
CAR-T Cells for the Treatment of Lung Cancer

 ,
,  , , , , , , , , ,
, , , , , , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
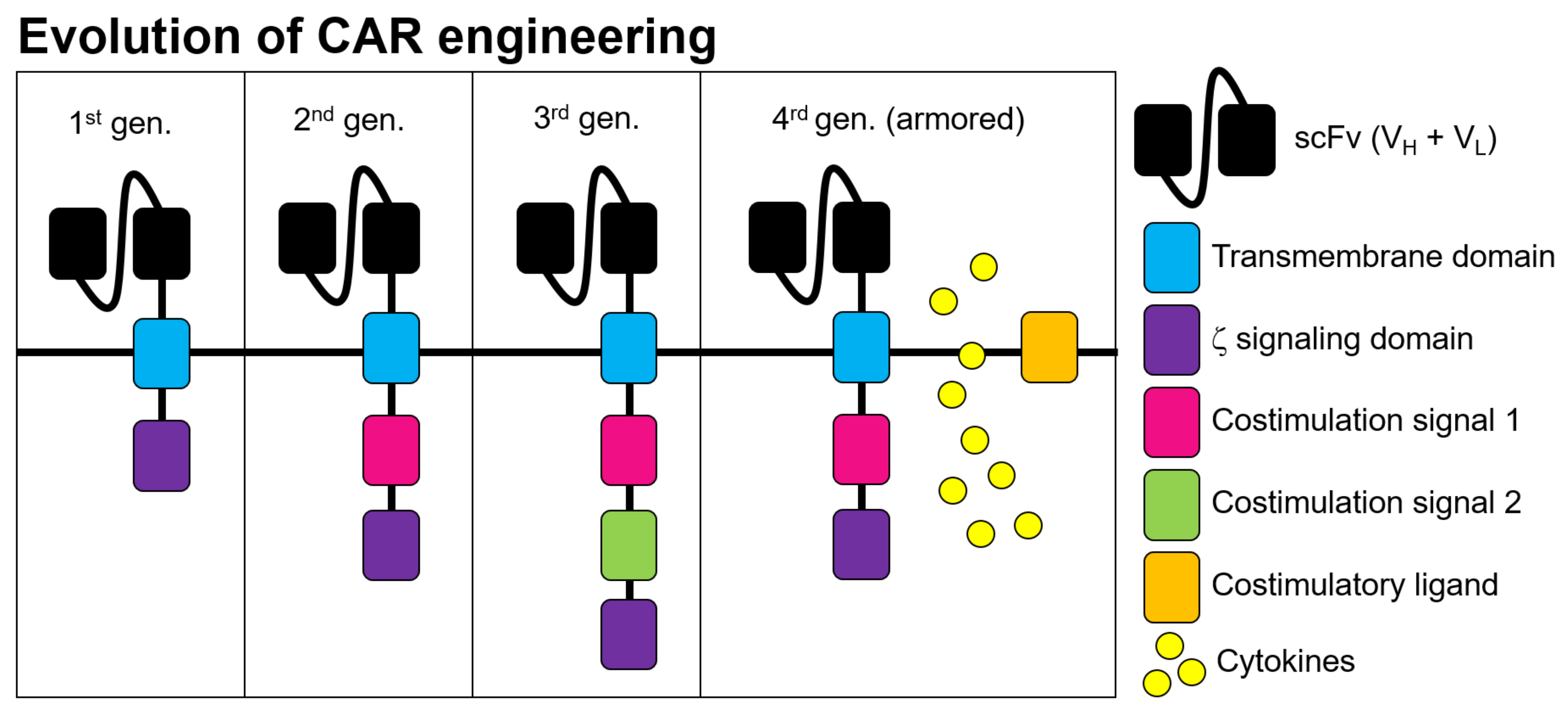
2. Pre-Clinical Development of CAR-T Cells
3. Clinical Development of CAR-T Cell Therapies
3.1. Hematological Malignancies
3.2. Solid Tumors
3.3. Current Development of CAR-T Clinical Trials for Lung Cancer
4. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Chocarro, L.; Blanco, E.; Zuazo, M.; Arasanz, H.; Bocanegra, A.; Fernandez-Rubio, L.; Morente, P.; Fernandez-Hinojal, G.; Echaide, M.; Garnica, M.; et al. Understanding LAG-3 Signaling. Int. J. Mol. Sci. 2021, 22, 5282. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Arasanz, H.; Chocarro, L.; Bocanegra, A.; Zuazo, M.; Fernandez-Hinojal, G.; Blanco, E.; Vera, R.; Escors, D.; Kochan, G. Systemic Blood Immune Cell Populations as Biomarkers for the Outcome of Immune Checkpoint Inhibitor Therapies. Int. J. Mol. Sci. 2020, 21, 2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Chocarro, L.; Blanco, E.; Martinez, M.; Morilla, I.; Fernandez, G.; Teijeira, L.; Morente, P.; et al. Hyperprogressive Disease: Main Features and Key Controversies. Int. J. Mol. Sci. 2021, 22, 3736. [Google Scholar] [CrossRef]
- Arasanz, H.; Zuazo, M.; Bocanegra, A.; Gato, M.; Martinez-Aguillo, M.; Morilla, I.; Fernandez, G.; Hernandez, B.; Lopez, P.; Alberdi, N.; et al. Early detection of hyperprogressive disease in non-small cell lung cancer by monitoring of systemic T cell dynamics. Cancers 2020, 12, 344. [Google Scholar] [CrossRef] [Green Version]
- Onesti, C.E.; Freres, P.; Jerusalem, G. Atypical patterns of response to immune checkpoint inhibitors: Interpreting pseudoprogression and hyperprogression in decision making for patients’ treatment. J. Thorac. Dis. 2019, 11, 35–38. [Google Scholar] [CrossRef]
- Saada-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Hart, D.P.; Xue, S.A.; Cesco-Gaspere, M.; Stauss, H.J. T-cell receptor gene therapy for cancer: The progress to date and future objectives. Expert Opin. Biol. Ther. 2007, 7, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Perro, M.; Tsang, J.; Xue, S.A.; Escors, D.; Cesco-Gaspere, M.; Pospori, C.; Gao, L.; Hart, D.; Collins, M.; Stauss, H.; et al. Generation of multi-functional antigen-specific human T-cells by lentiviral TCR gene transfer. Gene 2010, 17, 721–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauss, H.J.; Cesco-Gaspere, M.; Thomas, S.; Hart, D.P.; Xue, S.A.; Holler, A.; Wright, G.; Perro, M.; Little, A.M.; Pospori, C.; et al. Monoclonal T-cell receptors: New reagents for cancer therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1744–1750. [Google Scholar] [CrossRef]
- Escors, D.; Breckpot, K. Lentiviral Vectors in Gene Therapy: Their Current Status and Future Potential. Arch Immunol. Ther. Exp. 2010, 58, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Hartmann, J.; Schussler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Westin, J. CAR T Cells. Adv. Exp. Med. Biol. 2021, 1342, 297–317. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Subklewe, M.; von Bergwelt-Baildon, M.; Humpe, A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus. Med. Hemother. Off. Organ Dtsch. Ges. Transfus. Immunhamatol. 2019, 46, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Y.; Shao, Y.; Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 2020, 13, 54. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Latouche, J.B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Riviere, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Wang, K.; Wei, C.; Feng, D.; Liu, Y.; He, Q.; Xu, X.; Wang, C.; Zhao, S.; Lv, L.; et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front. Immunol. 2021, 12, 609421. [Google Scholar] [CrossRef]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2019, 129, 3464. [Google Scholar] [CrossRef]
- Zah, E.; Lin, M.Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [Green Version]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Wang, Z.; Wang, Y.; Liu, Y.; Dai, H.; Tong, C.; Guo, Y.; Guo, B.; Ti, D.; Han, X.; et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, C.H.; van Elzakker, P.; Langeveld, S.C.; Sleijfer, S.; Gratama, J.W. Process validation and clinical evaluation of a protocol to generate gene-modified T lymphocytes for imunogene therapy for metastatic renal cell carcinoma: GMP-controlled transduction and expansion of patient’s T lymphocytes using a carboxy anhydrase IX-specific scFv transgene. Cytotherapy 2006, 8, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef]
- Till, B.G.; Jensen, M.C.; Wang, J.; Chen, E.Y.; Wood, B.L.; Greisman, H.A.; Qian, X.; James, S.E.; Raubitschek, A.; Forman, S.J.; et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008, 112, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Yeku, O.; Li, X.; Brentjens, R.J. Adoptive T-Cell Therapy for Solid Tumors. Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncology. Annu. Meet. 2017, 37, 193–204. [Google Scholar] [CrossRef]
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl. Res. 2017, 187, 1–10. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. CCS 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Blanco, E.; Ibanez-Vea, M.; Hernandez, C.; Drici, L.; Martinez de Morentin, X.; Gato, M.; Ausin, K.; Bocanegra, A.; Zuazo, M.; Chocarro, L.; et al. A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets. J. Pers. Med. 2021, 11, 542. [Google Scholar] [CrossRef]
- Ortiz-Espinosa, S.; Morales, X.; Senent, Y.; Alignani, D.; Tavira, B.; Macaya, I.; Ruiz, B.; Moreno, H.; Remirez, A.; Sainz, C.; et al. Complement C5a induces the formation of neutrophil extracellular traps by myeloid-derived suppressor cells to promote metastasis. Cancer Lett. 2022, 529, 70–84. [Google Scholar] [CrossRef]
- Ibanez-Vea, M.; Zuazo, M.; Gato, M.; Arasanz, H.; Fernandez-Hinojal, G.; Escors, D.; Kochan, G. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Current Knowledge and Future Perspectives. Arch Immunol. Ther. Exp. 2017, 66, 113–123. [Google Scholar] [CrossRef]
- Gato-Canas, M.; Martinez de Morentin, X.; Blanco-Luquin, I.; Fernandez-Irigoyen, J.; Zudaire, I.; Liechtenstein, T.; Arasanz, H.; Lozano, T.; Casares, N.; Chaikuad, A.; et al. A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget 2015, 6, 27160–27175. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Gato, M.; Caliendo, F.; Kochan, G.; Blanco-Luquin, I.; Van der Jeught, K.; Arce, F.; Guerrero-Setas, D.; Fernandez-Irigoyen, J.; et al. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget 2014, 5, 7843–7857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Specht, J.M.; Lee, S.; Turtle, C.J.; Berger, C.; Baladrishnan, A.; Srivastava, S.; Voillet, V.; Veatch, J.; Gooley, T.; Mullane, E.; et al. A phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res. 2018, 78, CT131. [Google Scholar]
- Specht, J.M.; Lee, S.; Turtle, C.; Berger, C.; Veatch, J.; Gooley, T.; Mullane, E.; Chaney, C.; Riddel, S.R.; Maloney, D.G. Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, TPS79. [Google Scholar] [CrossRef]
- Haanen, J.; Mackensen, A.; Koenecke, C.; Alsdorf, W.; Desuki, A.; Wagner-Drouet, E.; Heudobler, D.; Borchmann, P.; Wiegert, E.; Schultz, C.; et al. LBA1-BNT211: A Phase I/II trial to evaluate safety and efficacy of CLDN6 CAR-T cells and CARVac-mediated in vivo expansion in patients with CLDN6+ advanced solid tumors. Ann. Oncol. 2021, 32, S1392–S1397. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, S.; Zhong, S.; An, H.; Yin, H.; McGowan, E.E. 35O-Phase I clinical trial of PD-1 knockout anti-MUC1 CAR-T cells in the treatment of patients with non-small cell lung cancer. Ann. Oncol. 2019, 30, xi12. [Google Scholar]
- Xia, Y.; Tian, X.; Wang, J.; Qiao, D.; Liu, X.; Xiao, L.; Liang, W.; Ban, D.; Chu, J.; Yu, J.; et al. Treatment of metastatic non-small cell lung cancer with NY-ESO-1 specific TCR engineered-T cells in a phase I clinical trial: A case report. Oncol. Lett. 2018, 16, 6998–7007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, R.; Shah, P.D.; Hamid, O.; Garfall, A.L.; Posey, A.; Bishop, M.R.; Blumenschein, G.R.; Johnson, M.L.; Lee, S.; Luke, J.J.; et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J. Clin. Oncol. 2021, 39, e14513. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Saltos, A.; Khalil, F.; Smith, M.; Li, J.; Schell, M.; Antonia, S.J.; Gray, J.E. Clinical associations of mucin 1 in human lung cancer and precancerous lesions. Oncotarget 2018, 9, 35666–35675. [Google Scholar] [CrossRef]
- Xu, T.; Li, D.; Wang, H.; Zheng, T.; Wang, G.; Xin, Y. MUC1 downregulation inhibits non-small cell lung cancer progression in human cell lines. Exp. Ther. Med. 2017, 14, 4443–4447. [Google Scholar] [CrossRef]
- Thomas, A. Should anti-mesothelin therapies be explored in lung cancer? Expert Rev. Anticancer Ther. 2016, 16, 677–679. [Google Scholar] [CrossRef]
- Bocanegra, A.; Fernandez-Hinojal, G.; Zuazo-Ibarra, M.; Arasanz, H.; Garcia-Granda, M.J.; Hernandez, C.; Ibanez, M.; Hernandez-Marin, B.; Martinez-Aguillo, M.; Lecumberri, M.J.; et al. PD-L1 Expression in Systemic Immune Cell Populations as a Potential Predictive Biomarker of Responses to PD-L1/PD-1 Blockade Therapy in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1631. [Google Scholar] [CrossRef] [Green Version]
- Zuazo, M.; Arasanz, H.; Fernandez-Hinojal, G.; Garcia-Granda, M.J.; Gato, M.; Bocanegra, A.; Martinez, M.; Hernandez, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl. Med 2012, 4, 127ra137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4828–4836. [Google Scholar] [CrossRef] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Liu, M.; Wang, X.; Li, W.; Yu, X.; Flores-Villanueva, P.; Xu-Monette, Z.Y.; Li, L.; Zhang, M.; Young, K.H.; Ma, X.; et al. Targeting PD-L1 in non-small cell lung cancer using CAR T cells. Oncogenesis 2020, 9, 72. [Google Scholar] [CrossRef]
- Chen, X.; Amar, N.; Zhu, Y.; Wang, C.; Xia, C.; Yang, X.; Wu, D.; Feng, M. Combined DLL3-targeted bispecific antibody with PD-1 inhibition is efficient to suppress small cell lung cancer growth. J. Immunother. Cancer 2020, 8, e000785. [Google Scholar] [CrossRef]
- Sui, H.; Ma, N.; Wang, Y.; Li, H.; Liu, X.; Su, Y.; Yang, J. Anti-PD-1/PD-L1 Therapy for Non-Small-Cell Lung Cancer: Toward Personalized Medicine and Combination Strategies. J. Immunol. Res. 2018, 2018, 6984948. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Zhao, R.; Chen, D.; Wei, X.; Wu, Q.; Long, Y.; Jiang, Z.; Li, Y.; Wu, H.; Zhang, X.; et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark. Res. 2020, 8, 19. [Google Scholar] [CrossRef]
- Paulsen, E.E.; Kilvaer, T.K.; Rakaee, M.; Richardsen, E.; Hald, S.M.; Andersen, S.; Busund, L.T.; Bremnes, R.M.; Donnem, T. CTLA-4 expression in the non-small cell lung cancer patient tumor microenvironment: Diverging prognostic impact in primary tumors and lymph node metastases. Cancer Immunol. Immunother. CII 2017, 66, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, S.; Hai, J.; Wang, X.; Chen, T.; Quinn, M.M.; Gao, P.; Zhang, Y.; Ji, H.; Cross, D.A.E.; et al. Targeting HER2 Aberrations in Non-Small Cell Lung Cancer with Osimertinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2594–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Tumbrink, H.L.; Heimsoeth, A.; Sos, M.L. The next tier of EGFR resistance mutations in lung cancer. Oncogene 2021, 40, 1–11. [Google Scholar] [CrossRef]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Yu, X.; Li, Y.; Chen, S.W.; Shi, Y.; Xu, F. Differential expression of glypican-3 (GPC3) in lung squamous cell carcinoma and lung adenocarcinoma and its clinical significance. Genet. Mol. Res. 2015, 14, 10185–10192. [Google Scholar] [CrossRef]
- Ning, J.; Jiang, S.; Li, X.; Wang, Y.; Deng, X.; Zhang, Z.; He, L.; Wang, D.; Jiang, Y. GPC3 affects the prognosis of lung adenocarcinoma and lung squamous cell carcinoma. BMC Pulm. Med. 2021, 21, 199. [Google Scholar] [CrossRef]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, P.; Li, J.; Zhao, J.; Liu, C.; Yang, F.; Yang, D.; Gao, A.; Lin, W.; Ma, X.; et al. B7-H3 in combination with regulatory T cell is associated with tumor progression in primary human non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13987–13995. [Google Scholar] [PubMed]
- Sun, Y.; Wang, Y.; Zhao, J.; Gu, M.; Giscombe, R.; Lefvert, A.K.; Wang, X. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer 2006, 53, 143–151. [Google Scholar] [CrossRef]
- Ye, L.; Pu, C.; Tang, J.; Wang, Y.; Wang, C.; Qiu, Z.; Xiang, T.; Zhang, Y.; Peng, W. Transmembrane-4 L-six family member-1 (TM4SF1) promotes non-small cell lung cancer proliferation, invasion and chemo-resistance through regulating the DDR1/Akt/ERK-mTOR axis. Respir. Res. 2019, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Mattsson, J.S.; Edlund, K.; Lohr, M.; Jirstrom, K.; Berglund, A.; Botling, J.; Rahnenfuehrer, J.; Marincevic, M.; Ponten, F.; et al. Aberrantly activated claudin 6 and 18.2 as potential therapy targets in non-small-cell lung cancer. Int. J. Cancer 2014, 135, 2206–2214. [Google Scholar] [CrossRef]
- Esaki, N.; Ohkawa, Y.; Hashimoto, N.; Tsuda, Y.; Ohmi, Y.; Bhuiyan, R.H.; Kotani, N.; Honke, K.; Enomoto, A.; Takahashi, M.; et al. ASC amino acid transporter 2, defined by enzyme-mediated activation of radical sources, enhances malignancy of GD2-positive small-cell lung cancer. Cancer Sci. 2018, 109, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunnet, M.; Sorensen, J.B. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer 2012, 76, 138–143. [Google Scholar] [CrossRef]
- Arrieta, O.; Villarreal-Garza, C.; Martinez-Barrera, L.; Morales, M.; Dorantes-Gallareta, Y.; Pena-Curiel, O.; Contreras-Reyes, S.; Macedo-Perez, E.O.; Alatorre-Alexander, J. Usefulness of serum carcinoembryonic antigen (CEA) in evaluating response to chemotherapy in patients with advanced non small-cell lung cancer: A prospective cohort study. BMC Cancer 2013, 13, 254. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.H.; Heitkotter, B.; Schulze, A.B.; Schliemann, C.; Steinestel, K.; Trautmann, M.; Marra, A.; Hillejan, L.; Mohr, M.; Evers, G.; et al. Prostate specific membrane antigen (PSMA) expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0186280. [Google Scholar] [CrossRef]
- Wang, H.L.; Wang, S.S.; Song, W.H.; Pan, Y.; Yu, H.P.; Si, T.G.; Liu, Y.; Cui, X.N.; Guo, Z. Expression of prostate-specific membrane antigen in lung cancer cells and tumor neovasculature endothelial cells and its clinical significance. PLoS ONE 2015, 10, e0125924. [Google Scholar] [CrossRef]
- Miyake, M.; Taki, T.; Hitomi, S.; Hakomori, S. Correlation of expression of H/Le(y)/Le(b) antigens with survival in patients with carcinoma of the lung. N. Engl. J. Med. 1992, 327, 14–18. [Google Scholar] [CrossRef]
- Westwood, J.A.; Murray, W.K.; Trivett, M.; Haynes, N.M.; Solomon, B.; Mileshkin, L.; Ball, D.; Michael, M.; Burman, A.; Mayura-Guru, P.; et al. The Lewis-Y carbohydrate antigen is expressed by many human tumors and can serve as a target for genetically redirected T cells despite the presence of soluble antigen in serum. J. Immunother. 2009, 32, 292–301. [Google Scholar] [CrossRef]
- Yoshida, S.; Fukumoto, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. Ganglioside G(D2) in small cell lung cancer cell lines: Enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 2001, 61, 4244–4252. [Google Scholar] [PubMed]
- Lee, L.; Wang, R.F.; Wang, X.; Mixon, A.; Johnson, B.E.; Rosenberg, S.A.; Schrump, D.S. NY-ESO-1 may be a potential target for lung cancer immunotherapy. Cancer J. Sci. Am. 1999, 5, 20–25. [Google Scholar] [PubMed]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schottle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; Konig, K.; Meder, L.; et al. Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, S.; Ohsaki, Y.; Nakanishi, K.; Toyoshima, E.; Kikuchi, K. Human small cell lung cancer cells express functional VEGF receptors, VEGFR-2 and VEGFR-3. Lung Cancer 2004, 46, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanipakdel, A.; Seilanian Toussi, M.; Rezazadeh, F.; Mohamadian Roshan, N.; Javadinia, S.A. Overexpression of cancer-testis antigen melanoma-associated antigen A1 in lung cancer: A novel biomarker for prognosis, and a possible target for immunotherapy. J. Cell. Physiol. 2019, 234, 12080–12086. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Fan, W.; Hu, H.; Zhang, L.; Michel, J.; Wu, Y.; Wang, J.; Jia, L.; Tang, X.; Xu, L.; et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J. Hematol. Oncol. 2019, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, M.; Kageyama, S.; Miyahara, Y.; Ishikawa, T.; Ueda, S.; Soga, N.; Naota, H.; Mukai, K.; Harada, N.; Ikeda, H.; et al. MAGE-A4, NY-ESO-1 and SAGE mRNA expression rates and co-expression relationships in solid tumours. BMC Cancer 2020, 20, 606. [Google Scholar] [CrossRef]
- Schooten, E.; Di Maggio, A.; van Bergen En Henegouwen, P.M.P.; Kijanka, M.M. MAGE-A antigens as targets for cancer immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyuno, D.; Takasawa, A.; Takasawa, K.; Ono, Y.; Aoyama, T.; Magara, K.; Nakamori, Y.; Takemasa, I.; Osanai, M. Claudin-18.2 as a therapeutic target in cancers: Cumulative findings from basic research and clinical trials. Tissue Barriers 2021, 10, 1967080. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Koslowski, M.; Dhaene, K.; Usener, D.; Brandenburg, G.; Seitz, G.; Huber, C.; Tureci, O. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 7624–7634. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Horie, M.; Nagase, T. TGF-beta Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Horie, M.; Micke, P.; Nagase, T. The Role of TGF-beta Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 3611. [Google Scholar] [CrossRef] [Green Version]
- Pop, L.M.; Barman, S.; Shao, C.; Poe, J.C.; Venturi, G.M.; Shelton, J.M.; Pop, I.V.; Gerber, D.E.; Girard, L.; Liu, X.Y.; et al. A reevaluation of CD22 expression in human lung cancer. Cancer Res. 2014, 74, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Tuscano, J.M.; Kato, J.; Pearson, D.; Xiong, C.; Newell, L.; Ma, Y.; Gandara, D.R.; O’Donnell, R.T. CD22 antigen is broadly expressed on lung cancer cells and is a target for antibody-based therapy. Cancer Res. 2012, 72, 5556–5565. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhang, D.; Guo, Y.; Lu, B.; Zhao, Z.J.; Xu, X.; Chen, Y. Tyrosine Kinase ROR1 as a Target for Anti-Cancer Therapies. Front. Oncol. 2021, 11, 680834. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Jia, K.; Dziadziuszko, R.; Zhao, S.; Deng, J.; Wang, H.; Hirsch, F.R.; Zhou, C. OX40 and OX40L protein expression of tumor infiltrating lymphocytes in non-small cell lung cancer and its role in clinical outcome and relationships with other immune biomarkers. Transl. Lung Cancer Res. 2019, 8, 352–366. [Google Scholar] [CrossRef]
- Massarelli, E.; Lam, V.K.; Parra, E.R.; Rodriguez-Canales, J.; Behrens, C.; Diao, L.; Wang, J.; Blando, J.; Byers, L.A.; Yanamandra, N.; et al. High OX-40 expression in the tumor immune infiltrate is a favorable prognostic factor of overall survival in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 351. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kikuchi, H.; Shoji, T.; Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Kinoshita, I.; Dosaka-Akita, H.; Sakakibara-Konishi, J. DLL3 regulates the migration and invasion of small cell lung cancer by modulating Snail. Cancer Sci. 2019, 110, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.H.; Giffin, M.J.; Bailis, J.M.; Smit, M.D.; Carbone, D.P.; He, K. DLL3: An emerging target in small cell lung cancer. J. Hematol. Oncol. 2019, 12, 61. [Google Scholar] [CrossRef]
- He, X.; Wang, L.; Riedel, H.; Wang, K.; Yang, Y.; Dinu, C.Z.; Rojanasakul, Y. Mesothelin promotes epithelial-to-mesenchymal transition and tumorigenicity of human lung cancer and mesothelioma cells. Mol. Cancer 2017, 16, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, J.; Yang, X.; Yang, J.; Lu, P.; Zhao, L.; Li, B.; Pan, H.; Jiang, Z.; Shen, X.; et al. Chemokine Receptor CCR2b Enhanced Anti-tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-small-cell Lung Carcinoma Model. Front. Immunol. 2021, 12, 628906. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, H.S.; Cui, Z.Y.; Lee, H.S.; Ahn, J.S.; Park, C.K.; Park, K.; Ahn, M.J. Clinicopathological implications of EpCAM expression in adenocarcinoma of the lung. Anticancer Res. 2009, 29, 1817–1822. [Google Scholar]
- Pak, M.G.; Shin, D.H.; Lee, C.H.; Lee, M.K. Significance of EpCAM and TROP2 expression in non-small cell lung cancer. World J. Surg. Oncol. 2012, 10, 53. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chocarro, L.; Arasanz, H.; Fernández-Rubio, L.; Blanco, E.; Echaide, M.; Bocanegra, A.; Teijeira, L.; Garnica, M.; Morilla, I.; Martínez-Aguillo, M.; et al. CAR-T Cells for the Treatment of Lung Cancer. Life 2022, 12, 561. https://doi.org/10.3390/life12040561
Chocarro L, Arasanz H, Fernández-Rubio L, Blanco E, Echaide M, Bocanegra A, Teijeira L, Garnica M, Morilla I, Martínez-Aguillo M, et al. CAR-T Cells for the Treatment of Lung Cancer. Life. 2022; 12(4):561. https://doi.org/10.3390/life12040561
Chicago/Turabian StyleChocarro, Luisa, Hugo Arasanz, Leticia Fernández-Rubio, Ester Blanco, Miriam Echaide, Ana Bocanegra, Lucía Teijeira, Maider Garnica, Idoia Morilla, Maite Martínez-Aguillo, and et al. 2022. "CAR-T Cells for the Treatment of Lung Cancer" Life 12, no. 4: 561. https://doi.org/10.3390/life12040561
APA StyleChocarro, L., Arasanz, H., Fernández-Rubio, L., Blanco, E., Echaide, M., Bocanegra, A., Teijeira, L., Garnica, M., Morilla, I., Martínez-Aguillo, M., Piñeiro-Hermida, S., Ramos, P., Lasarte, J. J., Vera, R., Kochan, G., & Escors, D. (2022). CAR-T Cells for the Treatment of Lung Cancer. Life, 12(4), 561. https://doi.org/10.3390/life12040561