro-inflammatory synovial cd8 t cells expressing tissue-homing receptors in psoriatic arthritis. Nat. Commun. 2020, 11, 4767. [Google Scholar] [CrossRef]
- Reynolds, G.; Vegh, P.; Fletcher, J.; Poyner, E.F.M.; Stephenson, E.; Goh, I.; Botting, R.A.; Huang, N.; Olabi, B.; Dubois, A.; et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 2021, 371, eaba6500. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dramiński, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte Carlo feature selection for supervised classification. Bioinformatics 2007, 24, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Safavian, S.R.; Landgrebe, D. A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 1991, 21, 660–674. [Google Scholar] [CrossRef] [Green Version]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; pp. 1137–1143. [Google Scholar]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Chen, L.; Li, Z.; Zhang, S.; Zhang, Y.-H.; Huang, T.; Cai, Y.-D. Predicting rna 5-methylcytosine sites by using essential sequence features and distributions. BioMed Res. Int. 2022, 2022, 4035462. [Google Scholar] [CrossRef]
- Ding, S.; Wang, D.; Zhou, X.; Chen, L.; Feng, K.; Xu, X.; Huang, T.; Li, Z.; Cai, Y. Predicting heart cell types by using transcriptome profiles and a machine learning method. Life 2022, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, L.; Chen, L. Identification of protein functions in mouse with a label space partition method. Math. Biosci. Eng. 2022, 19, 3820–3842. [Google Scholar] [CrossRef]
- Chen, W.; Chen, L.; Dai, Q. Impt-fdnpl: Identification of membrane protein types with functional domains and a natural language processing approach. Comput. Math. Methods Med. 2021, 2021, 7681497. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, L. Identification of drug–disease associations by using multiple drug and disease networks. Curr. Bioinform. 2022, 17, 48–59. [Google Scholar] [CrossRef]
- Jia, Y.; Zhao, R.; Chen, L. Similarity-based machine learning model for predicting the metabolic pathways of compounds. IEEE Access 2020, 8, 130687–130696. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, L.; Lu, J. A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 2018, 306, 136–144. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Zhang, Y.-H.; Zeng, T.; Chen, L.; Huang, T.; Cai, Y.-D. Determining protein–protein functional associations by functional rules based on gene ontology and kegg pathway. Biochim. Biophys. Acta BBA Proteins Proteom. 2021, 1869, 140621. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Li, H.; Zeng, T.; Chen, L.; Li, Z.; Huang, T.; Cai, Y.D. Identifying transcriptomic signatures and rules for sars-cov-2 infection. Front. Cell Dev. Biol. 2021, 8, 627302. [Google Scholar] [CrossRef]
- Yuan, F.; Li, Z.; Chen, L.; Zeng, T.; Zhang, Y.-H.; Ding, S.; Huang, T.; Cai, Y.-D. Identifying the signatures and rules of circulating extracellular microrna for distinguishing cancer subtypes. Front. Genet. 2021, 12, 651610. [Google Scholar] [CrossRef] [PubMed]
- Jurman, G.; Riccadonna, S.; Furlanello, C. A comparison of mcc and cen error measures in multi-class prediction. PLoS ONE 2012, 7, e41882. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta BBA Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Gorodkin, J. Comparing two k-category assignments by a k-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef]
- Liu, H.; Hu, B.; Chen, L.; Lu, L. Identifying protein subcellular location with embedding features learned from networks. Curr. Proteom. 2021, 18, 646–660. [Google Scholar] [CrossRef]
- Boegel, S.; Löwer, M.; Bukur, T.; Sorn, P.; Castle, J.C.; Sahin, U. Hla and proteasome expression body map. BMC Med. Genom. 2018, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Schutt, C.; Mirizio, E.; Salgado, C.; Reyes-Mugica, M.; Wang, X.; Chen, W.; Grunwaldt, L.; Schollaert, K.L.; Torok, K.S. Transcriptomic evaluation of pediatric localized scleroderma skin with histological and clinical correlation. Arthritis Rheumatol. 2021, 73, 1921–1930. [Google Scholar] [CrossRef]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The hla genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Hubbard, M.; Jones, B.M. Use of cd45 fluorescence and side-scatter characteristics for gating lymphocytes when using the whole blood lysis procedure and flow cytometry. J. Int. Soc. Anal. Cytol. 1996, 26, 16–21. [Google Scholar] [CrossRef]
- Hermiston, M.L.; Xu, Z.; Weiss, A. Cd45: A critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 2003, 21, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Rheinländer, A.; Schraven, B.; Bommhardt, U. Cd45 in human physiology and clinical medicine. Immunol. Lett. 2018, 196, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Marques, M.R.; Nguyen, B.T.; Horner, J.S.; Papazoglu, C.; Bronson, R.T.; Mills, A.A.; Attardi, L.D. Perp is a p63-regulated gene essential for epithelial integrity. Cell 2005, 120, 843–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudry, V.G.; Ihrie, R.A.; Jacobs, S.B.; Nguyen, B.; Pathak, N.; Park, E.; Attardi, L.D. Loss of the desmosomal component perp impairs wound healing in vivo. Dermatol. Res. Pract. 2010, 2010, 759731. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Lullo, E.D.; Malatesta, M. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Na, N.; Zhang, X.; Zhao, Y. The biological function and significance of cd74 in immune diseases. Inflamm. Res. 2017, 66, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, M.R.; Patel, R.A.; Adem, S.; Deleon, N.M.D.; Shen, A.H.; Sokol, J.; Yen, S.; Chang, E.Y.; Nazerali, R.; Nguyen, D. The antifibrotic adipose-derived stromal cell: Grafted fat enriched with cd74+ adipose-derived stromal cells reduces chronic radiation-induced skin fibrosis. Stem Cells Transl. Med. 2020, 9, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Ascensión, A.M.; Fuertes-Álvarez, S.; Ibañez-Solé, O.; Izeta, A.; Araúzo-Bravo, M.J. Human dermal fibroblast subpopulations are conserved across single-cell rna sequencing studies. J. Investig. Dermatol. 2020, 141, 1735–1744. [Google Scholar] [CrossRef]
- Zukauskas, A.; Merley, A.; Li, D.; Ang, L.-H.; Sciuto, T.E.; Salman, S.; Dvorak, A.M.; Dvorak, H.F.; Jaminet, S.-C.S. Tm4sf1: A tetraspanin-like protein necessary for nanopodia formation and endothelial cell migration. Angiogenesis 2011, 14, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Has, C.; Bruckner-Tuderman, L. Molecular and diagnostic aspects of genetic skin fragility. J. Dermatol. Sci. 2006, 44, 129–144. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, P.; Zhao, M.; Liang, G.; Yin, H.; Zhang, G.; Wen, H.; Lu, Q. Demethylation of the fcer1g promoter leads to fcεri overexpression on monocytes of patients with atopic dermatitis. Allergy 2012, 67, 424–430. [Google Scholar] [CrossRef]
- Schäfer, B.; Piliponsky, A.M.; Oka, T.; Song, C.H.; Gerard, N.P.; Gerard, C.; Tsai, M.; Kalesnikoff, J.; Galli, S.J. Mast cell anaphylatoxin receptor expression can enhance ige-dependent skin inflammation in mice. J. Allergy Clin. 2013, 131, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, R.; Smith, C.; DuHadaway, J.B.; Chandler, P.; Baban, B.; Merlo, L.M.; Pigott, E.; Keough, M.P.; Rust, S.; Mellor, A.L. Ido2 is critical for ido1-mediated t-cell regulation and exerts a non-redundant function in inflammation. Int. Immunol. 2014, 26, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Ando, T.; Ogiso, H.; Arioka, Y.; Saito, K.; Seishima, M. Inhibition of indoleamine 2, 3-dioxygenase activity accelerates skin wound healing. Biomaterials 2015, 53, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Llamas-Velasco, M.; Bonay, P.; Concha-Garzón, M.J.; Corvo-Villén, L.; Vara, A.; Cibrian, D.; Sanguino-Pascual, A.; Sánchez-Madrid, F.; Fuente, H.D.l.; Dauden, E. Immune cells from patients with psoriasis are defective in inducing indoleamine 2, 3-dioxygenase expression in response to inflammatory stimuli. Br. J. Dermatol. 2017, 176, 695–704. [Google Scholar] [CrossRef]
- Staudacher, A.; Hinz, T.; Novak, N.; Bubnoff, D.v.; Bieber, T. Exaggerated ido 1 expression and activity in langerhans cells from patients with atopic dermatitis upon viral stimulation: A potential predictive biomarker for high risk of eczema herpeticum. Allergy 2015, 70, 1432–1439. [Google Scholar] [CrossRef] [Green Version]
- Lowes, M.A.; Chamian, F.; Abello, M.V.; Fuentes-Duculan, J.; Lin, S.-L.; Nussbaum, R.; Novitskaya, I.; Carbonaro, H.; Cardinale, I.; Kikuchi, T. Increase in tnf-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-cd11a). Proc. Natl. Acad. Sci. USA 2005, 102, 19057–19062. [Google Scholar] [CrossRef] [Green Version]
- Koga, T.; Duan, H.; Urabe, K.; Furue, M. In situ localization of cd83-positive dendritic cells in psoriatic lesions. Dermatology 2002, 204, 100–103. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Lowes, M.A.; Fuentes-Duculan, J.; Whynot, J.; Novitskaya, I.; Cardinale, I.; Haider, A.; Khatcherian, A.; Carucci, J.A.; Bergman, R. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J. Allergy Clin. Immunol. 2007, 119, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.A.; Huchenq, A.; Kezic, S.; Serre, G.; Jonca, N. Mice deficient for the epidermal dermokine β and γ isoforms display transient cornification defects. J. Cell Sci. 2014, 127, 2862–2872. [Google Scholar]
- Tokuriki, A.; Chino, T.; Luong, V.H.; Oyama, N.; Higashi, K.; Saito, K.; Hasegawa, M. Dermokine β/γ deficiency enhances imiquimod-induced psoriasis-like inflammation. J. Dermatol. Sci. 2016, 84, e161. [Google Scholar] [CrossRef]
- Basile, G.d.S.; Geissmann, F.; Flori, E.; Uring-Lambert, B.; Soudais, C.; Cavazzana-Calvo, M.; Durandy, A.; Jabado, N.; Fischer, A.; Deist, F.L. Severe combined immunodeficiency caused by deficiency in either the δ or the ε subunit of cd3. J. Clin. Investig. 2004, 114, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Puel, A.; Ziegler, S.F.; Buckley, R.H.; Leonard, W.J. Defective il7r expression in t-b+ nk+ severe combined immunodeficiency. Nat. Genet. 1998, 20, 394–397. [Google Scholar] [CrossRef]
- Liu, X.; Leung, S.; Wang, C.; Tan, Z.; Wang, J.; Guo, T.B.; Fang, L.; Zhao, Y.; Wan, B.; Qin, X. Crucial role of interleukin-7 in t helper type 17 survival and expansion in autoimmune disease. Nat. Med. 2010, 16, 191–197. [Google Scholar] [CrossRef]
- Jung, H.; Kim, M.J.; Kim, D.O.; Kim, W.S.; Yoon, S.-J.; Park, Y.-J.; Yoon, S.R.; Kim, T.-D.; Suh, H.-W.; Yun, S. Txnip maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013, 18, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulski, J.K.; Kenworthy, W.; Bellgard, M.; Taplin, R.; Okamoto, K.; Oka, A.; Mabuchi, T.; Ozawa, A.; Tamiya, G.; Inoko, H. Gene expression profiling of japanese psoriatic skin reveals an increased activity in molecular stress and immune response signals. J. Mol. Med. 2005, 83, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Llamas-Velasco, M.; Reolid, A.; Sanz-García, A.; Alonso-Guirado, L.; García-Martínez, J.; Sánchez-Jiménez, P.; Muñoz-Aceituno, E.; Daudén, E.; Abad-Santos, F.; Ovejero-Benito, M. Methylation in psoriasis. Does sex matter? J. Eur. Acad. Dermatol. Venereol. 2020, 35, e161–e163. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, H.; Hou, Y.; Wei, T.; Liu, J. Plasmalemma vesicle-associated protein: A crucial component of vascular homeostasis. Exp. Ther. Med. 2016, 12, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Herrnberger, L.; Seitz, R.; Kuespert, S.; Bösl, M.R.; Fuchshofer, R.; Tamm, E.R. Lack of endothelial diaphragms in fenestrae and caveolae of mutant plvap-deficient mice. Histochem. Cell Biol. 2012, 138, 709–724. [Google Scholar] [CrossRef]
- Strickland, L.A.; Jubb, A.M.; Hongo, J.A.; Zhong, F.; Burwick, J.; Fu, L.; Frantz, G.D.; Koeppen, H. Plasmalemmal vesicle-associated protein (plvap) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-a (vegf). J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2005, 206, 466–475. [Google Scholar] [CrossRef]
- Ekelund, E.; Sääf, A.; Tengvall-Linder, M.; Melen, E.; Link, J.; Barker, J.; Reynolds, N.J.; Meggitt, S.J.; Kere, J.; Wahlgren, C.-F. Elevated expression and genetic association links the socs3 gene to atopic dermatitis. Am. J. Hum. Genet. 2006, 78, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, Y.; Bae, S.J.; Katayama, I. Overexpression of the suppressor of cytokine signalling 3 (socs3) in severe atopic dermatitis. Clin. Exp. Dermatol. Exp. Dermatol. 2006, 31, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The cxcl8/il-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Marriott, H.M.; Gascoyne, K.A.; Gowda, R.; Geary, I.; Nicklin, M.J.; Iannelli, F.; Pozzi, G.; Mitchell, T.J.; Whyte, M.K.; Sabroe, I. Interleukin-1β regulates cxcl8 release and influences disease outcome in response to streptococcus pneumoniae, defining intercellular cooperation between pulmonary epithelial cells and macrophages. Infect. Immun. 2012, 80, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Ha, H.; Debnath, B.; Neamati, N. Role of the cxcl8-cxcr1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543. [Google Scholar] [CrossRef] [PubMed]
- Homey, B.; Meller, S. Chemokines and other mediators as therapeutic targets in psoriasis vulgaris. Clin. Dermatol. 2008, 26, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Bruch-Gerharz, D.; Fehsel, K.; Suschek, C.; Michel, G.; Ruzicka, T.; Kolb-Bachofen, V. A proinflammatory activity of interleukin 8 in human skin: Expression of the inducible nitric oxide synthase in psoriatic lesions and cultured keratinocytes. J. Exp. Med. 1996, 184, 2007–2012. [Google Scholar] [CrossRef]
- Carrier, Y.; Ma, H.-L.; Ramon, H.E.; Napierata, L.; Small, C.; O’toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M. Inter-regulation of th17 cytokines and the il-36 cytokines in vitro and in vivo: Implications in psoriasis pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [Green Version]
- Hulshof, L.; Hack, D.; Hasnoe, Q.; Dontje, B.; Jakasa, I.; Riethmüller, C.; McLean, W.; Aalderen, W.v.; Land, B.V.; Kezic, S. A minimally invasive tool to study immune response and skin barrier in children with atopic dermatitis. Br. J. Dermatol. 2019, 180, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-K.; Leung, K.M.-L.; Qiu, H.-N.; Chow, J.Y.-S.; Choi, A.O.K.; Lam, C.W.-K. Activation of eosinophils interacting with dermal fibroblasts by pruritogenic cytokine il-31 and alarmin il-33: Implications in atopic dermatitis. PLoS ONE 2012, 7, e29815. [Google Scholar] [CrossRef] [Green Version]
- Hoober, J.K. Asgr1 and its enigmatic relative, clec10a. Int. J. Mol. Sci. 2020, 21, 4818. [Google Scholar] [CrossRef]
- He, H.; Li, R.; Choi, S.; Zhou, L.; Pavel, A.; Estrada, Y.D.; Krueger, J.G.; Guttman-Yassky, E. Increased cardiovascular and atherosclerosis markers in blood of older patients with atopic dermatitis. Ann. Allergy Asthma Immunol. 2020, 124, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.K.; Wadsworth, M.H., II; Gierahn, T.M.; Do, T.; Weiss, D.; Andrade, P.R.; Ma, F.; Silva, B.J.d.A.; Shao, S.; Tsoi, L.C. Second-strand synthesis-based massively parallel scrna-seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity 2020, 53, 878–894. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Olesen, C.M.; Pavel, A.B.; Clausen, M.-L.; Wu, J.; Estrada, Y.; Zhang, N.; Agner, T.; Guttman-Yassky, E. Tape-strip proteomic profiling of atopic dermatitis on dupilumab identifies minimally invasive biomarkers. Front. Immunol. 2020, 11, 1768. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification Algorithm | Number of Features | ACC | MCC |
|---|---|---|---|
| Random forest | 795 | 0.951 | 0.949 |
| Random forest | 225 | 0.932 | 0.931 |
| Decision tree | 805 | 0.815 | 0.810 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Ding, S.; Wang, D.; Chen, L.; Feng, K.; Huang, T.; Li, Z.; Cai, Y. Identification of Cell Markers and Their Expression Patterns in Skin Based on Single-Cell RNA-Sequencing Profiles. Life 2022, 12, 550. https://doi.org/10.3390/life12040550
Zhou X, Ding S, Wang D, Chen L, Feng K, Huang T, Li Z, Cai Y. Identification of Cell Markers and Their Expression Patterns in Skin Based on Single-Cell RNA-Sequencing Profiles. Life. 2022; 12(4):550. https://doi.org/10.3390/life12040550
Chicago/Turabian StyleZhou, Xianchao, Shijian Ding, Deling Wang, Lei Chen, Kaiyan Feng, Tao Huang, Zhandong Li, and Yudong Cai. 2022. "Identification of Cell Markers and Their Expression Patterns in Skin Based on Single-Cell RNA-Sequencing Profiles" Life 12, no. 4: 550. https://doi.org/10.3390/life12040550
APA StyleZhou, X., Ding, S., Wang, D., Chen, L., Feng, K., Huang, T., Li, Z., & Cai, Y. (2022). Identification of Cell Markers and Their Expression Patterns in Skin Based on Single-Cell RNA-Sequencing Profiles. Life, 12(4), 550. https://doi.org/10.3390/life12040550