
Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component?


 ,
,  ,
, 
Abstract
1. Introduction
2. Literature Search Strategies and Data Collection
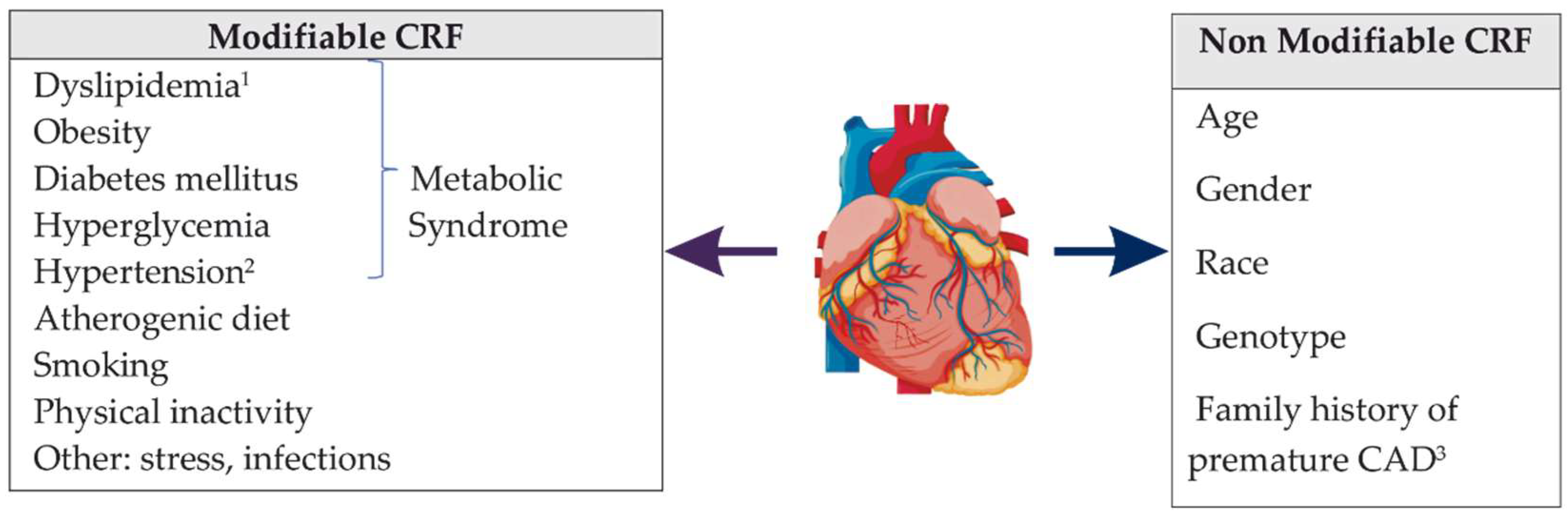
3. Phenotypic Variability of Coronary Artery Disease
4. Heritability of Atherosclerotic CAD
5. Monogenic Etiology of CAD/MI
5.1. Monogenic Lipid Disorders
5.1.1. Genetic Causes of Elevated Plasma LDL-Cholesterol Level
- a.
- Low-Density Lipoprotein Receptor (LDLR) Gene
- b.
- APOB Gene
- c.
- PCSK9 Gene
- d.
- LDLRAP1 Gene
5.1.2. Genetic Causes of Low Plasma HDL-Cholesterol Level
- a.
- APOA1 Gene
- b.
- ABC1 Gene
- c.
- LCAT Gene
5.1.3. Genetic Etiology of Hypertriglyceridemia
- a.
- LPL Gene
- b.
- APOC2 Gene
- c.
- ABCG5 and ABCG8 Genes
5.1.4. Familial Combined Hyperlipidemia and Familial Hypertriglyceridemia
The APOA1/C3/A4/A5 Gene Cluster and Lipid Metabolism
- a.
- Familial Combined Hyperlipidemia
- b.
- Familial Hypertriglyceridemia
5.1.5. Atherosclerosis Susceptibility/Atherogenic Lipoprotein Phenotype
5.2. Other Monogenic CAD
5.2.1. MEF2A Gene
5.2.2. ST6GALNAC5 Gene
5.2.3. CYP27A1 Gene
5.2.4. LRP6 Gene
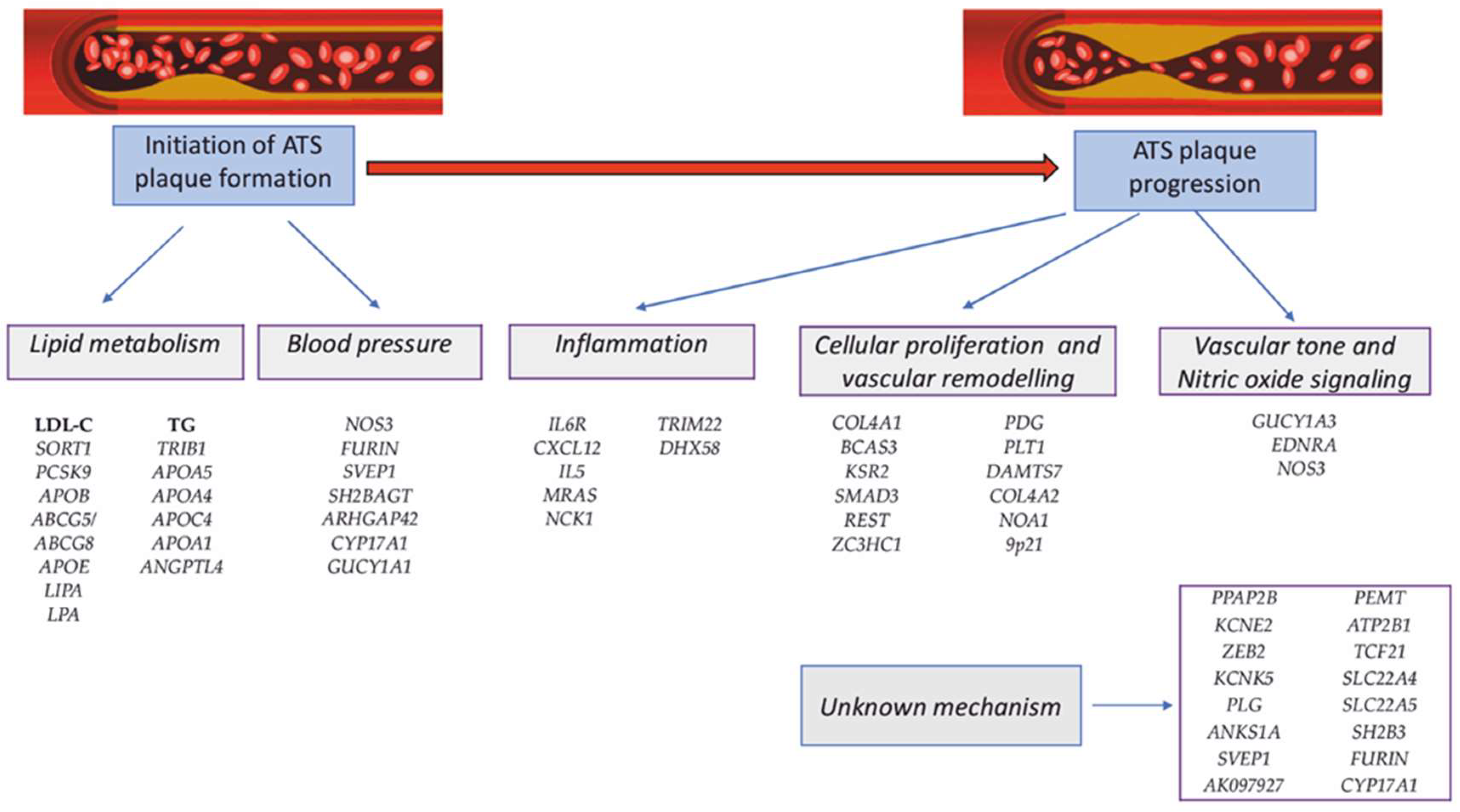
6. Polygenic CAD: Genes and Polymorphisms Associated with CAD
6.1. Polymorphism of Genes Involved in Lipid Metabolism: A Novel View
6.1.1. Polymorphisms of APOE Gene and CAD
6.1.2. Polymorphisms of APOB Gene and CAD
6.1.3. Polymorphisms of LPL Gene and Its Modulators Associated with High Risk of CAD
6.1.4. Polymorphism of OLR1 (LOX1) Gene and CAD
6.1.5. Other Genetic Polymorphisms Involved in Lipid Metabolism Associated with Increased Risk of CAD
- a.
- SORT1 Gene
- b.
- TRIB1 Gene
6.2. Polymorphism of Genes Involved in Vascular Homeostasis
6.2.1. Genes Involved in the Function of Vascular Smooth Muscle Cells (VSMc)
- a.
- Endothelial Cell Nitric Oxide Synthase 3 (NOS3) Gene Polymorphism
- b.
- TCF21 Gene Polymorphysm
- c.
- ADAMTS7 Gene Polymorphism
- d.
- HHIPL1 Gene Polymorphism
6.2.2. Genes Involved in Blood Pressure Regulation
- a.
- Angiotensin Converting Enzyme (ACE), Angiotensin II Type I Receptor (AGTR1) and Angiotensinogen (AGT) Genes Polymorphism
- b.
- CYP11B2 Gene Polymorphism
6.3. Genes Associated with Vascular Hemostasis: Role of Hemostatic Gene Polymorphisms in CAD
6.3.1. ITGA2 Gene
6.3.2. Glycoprotein IIb/IIIa Platelet Receptor Genes (ITGB2 and ITGB3 Gene Polymorphisms)
6.3.3. Plasminogen Activator Inhibitor 1 (PAI-1) Gene Polymorphism
6.3.4. Thrombospondin (TBHS) Genes Polymorphisms
6.3.5. Factor V Leiden (F5) Allele Arg506Gln and Prothrombin (F2) Variant G20210A
6.4. Metabolic Factors: Hyperhomocysteinemia (MTHFR Gene Polymorphism)
6.5. Genes Associated with Inflammation: IL6 Gene Polymorphism
6.6. Other Susceptibility Loci for CAD
7. Discussion
7.1. Challenges for the Future in the Post-GWAS Era
7.2. Translating the Results of GWASs into Clinical Practice and the Importance of Polygenic Risk Scores (PRS) for Prevention of CAD
7.3. Prophylactic Measures in Families at High Risk for CAD
7.4. Genetic Counseling in Families at High Risk for CAD
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- European Heart References Network (EHN). European Cardiovascular Disease Statistics. 2017. Available online: https://ehnheart.org/cvd-statistics.html/ (accessed on 21 January 2022).
- Heart Disease Statistics 2022. Gerardo Sison Editor. Available online: https://www.singlecare.com/blog/news/heart-disease-statistics/ (accessed on 21 January 2022).
- Muse, E.D.; Chen, S.F.; Torkamani, A. Monogenic and Polygenic Models of Coronary Artery Disease. Curr. Cardiol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, M.A.; Wold, L.E.; Kloner, R.A. Genetic contributors toward increased risk for ischemic heart disease. J. Mol. Cell. Cardiol. 2005, 39, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, M.T. Genetic evaluation for coronary artery disease. Genet. Med. 2003, 5, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wiernek, S.; Evans, J.P.; Runge, M.S. Genetics of coronary artery disease and myocardial infarction. World J. Cardiol. 2016, 8, 1–23. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020, 41, 111–188, Erratum in Eur. Heart J. 2020, 41, 4255. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. ESC Scientific Document Group. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies with the special contribution of the European Association of Preventive Cardiology (EAPC). Rev. Esp. Cardiol. 2022, 75, 429. [Google Scholar] [CrossRef]
- Fedele, F.; Pucci, M.; Severino, P. Genetic Polymorphisms and Ischemic Heart Disease. In Genetic Polymorphisms; Parine, N.R., Ed.; IntechOpen: London, UK, 2017; pp. 205–219. [Google Scholar] [CrossRef]
- Elosua, R.; Sayols-Baixeras, S. The Genetics of Ischemic Heart Disease: From Current Knowledge to Clinical Implications. Rev. Esp. Cardiol. 2017, 70, 754–762. (In English) [Google Scholar] [CrossRef]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, 1269–1324, Erratum in Hypertension 2018, 71, e136–e139; Erratum in Hypertension 2018, 72, e33. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Pechlivanis, S.; Lehmann, N.; Hoffmann, P.; Nöthen, M.M.; Jöckel, K.H.; Erbel, R.; Moebus, S. Risk prediction for coronary heart disease by a genetic risk score—Results from the Heinz Nixdorf Recall study. BMC Med. Genet. 2020, 21, 178. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. ESC Scientific Document Group. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477, Erratum in Eur. Heart J. 2020, 41, 4242. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.C.; Gerhardt, T.E.; Kwon, E. Risk Factors for Coronary Artery Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554410/ (accessed on 22 January 2022).
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org (accessed on 14 February 2022).
- Gabcova-Balaziova, D.; Stanikova, D.; Vohnout, B.; Huckova, M.; Stanik, J.; Klimes, I.; Raslova, K.; Gasperikova, D. Molecular-genetic aspects of familial hypercholesterolemia. Endocr. Regul. 2015, 49, 164–181. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.S.; Rabès, J.H.; Boileau, C.R. Genetic Testing in Familial Hypercholesterolemia: Strengthening the Tools, Reinforcing Efforts, and Diagnosis. JACC Basic Transl. Sci. 2021, 6, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Soria, L.F.; Ludwig, E.H.; Clarke, H.R.; Vega, G.L.; Grundy, S.M.; McCarthy, B.J. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc. Natl. Acad. Sci. USA 1989, 86, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.H.; Miserez, A.R.; Ahmad, Z.; Andersen, R.L. Familial defective apolipoprotein B-100: A review. J. Clin. Lipidol. 2016, 10, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Pullinger, C.R.; Hennessy, L.K.; Chatterton, J.E.; Liu, W.; Love, J.A.; Mendel, C.M.; Frost, P.H.; Malloy, M.J.; Schumaker, V.N.; Kane, J.P. Familial ligand-defective apolipoprotein B. Identification of a new mutation that decreases LDL receptor binding affinity. J. Clin. Investig. 1995, 95, 1225–1234. [Google Scholar] [CrossRef]
- Thomas, E.R.; Atanur, S.S.; Norsworthy, P.J.; Encheva, V.; Snijders, A.P.; Game, L.; Vandrovcova, J.; Siddiq, A.; Seed, M.; Soutar, A.K.; et al. Identification and biochemical analysis of a novel APOB mutation that causes autosomal dominant hypercholesterolemia. Mol. Genet. Genom. Med. 2013, 1, 155–161. [Google Scholar] [CrossRef]
- Schmidt, R.J.; Beyer, T.P.; Bensch, W.R.; Qian, Y.-W.; Lin, A.; Kowala, M.; Alborn, W.E.; Konrad, R.J.; Cao, G. Secreted proprotein convertase subtilisin/kexin type 9 reduces both hepatic and extrahepatic low-density lipoprotein receptors in vivo. Biochem. Biophys. Res. Commun. 2008, 370, 634–640. [Google Scholar] [CrossRef]
- Abifadel, M.; Rabès, J.P.; Devillers, M.; Munnich, A.; Erlich, D.; Junien, C.; Varret, M.; Boileau, C. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 2009, 30, 520–529. [Google Scholar] [CrossRef]
- Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; Schunkert, H.; et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 1, 334–341. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Chuan, J.; Qian, Z.; Zhang, Y.; Tong, R.; Peng, M. The association of the PCSK9 rs562556 polymorphism with serum lipids level: A meta-analysis. Lipids Health Dis. 2019, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- D’Erasmo, L.; Di Costanzo, A.; Arca, M. Autosomal recessive hypercholesterolemia: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Zuliani, G.; Wilund, K.; Campagna, F.; Fellin, R.; Bertolini, S.; Calandra, S.; Ricci, G.; Glorioso, N.; Maioli, M.; et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: A clinical and molecular genetic analysis. Lancet 2002, 359, 841–847. [Google Scholar] [CrossRef]
- Novo, S. Low HDL-cholesterol concentrations cause atherosclerotic disease to develop. ESC European Society of Cardiology. E-J. ESC Counc. Cardiol. Pract. 2009, 7, 32. Available online: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-7/Low-HDL-cholesterol-concentrations-cause-atherosclerotic-disease-to-develop (accessed on 26 January 2022).
- Rader, D.J.; deGoma, E.M. Approach to the patient with extremely low HDL-cholesterol. J. Clin. Endocrinol. Metab. 2012, 97, 3399–3407. [Google Scholar] [CrossRef]
- Yamakawa-Kobayashi, K.; Yanagi, H.; Fukayama, H.; Hirano, C.; Shimakura, Y.; Yamamoto, N.; Arinami, T.; Tsuchiya, S.; Hamaguchi, H. Frequent occurrence of hypoalphalipoproteinemia due to mutant apolipoprotein A-I gene in the population: A population-based survey. Hum. Molec. Genet. 1999, 8, 331–336. [Google Scholar] [CrossRef]
- Haase, C.L.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Population-based resequencing of APOA1 in 10,330 individuals: Spectrum of genetic variation, phenotype, and comparison with extreme phenotype approach. PLoS Genet. 2012, 8, e1003063. [Google Scholar] [CrossRef]
- Bielicki, J.K.; Oda, M.N. Apolipoprotein A-I(Milano) and apolipoprotein A-I(Paris) exhibit an antioxidant activity distinct from that of wild-type apolipoprotein A-I. Biochemistry 2002, 41, 2089–2096. [Google Scholar] [CrossRef]
- Koseki, M.; Yamashita, S.; Ogura, M.; Ishigaki, Y.; Ono, K.; Tsukamoto, K.; Hori, M.; Matsuki, K.; Yokoyama, S.; Harada-Shiba, M. Current Diagnosis and Management of Tangier Disease. J. Atheroscler. Thromb. 2021, 28, 802–810. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff. Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ABCA1/ (accessed on 26 January 2022).
- Mokuno, J.; Hishida, A.; Morita, E.; Sasakabe, T.; Hattori, Y.; Suma, S.; Okada, R.; Kawai, S.; Naito, M.; Wakai, K. ATP-binding cassette transporter A1 (ABCA1) R219K (G1051A, rs2230806) polymorphism and serum high-density lipoprotein cholesterol levels in a large Japanese population: Cross-sectional data from the Daiko Study. Endocr. J. 2015, 62, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Elías-López, D.; Martagón, A.J.; Pérez-Méndez, O.A.; Sánchez, M.L.O.; Segura, Y.; Tusié, M.; Aguilar-Salinas, C.A. LCAT deficiency: A systematic review with the clinical and genetic description of Mexican kindred. Lipids Health Dis. 2021, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, N. Familial LCAT deficiency and fish-eye disease. J. Inherit. Metab. Dis. 1988, 11, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Gehrisch, S. Common mutations of the lipoprotein lipase gene and their clinical significance. Curr. Atheroscler. Rep. 1999, 1, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.W.; Breckenridge, W.C.; Little, J.A. Inheritance of apolipoprotein C-II deficiency with hypertriglyceridemia and pancreatitis. N. Engl. J. Med. 1978, 299, 1421–1424. [Google Scholar] [CrossRef]
- Yoo, E.G. Sitosterolemia: A review and update of pathophysiology, clinical spectrum, diagnosis, and management. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 7–14. [Google Scholar] [CrossRef]
- Wang, Z.; Caom, L.; Sum, Y.; Wang, G.; Wang, R.; Yu, Z.; Bai, X.; Ruan, C. Specific macrothrombocytopenia/hemolytic anemia associated with sitosterolemia. Am. J. Hematol. 2014, 89, 320–324. [Google Scholar] [CrossRef]
- Liu, Y.; Ordovas, J.M.; Gao, G.; Province, M.; Straka, R.J.; Tsai, M.Y.; Lai, C.Q.; Zhang, K.; Borecki, I.; Hixson, J.E.; et al. Pharmacogenetic association of the APOA1/C3/A4/A5 gene cluster and lipid responses to fenofibrate: The genetics of lipid-lowering drugs and diet network study. Pharm. Genom. 2009, 19, 161–169. [Google Scholar] [CrossRef]
- Eichenbaum-Voline, S.; Olivier, M.; Jones, E.L.; Naoumova, R.P.; Jones, B.; Gau, B.; Patel, H.N.; Seed, M.; Betteridge, D.J.; Galton, D.J.; et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arter. Thromb. Vasc. Biol. 2004, 24, 167–174. [Google Scholar] [CrossRef]
- Timpson, N.J.; Walter, K.; Min, J.L.; Tachmazidou, I.; Malerba, G.; Shin, S.Y.; Chen, L.; Futema, M.; Southam, L.; Iotchkova, V.; et al. UK1OK Consortium Members. A rare variant in APOC3 is associated with plasma triglyceride and VLDL levels in Europeans. Nat. Commun. 2014, 5, 4871. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, E.; Esfehani, R.J.; Sahebkar, A.; Parizadeh, S.M.; Rostami, D.; Mirinezhad, M.; Poursheikhani, A.; Mobarhan, M.G.; Pasdar, A. Familial combined hyperlipidemia: An overview of the underlying molecular mechanisms and therapeutic strategies. IUBMB Life 2019, 71, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.N.; Heiss, G.; Ellison, R.C.; Province, M.A.; Pankow, J.S.; Eckfeldt, J.H.; Hunt, S.C. Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: A case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation 2003, 108, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bogari, N.M.; Aljohani, A.; Amin, A.A.; Al-Allaf, F.A.; Dannoun, A.; Taher, M.M.; Elsayed, A.; Rednah, D.I.; Elkhatee, O.; Porqueddu, M.; et al. A genetic variant c.553G>T (rs2075291) in the apolipoprotein A5 gene is associated with altered triglycerides levels in coronary artery disease (CAD) patients with lipid lowering drug. BMC Cardiovasc. Disord. 2019, 19, 2. [Google Scholar] [CrossRef]
- Kao, J.T.; Wen, H.C.; Chien, K.L.; Hsu, H.C.; Lin, S.W. A novel genetic variant in the apolipoprotein A5 gene is associated with hypertriglyceridemia. Hum. Mol. Genet. 2003, 12, 2533–2539. [Google Scholar] [CrossRef]
- Do, R.; Project, N.E.S.; Stitziel, N.; Won, H.-H.; Jørgensen, A.B.; Duga, S.; Merlini, P.A.; Kiezun, A.; Farrall, M.; NHLBI Exome Sequencing Project; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef]
- Genest, J.J., Jr.; Martin-Munley, S.S.; McNamara, J.R.; Ordovas, J.M.; Jenner, J.; Myers, R.H.; Silberman, S.R.; Wilson, P.W.; Salem, D.N.; Schaefer, E.J. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation 1992, 85, 2025–2033. [Google Scholar] [CrossRef]
- Austin, M.A.; Breslow, J.L.; Hennekens, C.H.; Buring, J.E.; Willett, W.C.; Krauss, R.M. Low-Density Lipoprotein Subclass Patterns and Risk of Myocardial Infarction. JAMA 1988, 260, 1917–1921. [Google Scholar] [CrossRef]
- Nishina, P.M.; Johnson, J.P.; Naggert, J.K.; Krauss, R.M. Linkage of atherogenic lipoprotein phenotype to the low density lipoprotein receptor locus on the short arm of chromosome 19. Proc. Natl. Acad. Sci. USA 1992, 89, 708–712. [Google Scholar] [CrossRef]
- Rotter, J.I.; Bu, X.; Cantor, R.M.; Warden, C.H.; Brown, J.; Gray, R.J.; Blanche, P.J.; Krauss, R.M.; Lusis, A.J. Multilocus genetic determinants of LDL particle size in coronary artery disease families. Am. J. Hum. Genet. 1996, 58, 585–594. [Google Scholar]
- Srisawasdi, P.; Rodcharoen, P.; Vanavanan, S.; Chittamma, A.; Sukasem, C.; Na Nakorn, C.; Dejthevaporn, C.; Kroll, M.H. Association of CETP Gene Variants with Atherogenic Dyslipidemia Among Thai Patients Treated with Statin. Pharmgenom. Pers. Med. 2021, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Allayee, H.; Aouizerat, B.E.; Cantor, R.M.; Dallinga-Thie, G.M.; Krauss, R.M.; Lanning, C.D.; Rotter, J.I.; Lusis, A.J.; de Bruin, T.W. Families with familial combined hyperlipidemia and families enriched for coronary artery disease share genetic determinants for the atherogenic lipoprotein phenotype. Am. J. Hum. Genet. 1998, 63, 577–585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Omidi, S.; Ghasemi, S.; Kalayinia, S. Is There Any Association Between the MEF2A Gene Changes and Coronary Artery Disease? Acta Med. Iran. 2020, 58, 366–375. [Google Scholar] [CrossRef]
- Wang, L.; Fan, C.; Topol, S.E.; Topol, E.J.; Wang, Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science 2003, 302, 1578–1581. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.C.; Wang, W. Association of MEF2A gene 3’UTR mutations with coronary artery disease. Genet. Mol. Res. 2015, 14, 11073–11078. [Google Scholar] [CrossRef]
- Liu, B.; Pang, L.; Ji, Y.; Fang, L.; Tian, C.W.; Chen, J.; Chen, C.; Zhong, Y.; Ou, W.C.; Xiong, Y.; et al. MEF2A Is the Trigger of Resveratrol Exerting Protection on Vascular Endothelial Cell. Front. Cardiovasc. Med. 2022, 8, 775392. [Google Scholar] [CrossRef]
- InanlooRahatloo, K.; Parsa, A.F.; Huse, K.; Rasooli, P.; Davaran, S.; Platzer, M.; Kramer, M.; Fan, J.B.; Turk, C.; Amini, S.; et al. Mutation in ST6GALNAC5 identified in family with coronary artery disease. Sci. Rep. 2014, 4, 3595. [Google Scholar] [CrossRef]
- Cheeseman, J.; Kuhnle, G.; Stafford, G.; Gardner, R.A.; Spencer, D.I.; Osborn, H.M. Sialic acid as a potential biomarker for cardiovascular disease, diabetes and cancer. Biomark. Med. 2021, 15, 911–928. [Google Scholar] [CrossRef]
- Inanloorahatloo, K.; Zand Parsa, A.F.; Huse, K.; Rasooli, P.; Davaran, S.; Platzer, M.; Fan, J.B.; Amini, S.; Steemers, F.; Elahi, E. Mutation in CYP27A1 identified in family with coronary artery disease. Eur. J. Med. Genet. 2013, 56, 655–660. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Y.; Wu, H.; Sun, Y.M.; Cai, Y.H.; Wu, J.J.; Wang, J.; Gong, L.Y.; Ding, Z.T. Clinical and molecular genetic features of cerebrotendinous xanthomatosis patients in Chinese families. Metab. Brain Dis. 2017, 32, 1609–1618. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, J.J.; Lee, Y.F.; Wang, P.W.; Pan, T.L.; Chang, W.N.; Tsai, M.H. Clinical and molecular genetic features of cerebrotendinous xanthomatosis in Taiwan: Report of a novel CYP27A1 mutation and literature review. J. Clin. Lipidol. 2019, 13, 954–959.e1. [Google Scholar] [CrossRef] [PubMed]
- Rashvand, Z.; Kahrizi, K.; Najmabadi, H.; Najafipour, R.; Omrani, M.D. Clinical and Genetic Characteristics of Splicing Variant in CYP27A1 in an Iranian Family with Cerebrotendinous xanthomatosis. Iran. Biomed. J. 2021, 25, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, A.; Mani, M.A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Q.J.; Chen, M.Z.; Li, L.; Zhang, K.; Cheng, G.H.; Ma, L.; Gong, Y.Q. Association of common polymorphisms in the LRP6 gene with sporadic coronary artery disease in a Chinese population. Chin. Med. J. 2012, 125, 444–449. [Google Scholar]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. WTCCC and the Cardiogenics Consortium Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Wang, F.; Xu, C.Q.; He, Q.; Cai, J.P.; Li, X.C.; Wang, D.; Xiong, X.; Liao, Y.H.; Zeng, Q.T.; Yang, Y.Z.; et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat. Genet. 2011, 43, 345–349. [Google Scholar] [CrossRef]
- IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011, 7, e1002260, Erratum in PLoS Genet. 2012, 8, 2–12. [Google Scholar] [CrossRef]
- Koyama, S.; Ito, K.; Terao, C.; Akiyama, M.; Horikoshi, M.; Momozawa, Y.; Matsunaga, H.; Ieki, H.; Ozaki, K.; Onouchi, Y.; et al. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat. Genet. 2020, 52, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Ito, K.; Akiyama, M.; Takahashi, A.; Koyama, S.; Nomura, S.; Ieki, H.; Ozaki, K.; Onouchi, Y.; Sakaue, S.; et al. Transethnic Meta-Analysis of Genome-Wide Association Studies Identifies Three New Loci and Characterizes Population-Specific Differences for Coronary Artery Disease. Circ. Genom. Precis. Med. 2020, 13, e002670. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Schunkert, H. Coronary Artery Disease Genetics Enlightened by Genome-Wide Association Studies. JACC Basic Transl. Sci. 2021, 6, 610–623. [Google Scholar] [CrossRef]
- Shadrina, A.S.; Shashkova, T.I.; Torgasheva, A.A.; Sharapov, S.Z.; Klarić, L.; Pakhomov, E.D.; Alexeev, D.G.; Wilson, J.F.; Tsepilov, Y.A.; Joshi, P.K.; et al. Prioritization of causal genes for coronary artery disease based on cumulative evidence from experimental and in silico studies. Sci. Rep. 2020, 10, 10486. [Google Scholar] [CrossRef]
- Chen, Z.; Schunkert, H. Genetics of coronary artery disease in the post-GWAS era. J. Intern. Med. 2021, 290, 980–992. [Google Scholar] [CrossRef]
- LeBlanc, M.; Zuber, V.; Andreassen, B.K.; Witoelar, A.; Zeng, L.; Bettella, F.; Wang, Y.; McEvoy, L.K.; Thompson, W.K.; Schork, A.J.; et al. Identifying Novel Gene Variants in Coronary Artery Disease and Shared Genes with Several Cardiovascular Risk Factors. Circ. Res. 2016, 118, 83–94. [Google Scholar] [CrossRef]
- Ghosh, S.; Vivar, J.; Nelson, C.P.; Willenborg, C.; Segrè, A.V.; Mäkinen, V.P.; Nikpay, M.; Erdmann, J.; Blankenberg, S.; O’Donnell, C.; et al. Systems Genetics Analysis of Genome-Wide Association Study Reveals Novel Associations Between Key Biological Processes and Coronary Artery Disease. Arteriosclerosis, thrombosis, and vascular biolog. Arter. Thromb. Vasc. Biol. 2015, 35, 1712–1722. [Google Scholar] [CrossRef]
- Won, H.H.; Natarajan, P.; Dobbyn, A.; Jordan, D.M.; Roussos, P.; Lage, K.; Raychaudhuri, S.; Stahl, E.; Do, R. Disproportionate Contributions of Select Genomic Compartments and Cell Types to Genetic Risk for Coronary Artery Disease. PLoS Genet. 2015, 11, e1005622. [Google Scholar] [CrossRef]
- Blum, C.B. Type III Hyperlipoproteinemia: Still Worth Considering? Prog. Cardiovasc. Dis. 2016, 59, 119–124. [Google Scholar] [CrossRef]
- Atis, O.; Sahin, S.; Ceyhan, K.; Ozyurt, H.; Akbas, A.; Benli, I. The Distribution of Apolipoprotein E Gene Polymorphism and Apolipoprotein E Levels among Coronary Artery Patients Compared to Controls. Eurasian J. Med. 2016, 48, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Eichne, J.E.; Dunn, S.T.; Perveen, G.; Thompson, D.M.; Stewart, K.E.; Stroehla, B.C. Apolipoprotein E polymorphism and cardiovascular disease: A HuGE review. Am. J. Epidemiol. 2002, 155, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, L.U.; Jeune, B.; Ranberg, K.A.; Nybo, H.; Vaupel, J.W. Estimation of apolipoprotein E genotype-specific relative mortality risks from the distribution of genotypes in centenarians and middle-aged men: Apolipoprotein E gene is a “frailty gene”, not a “longevity gene”. Genet. Epidemiol. 2000, 19, 202–210. [Google Scholar] [CrossRef]
- Humphries, S.E.; Talmud, P.J.; Hawe, E.; Bolla, M.; Day, I.N.; Miller, G.J. Apolipoprotein E4 and coronary heart disease in middle-aged men who smoke: A prospective study. Lancet 2001, 358, 115–119. [Google Scholar] [CrossRef]
- Talmud, P.J.; Lewis, S.J.; Hawe, E.; Martin, S.; Acharya, J.; Marmot, M.G.; Humphries, S.E.; Brunner, E.J. No APOEepsilon4 effect on coronary heart disease risk in a cohort with low smoking prevalence: The Whitehall II study. Atherosclerosis 2004, 177, 105–112. [Google Scholar] [CrossRef]
- Chiodini, B.D.; Barlera, S.; Franzosi, M.G.; Beceiro, V.L.; Introna, M.; Tognoni, G. APO B gene polymorphisms and coronary artery disease: A meta-analysis. Atherosclerosis 2003, 167, 355–366. [Google Scholar] [CrossRef]
- Hegele, R.A.; Huang, L.S.; Herbert, P.N.; Blum, C.B.; Buring, J.E.; Hennekens, C.H.; Breslow, J.L. Apolipoprotein B-gene DNA polymorphisms associated with myocardial infarction. N. Engl. J. Med. 1986, 315, 1509–1515. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, J.; Tan, Y.; Feng, M.; Qin, J.; Lin, M.; Zhao, X.; Zhao, X.; Liang, Y.; Zhang, N.; et al. Association between apolipoprotein B EcoRI polymorphisms and coronary heart disease: A meta-analysis. Wien. Klin. Wochenschr. 2016, 128, 890–897. [Google Scholar] [CrossRef]
- He, K.; Zhu, Z.; Chen, Y. Lipoprotein Lipase Gene Polymorphisms Are Associated with Myocardial Infarction Risk: A Meta-Analysis. Genet. Test. Mol. Biomark. 2021, 25, 434–444. [Google Scholar] [CrossRef]
- Ma, W.Q.; Wang, Y.; Han, X.Q.; Zhu, Y.; Liu, N.F. Associations between LPL gene polymorphisms and coronary artery disease: Evidence based on an updated and cumulative meta-analysis. Biosci. Rep. 2018, 38, BSR20171642. [Google Scholar] [CrossRef]
- Talmud, P.J.; Bujac, S.R.; Hall, S.; Miller, G.J.; Humphries, S.E. Substitution of asparagine for aspartic acid at residue 9 (D9N) of lipoprotein lipase markedly augments risk of ischaemic heart disease in male smokers. Atherosclerosis 2000, 149, 75–81. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.W.; Konrad, R.J. ApoA5 lowers triglyceride levels via suppression of ANGPTL3/8-mediated LPL inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef] [PubMed]
- Trabetti, E.; Biscuola, M.; Cavallari, U.; Malerba, G.; Girelli, D.; Olivieri, O.; Martinelli, N.; Corrocher, R.; Pignatti, P.F. On the association of the oxidised LDL receptor 1 (OLR1) gene in patients with acute myocardial infarction or coronary artery disease. Eur. J. Hum. Genet. 2006, 14, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Salehipour, P.; Rezagholizadeh, F.; Mahdiannasser, M.; Kazerani, R.; Modarressi, M.H. Association of OLR1 gene polymorphisms with the risk of coronary artery disease: A systematic review and meta-analysis. Heart Lung 2021, 50, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.L.; Rohde, P.D.; Winther, S.; Breining, P.; Nissen, L.; Nykjaer, A.; Bøttcher, M.; Nyegaard, M.; Kjolby, M. Sortilin as a Biomarker for Cardiovascular Disease Revisited. Front. Cardiovasc. Med. 2021, 8, 652584. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, K.; Itoh, Y.; Inoue, Y.; Hayashi, H. TRB1 negatively regulates gluconeogenesis by suppressing the transcriptional activity of FOXO1. FEBS Lett. 2019, 593, 369–380. [Google Scholar] [CrossRef]
- Douvris, A.; Soubeyrand, S.; Naing, T.; Martinuk, A.; Nikpay, M.; Williams, A.; Buick, J.; Yauk, C.; McPherson, R. Functional analysis of the TRIB1 associated locus linked to plasma triglycerides and coronary artery disease. J. Am. Heart Assoc. 2014, 3, e000884, Erratum in J. Am. Heart Assoc. 2016, 5, e002056. [Google Scholar] [CrossRef]
- Aimo, A.; Botto, N.; Vittorini, S.; Emdin, M. Polymorphisms in the eNOS gene and the risk of coronary artery disease: Making the case for genome-wide association studies. Eur. J. Prev. Cardiol. 2019, 26, 157–159. [Google Scholar] [CrossRef]
- Li, X.; Lin, Y.; Zhang, R. Associations between endothelial nitric oxide synthase gene polymorphisms and the risk of coronary artery disease: A systematic review and meta-analysis of 132 case-control studies. Eur. J. Prev. Cardiol. 2019, 26, 160–170. [Google Scholar] [CrossRef]
- Gholami, M.; Amoli, M.M.; Sharifi, F.; Khoshnevisan, K. Comments on and assessments of ‘Associations between endothelial nitric oxide synthase gene polymorphisms and the risk of coronary artery disease: A systematic review and meta-analysis of 132 case-control studies. Eur. J. Prev. Cardiol. 2020, 27, 660–663. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wirka, R.; Nguyen, T.; Nagao, M.; Cheng, P.; Miller, C.L.; Kim, J.B.; Pjanic, M.; Quertermous, T. TCF21 and AP-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 2019, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.; Palma Dos Reis, R.; Rodrigues, R.; Sousa, A.C.; Gomes, S.; Borges, S.; Ornelas, I.; Freitas, A.I.; Guerra, G.; Henriques, E.; et al. Association of ADAMTS7 gene polymorphism with cardiovascular survival in coronary artery disease. Physiol. Genom. 2016, 48, 810–815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aravani, D.; Morris, G.E.; Jones, P.D.; Tattersall, H.K.; Karamanavi, E.; Kaiser, M.A.; Kostogrys, R.B.; Ghaderi Najafabadi, M.; Andrews, S.L.; Nath, M.; et al. HHIPL1, a Gene at the 14q32 Coronary Artery Disease Locus, Positively Regulates Hedgehog Signaling and Promotes Atherosclerosis. Circulation 2019, 140, 500–513. [Google Scholar] [CrossRef]
- Amara, A.; Mrad, M.; Sayeh, A.; Lahideb, D.; Layouni, S.; Haggui, A.; Fekih-Mrissa, N.; Haouala, H.; Nsiri, B. The Effect of ACE I/D Polymorphisms Alone and with Concomitant Risk Factors on Coronary Artery Disease. Clin. Appl. Thromb. Hemost. 2018, 24, 157–163. [Google Scholar] [CrossRef]
- Lindpaintner, K.; Lee, M.; Larson, M.G.; Rao, V.S.; Pfeffer, M.A.; Ordovas, J.M.; Schaefer, E.J.; Wilson, A.F.; Wilson, P.W.; Vasan, R.S.; et al. Absence of association or genetic linkage between the angiotensin-converting-enzyme gene and left ventricular mass. N. Engl. J. Med. 1996, 334, 1023–1028. [Google Scholar] [CrossRef]
- Keavney, B.; McKenzie, C.; Parish, S.; Palmer, A.; Clark, S.; Youngman, L.; Delépine, M.; Lathrop, M.; Peto, R.; Collins, R. Large-scale test of hypothesised associations between the angiotensin-converting-enzyme insertion/deletion polymorphism and myocardial infarction in about 5000 cases and 6000 controls. International Studies of Infarct Survival (ISIS) Collaborators. Lancet 2000, 355, 434–442. [Google Scholar] [CrossRef]
- Borai, I.H.; Hassan, N.S.; Shaker, O.G.; Ashour, E.E.; Badrawy, M.E.I.; Olfat, M.; Fawzi, L.; Mageed, L. Synergistic effect of ACE and AGT genes in coronary artery disease. J. Basic Appl. Sci. 2018, 7, 111–117. [Google Scholar] [CrossRef]
- Feng, X.; Zheng, B.S.; Shi, J.J.; Qian, J.; He, W.; Zhou, H.F. A systematic review and meta-analysis of the association between angiotensin II type 1 receptor A1166C gene polymorphism and myocardial infarction susceptibility. J. Renin-Angiotensin-Aldosterone Syst. 2014, 15, 307–315. [Google Scholar] [CrossRef]
- Liu, D.X.; Zhang, Y.Q.; Hu, B.; Zhang, J.; Zhao, Q. Association of AT1R polymorphism with hypertension risk: An update meta-analysis based on 28,952 subjects. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 898–909. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, M.; Miao, S.; Xi, B. Polymorphisms of three genes (ACE, AGT and CYP11B2) in the renin-angiotensinaldosterone system are not associated with blood pressure salt sensitivity: A systematic meta-analysis. Blood Press. 2016, 25, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Liu, D.; Yuan, K.; Zhu, G.; Qi, X. Association of -344C/T polymorphism in the aldosterone synthase (CYP11B2) gene with cardiac and cerebrovascular events in Chinese patients with hypertension. J. Int. Med. Res. 2020, 48, 300060520949409. [Google Scholar] [CrossRef] [PubMed]
- Hautanen, A.; Toivanen, P.; Mänttäri, M.; Tenkanen, L.; Kupari, M.; Manninen, V.; Kayes, K.M.; Rosenfeld, S.; White, P.C. Joint effects of an aldosterone synthase (CYP11B2) gene polymorphism and classic risk factors on risk of myocardial infarction. Circulation 1999, 100, 2213–2218. [Google Scholar] [CrossRef] [PubMed]
- Kroll, H.; Gardemann, A.; Fechter, A.; Haberbosch, W.; Santoso, S. The impact of the glycoprotein Ia collagen receptor subunit A1648G gene polymorphism on coronary artery disease and acute myocardial infarction. Thromb. Haemost. 2000, 83, 392–396. [Google Scholar]
- Santoso, S.; Kunicki, T.J.; Kroll, H.; Haberbosch, W.; Gardemann, A. Association of the platelet glycoprotein Ia C807T gene polymorphism with nonfatal myocardial infarction in younger patients. Blood 1999, 93, 2449–2453. [Google Scholar] [CrossRef]
- Reiner, A.P.; Schwartz, S.M.; Kumar, P.N.; Rosendaal, F.R.; Pearce, R.M.; Aramaki, K.M.; Psaty, B.M.; Siscovick, D.S. Platelet glycoprotein IIb polymorphism, traditional risk factors and non-fatal myocardial infarction in young women. Br. J. Haematol. 2001, 112, 632–636. [Google Scholar] [CrossRef]
- Hato, T.; Minamoto, Y.; Fukuyama, T.; Fujit, S. Polymorphisms of HPA-1 through 6 on platelet membrane glycoprotein receptors are not a genetic risk factor for myocardial infarction in the Japanese population. Am. J. Cardiol. 1997, 80, 1222–1224. [Google Scholar] [CrossRef]
- Floyd, C.N.; Mustafa, A.; Ferro, A. The PlA1/A2 polymorphism of glycoprotein IIIa as a risk factor for myocardial infarction: A meta-analysis. PLoS ONE 2014, 9, e101518. [Google Scholar] [CrossRef]
- Bojesen, S.E.; Juul, K.; Schnohr, P.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Copenhagen City Heart Study. Platelet glycoprotein IIb/IIIa Pl(A2)/Pl(A2) homozygosity associated with risk of ischemic cardiovascular disease and myocardial infarction in young men: The Copenhagen City Heart Study. J. Am. Coll. Cardiol. 2003, 42, 661–667. [Google Scholar] [CrossRef]
- Liang, Z.; Jiang, W.; Ouyang, M.; Yang, K. PAI-1 4G/5G polymorphism and coronary artery disease risk: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2097–2107. [Google Scholar]
- Khaki-Khatibi, F.; Karimian, A. Association of PAI-1 serum levels and polymorphism of gene of Plasminogen Activator Inhibitor-1 in patient of Non-diabetic and Non-smoker with Coronary Artery Disease. Med. J. Tabriz Univ. Med. Sci. 2018, 40, 43–48. [Google Scholar]
- Zhang, X.J.; Wei, C.Y.; Li, W.B.; Zhang, L.L.; Zhou, Y.; Wang, Z.H.; Tang, M.X.; Zhang, W.; Zhang, Y.; Zhong, M. Association between single nucleotide polymorphisms in thrombospondins genes and coronary artery disease: A meta-analysis. Thromb. Res. 2015, 136, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ercan, B.; Tamer, L.; Sucu, N.; Pekdemir, H.; Camsari, A.; Atik, U. Factor VLeiden and prothrombin G20210A gene polymorphisms in patients with coronary artery disease. Yonsei Med. J. 2008, 49, 237–243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, S.J.; Ebrahim, S.; Smith, G.D. Meta-analysis of MTHFR 677C->T polymorphism and coronary heart disease: Does totality of evidence support causal role for homocysteine and preventive potential of folate? BMJ 2005, 331, 1053. [Google Scholar] [CrossRef]
- Brattström, L.; Wilcken, D.E.; Ohrvik, J.; Brudin, L. Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: The result of a meta-analysis. Circulation 1998, 98, 2520–2526. [Google Scholar] [CrossRef]
- Nedelcu, C.; Ionescu, M.; Pantea-Stoian, A.; Niţă, D.; Petcu, L.; Mazilu, L.; Suceveanu, A.I.; Tuţă, L.A.; Parepa, I.R. Correlation between plasma homocysteine and first myocardial infarction in young patients: Case-control study in Constanta County, Romania. Exp. Ther. Med. 2021, 21, 101. [Google Scholar] [CrossRef]
- Klerk, M.; Verhoef, P.; Clarke, R.; Blom, H.J.; Kok, F.J.; Schouten, E.G. MTHFR Studies Collaboration Group. MTHFR 677C-->T polymorphism and risk of coronary heart disease: A meta-analysis. JAMA 2002, 288, 2023–2031. [Google Scholar] [CrossRef]
- Kelberman, D.; Hawe, E.; Luong, L.A.; Mohamed-Ali, V.; Lundman, P.; Tornvall, P.; Aillaud, M.F.; Juhan-Vague, I.; Yudkin, J.S.; Margaglione, M.; et al. HIFMECH study group. Effect of Interleukin-6 promoter polymorphisms in survivors of myocardial infarction and matched controls in the North and South of Europe. The HIFMECH Study. Thromb. Haemost. 2004, 92, 1122–1128. [Google Scholar] [CrossRef]
- Gudmundsson, G.; Matthiasson, S.E.; Arason, H.; Johannsson, H.; Runarsson, F.; Bjarnason, H.; Helgadottir, K.; Thorisdottir, S.; Ingadottir, G.; Lindpaintner, K.; et al. Localization of a gene for peripheral arterial occlusive disease to chromosome 1p31. Am. J. Hum. Genet. 2002, 70, 586–592. [Google Scholar] [CrossRef]
- Pajukanta, P.; Cargill, M.; Viitanen, L.; Nuotio, I.; Kareinen, A.; Perola, M.; Terwilliger, J.D.; Kempas, E.; Daly, M.; Lilja, H.; et al. Two loci on chromosomes 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am. J. Hum. Genet. 2000, 67, 1481–1493. [Google Scholar] [CrossRef]
- Harrap, S.B.; Zammit, K.S.; Wong, Z.Y.; Williams, F.M.; Bahlo, M.; Tonkin, A.M.; Anderson, S.T. Genome-wide linkage analysis of the acute coronary syndrome suggests a locus on chromosome 2. Arter. Thromb. Vasc. Biol. 2002, 22, 874–878. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lange, L.A.; Lange, E.M.; Bielak, L.F.; Langefeld, D.; Kardia, S.L.; Royston, P.; Turner, S.T.; Sheedy, P.F., 2nd; Boerwinkle, E.; Peyser, P.A. Autosomal genome-wide scan for coronary artery calcification loci in sibships at high risk for hypertension. Arter. Thromb. Vasc. Biol. 2002, 22, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Yin, W.L.; Peng, L.; Zhou, X.H.; Zhou, P.; Xuan, S.X.; Luo, Y.; Chen, C.; Cheng, B.; Lin, J.D.; et al. Association of Aldehyde Dehydrogenase 2 Gene Polymorphism with Myocardial Infarction. J. Inflamm. Res. 2021, 14, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Stevens, H.Y.; Melchior, B.; Bell, K.S.; Yun, S.; Yeh, J.C.; Frangos, J.A. PECAM-1 is a critical mediator of atherosclerosis. Dis. Model. Mech. 2008, 1, 175–181. [Google Scholar] [CrossRef]
- Thomas, C.B.; Cohen, B.H. The familial occurrence of hypertension and coronary artery disease, with observations concerning obesity and diabetes. Ann. Intern. Med. 1955, 42, 90–127. [Google Scholar] [CrossRef]
- Brown, D.; Giles, W.H.; Burke, W.; Greenlund, K.J.; Croft, J.B. Familial aggregation of early-onset myocardial infarction. Community Genet. 2002, 5, 232–238. [Google Scholar] [CrossRef]
- Schildkraut, J.M.; Myers, R.H.; Cupples, L.A.; Kiely, D.K.; Kannel, W.B. Coronary risk associated with age and sex of parental heart disease in the Framingham Study. Am. J. Cardiol. 1989, 64, 555–559. [Google Scholar] [CrossRef]
- Chacko, M.; Sarma, P.S.; Harikrishnan, S.; Zachariah, G.; Jeemon, P. Family history of cardiovascular disease and risk of premature coronary heart disease: A matched case-control study. Wellcome Open Res. 2020, 5, 70. [Google Scholar] [CrossRef]
- Zdravkovic, S.; Wienke, A.; Pedersen, N.L.; Marenberg, M.E.; Yashin, A.I.; De Faire, U. Heritability of death from coronary heart disease: A 36-year follow-up of 20966 Swedish twins. J. Intern. Med. 2002, 252, 247–254. [Google Scholar] [CrossRef]
- Wienke, A.; Holm, N.V.; Skytthe, A.; Yashin, A.I. The heritability of mortality due to heart diseases: A correlated frailty model applied to Danish twins. Twin Res. 2001, 4, 266–274. [Google Scholar] [CrossRef][Green Version]
- Jansen, H.; Samani, N.J.; Schunkert, H. Mendelian randomization studies in coronary artery disease. Eur. Heart J. 2014, 35, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, E.W.; Thomassen, J.Q.; Frikke-Schmidt, R. HDL cholesterol concentrations and risk of atherosclerotic cardiovascular disease—Insights from randomized clinical trials and human genetics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159063. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: From research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Natarajanm, P. Clinical utility of polygenic risk scores for coronary artery disease. Nat. Rev. Cardiol. 2021, 19, 291–301. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Genetic Transmission Monogenic/Polygenic | Gene(s) | Location/ Chromosome | Disease/CAD-S | Biochemical Changes | Method CGS/GWAS/LA/WES | References |
|---|---|---|---|---|---|---|
| Monogenic lipid CAD | ||||||
| LDLR | 19p13.2 | FH | ↑ LDL-C | CGS/GWAS | [4,6,16,17,18] | |
| APOB | 2p24.1 | FHCL2/FDB | ↑ LDL-C | CGS/GWAS | [4,16,19,20,21,22] | |
| PCSK9 | 1p34.1-p32 | HCHOLA3 | ↑ LDL-C | CGS/GWAS | [16,23,24,25,26,27] | |
| LDLRAP1 | 1p34-p35 | ARH | ↑ LDL-C | CGS | [16,28,29] | |
| APOAI | 11q23.3 | Apo AI deficiency, Apo-A1 and apo C-III combined deficiency | ↓ HDL-C | CGS | [4,16,30,31,32,33,34] | |
| ABCA1 | 9q31.1 | TGD | ↓ HDL-C | CGS/GWAS | [16,35,36,37] | |
| LCAT | 16q22.1 | LCAT deficiency | ↓ HDL-C | CGS/GWAS | [10,16,38,39] | |
| LPL | 8p21.3 | LPL deficiency/CHLF | ↑ TG, ↓ LDL-C, ↓ HDL-C | CGS | [16,40,41] | |
| APOC2 | 19q13.2 | HL Ib | ↑ TG | CGS | [16,42] | |
| ABCG5, ABCG8 | 2p21 | STSL | ↑ plant sterols | CGS | [6,43,44] | |
| APOA1/C3/A4/A5 | 11p14.1-q12.1, 1q21-23, 16q22-24.1 | FCHL | ↑ VLDL, ↑ LDL-C, ↑ ApoB, ↑ TG | LA/GWAS | [6,45,46,47,48,49,50] | |
| APOA5 | 11p14.1-q12.1, 15q11.2-q13.1, 8q11-q13 | FHTG | ↑ TG | LA/GWAS | [4,16,50,51,52,53] | |
| ATHS | 19p13.3-p13.2 | ATHS/ALP | ↑ LDL-C, ↑ TG, ↓ HDL-C | LA | [16,54,55,56,57,58] | |
| Other Monogenic CAD | ||||||
| MEF2A | 15q26 | ADCAD1 | CGS | [6,59,60,61,62] | ||
| ST6GALNAC5 | 1p31.1 | GWAS/WES | [16,63,64] | |||
| CYP27A1 | 2p35 | ↓ LDL, ↓ VLDL, ↑ HDL-C | ||||
| CTX | WES | [16,65,66,67,68] | ||||
| LRP6 | 12p13.2 | ↑ LDL-C, ↑ TG | CGS/GWAS | [16,69,70] | ||
| Polygenic CAD | ||||||
| CDKN2A, CDKN2B | 9p21 | CAD-S | GWAS | [8,10,71,72,73,74] | ||
| C6orf105 gene | 6p24.1 | CAD-S | GWAS | [6,75,76] | ||
| COL4A1/COL4A2, ZC3HC1, CYP17A1 | 13q34, 7q32.2, 10q24.32 | CAD-S | GWAS | [77,78] | ||
| CTSS | 1q21 | CAD-S | GWAS | [79] | ||
| WDR11-FGFR2 | 10q26 | CAD-S | GWAS | [79] | ||
| RDX-FDX1 | 11q22 | CAD-S | GWAS | [79] | ||
| PSRC1 | 1p13.3 | CAD-S | GWAS | [73,80] | ||
| MIA3 | 1q41 | CAD-S | GWAS | [73,80] | ||
| SMAD3 | 15q22.3 | CAD-S | GWAS | [73,81,82,83] | ||
| Polygenic lipid CAD | APOE, | 19q13.32, | CAD-S | GWAS | [84,85,86,87,88,89,90,91] | |
| APOB, | 2p24.1, | [6,21,92,93,94] | ||||
| LPL, | 8p21.3, | [80,95,96,97,98] | ||||
| OLR1 (LOX1), | 12p13.2, | [99,100] | ||||
| SORT1, | 1p13.3, | [101] | ||||
| TRIB1, | 8q24.13 | [80,102,103] | ||||
| Genes associated with vascular homeostasis | NOS3, | 7q36.1, | CAD-S | GWAS | [4,104,105,106] | |
| TCF21, | 6q23.2, | [80,107,108] | ||||
| ADAMTS7, | 15q25.1, | [80,109] | ||||
| HHIPL1, | 14q32, | [110] | ||||
| ACE, | 17q23.3, | [80,111,112,113,114] | ||||
| AGT, | 1q42.2, | [114] | ||||
| AGT1R, | 3q23, | [115,116] | ||||
| CYP11B2 | 8q24.3 | [117,118,119] | ||||
| Genes associated with vascular hemostasis | ITGA2, | 5q11.2, | CAD-S | GWAS | [4,120,121] | |
| ITGB2, | 21q22.3, | [122,123] | ||||
| ITGB3, | 17q21.32, | [124,125] | ||||
| PAI-1, | 7q22.1, | [126,127] | ||||
| THBS (1, 2, 4), | 15q14, 6q27, 5q14.1 | [128] | ||||
| F5 Leiden (Arg506Gln), | 1q24.2, | [4,129] | ||||
| F2 gene (Arg 506Gln) | 11p11.2 | [4,129] | ||||
| HHcy | MTHFR | 1p36.22 | CAD-S | GWAS | [130,131,132,133] | |
| Inflammation | IL-6 | 7p15.3 | CAD-S | GWAS | [4,80,134] | |
| Other genes | POAD | 1p31 | CAD-S | GWAS | [135] | |
| CHDS2, | 2q21.2-q22, | [136] | ||||
| CHDS3, | Xq23-q26, | [16,136] | ||||
| AGTR2, | Xq23, | [136] | ||||
| IRS1, CAPN10, | 2q36-q37.3 | [5,137,138] | ||||
| HDLBPALDH2 | 12q24 | [139] | ||||
| Location/Chromosome | Gene (s) | SNPs | Risk Allele | Risk Allele Frequency |
|---|---|---|---|---|
| 1p32.3 | PCSK9 | rs112065101 | T/C | 0.848 |
| 1p32.3 | PPAP2B | rs9970807 | C/T | 0.915 |
| 1p13.3 | SORT1 | rs7528419 | A/G | 0.786 |
| 1q21 | CTSS | rs6587520 | T/C | 0.480 |
| 1q21.3 | IL6R | rs6689306 | A/G | 0.448 |
| 1q41 | MIA3 | rs67180937 | G/T | 0.663 |
| 2p24.1 | AK097927 | rs16986953 | A/G | 0.105 |
| 2p24.1 | APOB | chr2:21378433:D | D/I | 0.746 |
| 2p21 | ABCG5, ABCG8 | chr2:44074126:D | I/D | 0.745 |
| 2p11.2 | VAMP5-VAMP8-GCX | rs7568458 | A/T | 0.449 |
| 2q22.3 | ZEB2-ACO74093.1 | rs17678683 | G/T | 0.088 |
| 2q33.2 | WDR12 | chr2:203828796:I | I/D | 0.108 |
| 3q22.3 | MRAS | chr3:138099161:I | I/D | 0.163 |
| 4q31.22-q31.23 | EDNRA | rs4593108 | C/G | 0.795 |
| 4q32.1 | GUCY1A3 | rs72689147 | G/T | 0.817 |
| 4q12 | REST-NOA1 | rs17087335 | T/G | 0.210 |
| 5q31.1 | SLC22A4-SLC22A5 | rs273909 | G/A | 0.117 |
| 6p24.1 | ADTRP-C6orf105 | rs6903956 | A/G | 0.354 |
| 6p24.1 | PHACTR1 | rs9349379 | G/A | 0.432 |
| 6p21.31 | ANKS1A | rs17609940 | G/C | 0.824 |
| 6p21.2 | KCNK5 | rs56336142 | T/C | 0.807 |
| 6q23.2 | TCF21 | rs12202017 | A/G | 0.700 |
| 6q25.3 | SLC22A3-LPAL2-LPA | rs55730499 | T/C | 0.056 |
| 6q26 | PLG | rs4252185 | C/T | 0.060 |
| 7p21.1 | HDAC9 | rs2107595 | A/G | 0.200 |
| 7q22.3 | BCAP29 | rs10953541 | C/T | 0.783 |
| 7q34 | ZC3HC1 (PARP12) | rs11556924 | C/T | 0.687 |
| 7q36.1 | NOS3 | rs17087335 | T/C | 0.060 |
| 8p21.3 | LPL | rs264 | G/A | 0.853 |
| 8q24.13 | TRIB1 | rs2954029 | A/T | 0.551 |
| 9p21.3 | CDKN2BAS | rs2891168 | G/A | 0.489 |
| 9q34.2 | ABO | rs2519093 | T/C | 0.191 |
| 10p11.23 | KIAA1462 | rs2487928 | A/G | 0.418 |
| 10q11.21 | CXCL12 | rs1870634 | G/T | 0.637 |
| 10q23.31 | LIPA | rs1412444 | T/C | 0.369 |
| 10q24.32 | CYP17A1-CNNM2-NT5C2 | rs11191416 | T/G | 0.873 |
| 10q26 | WDR11-FGFR2 | rs2257129 | C/T | 0.900 |
| 11q22.3 | PDGFD | rs2128739 | A/C | 0.324 |
| 11q22 | RDX-FDX1 | rs10488763 | T/A | 0.180 |
| 11q23.3 | ZNF259-APOA5-APOA1 | rs964184 | G/C | 0.185 |
| 11p15.4 | SWAP70 | rs10840293 | A/G | 0.550 |
| 12q21.33 | ATP2B1 | rs2681472 | G/A | 0.201 |
| 12q24.12 | SH2B3 | rs3184504 | T/C | 0.422 |
| 12q24.22-q24.23 | KSR2 | rs1180803 | G/T | 0.360 |
| 13q12.3 | FLT1 | rs9319428 | A/G | 0.314 |
| 13q34 | COL4A1-COL4A2 | rs11838776 | A/G | 0.263 |
| 14q32 | HHIPL1 | rs10139550 | G/C | 0.423 |
| 15q25.1 | ADAMTS7 | rs4468572 | C/T | 0.586 |
| 15q26.1 | FURIN-FES | rs17514846 | A/C | 0.440 |
| 15q22.33 | SMAD3 | rs56062135 | C/T | 0.790 |
| 15q26.1 | MFGE8-ABHD2 | rs8042271 | G/A | 0.900 |
| 17p13.3 | SMG6 | rs216172 | C/G | 0.350 |
| 17p11.2 | RAI1-PEMT-RASD1 | rs12936587 | G/A | 0.611 |
| 17q21.32 | UBE2Z | rs46522 | T/C | 0.513 |
| 17q23.2 | BCAS3 | rs7212798 | C/T | 0.150 |
| 18q21.32 | PMAIP1-MC4R | rs663129 | A/G | 0.260 |
| 19p13.2 | LDLR | rs56289821 | G/A | 0.900 |
| 19q13.32 | APOE-APOC1 | rs4420638 | G/A | 0.166 |
| 19q13.11 | ZNF507-LOC400684 | rs12976411 | T/A | 0.090 |
| 21q22.11 | KCNE2 | rs28451064 | A/G | 0.121 |
| 22q11.23 | POM121L9P-ADORA2A | rs180803 | G/T | 0.970 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butnariu, L.I.; Florea, L.; Badescu, M.C.; Țarcă, E.; Costache, I.-I.; Gorduza, E.V. Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component? Life 2022, 12, 865. https://doi.org/10.3390/life12060865
Butnariu LI, Florea L, Badescu MC, Țarcă E, Costache I-I, Gorduza EV. Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component? Life. 2022; 12(6):865. https://doi.org/10.3390/life12060865
Chicago/Turabian StyleButnariu, Lăcrămioara Ionela, Laura Florea, Minerva Codruta Badescu, Elena Țarcă, Irina-Iuliana Costache, and Eusebiu Vlad Gorduza. 2022. "Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component?" Life 12, no. 6: 865. https://doi.org/10.3390/life12060865
APA StyleButnariu, L. I., Florea, L., Badescu, M. C., Țarcă, E., Costache, I.-I., & Gorduza, E. V. (2022). Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component? Life, 12(6), 865. https://doi.org/10.3390/life12060865