
Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants


Abstract
:1. Introduction
1.1. General Information
1.2. SARS-CoV-2 Virus
1.3. SARS-CoV-2 Mutation Rate
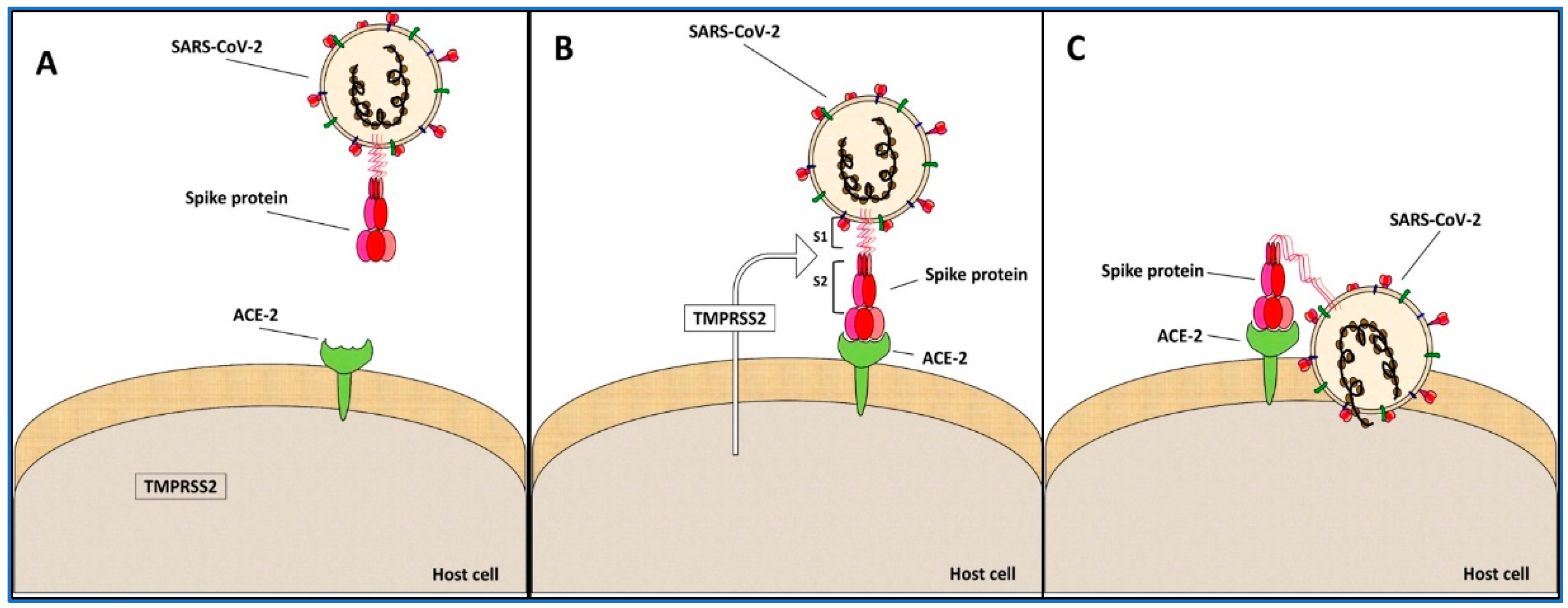
1.4. Spike Protein S
1.5. Spike Variants and Phylogenies
1.6. The Omicron Variants
2. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.C.; Phillips, E.J. Maintaining Safety with SARS-CoV-2 Vaccines. N. Engl. J. Med. 2020, 384, 643–649. [Google Scholar] [CrossRef]
- Fiolet, T.; Kherabi, Y.; MacDonald, C.-J.; Ghosn, J.; Peiffer-Smadja, N. Comparing COVID-19 vaccines for their characteristics, efficacy and effectiveness against SARS-CoV-2 and variants of concern: A narrative review. Clin. Microbiol. Infect. 2022, 28, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Caputo, E.; Mandrich, L. SARS-coV-2 infection: A case family report. Glob. J. Adv. Res. 2021, 8, 83–88. [Google Scholar]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef] [PubMed]
- Akram, F.; Haq, I.U.; Aqeel, A.; Ahmed, Z.; Shah, F.I.; Nawaz, A.; Zafar, J.; Sattar, R. Insights into the evolutionary and prophylactic analysis of SARS-CoV-2: A review. J. Virol. Methods 2022, 300, 114375. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based Covid-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; Wang-TO, K.K.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Rohaim, M.A.; El Naggar, R.F.; Clayton, E.; Munir, M. Structural and functional insights into non-structural proteins of coronaviruses. Microb. Pathog. 2021, 150, 104641. [Google Scholar] [CrossRef]
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef]
- Atzrodt, C.L.; Maknojia, I.; McCarthy, R.D.P.; Oldfield, T.M.; Po, J.; Ta, K.T.L.; Stepp, H.E.; Clements, T.P. A Guide to COVID-19: A global pandemic caused by the novel coronavirus SARS-CoV-2. FEBS J. 2020, 287, 3633–3650. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE-2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zè-Zè, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, G.; Huang, Y.-W. Modelling the load of SARS-CoV-2 virus in human expelled particles during coughing and speaking. PLoS ONE 2020, 15, e0241539. [Google Scholar] [CrossRef]
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 Variants, Vaccines, and Host Immunity. Front. Immunol. 2022, 12, 809244. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Braeye, T.; Catteau, L.; Brondeel, R.; van Loenhout, J.A.F.; Proesmans, K.; Cornelissen, L.; Van Oyen, H.; Stouten, V.; Hubin, P.; Billuart, M.; et al. Vaccine effectiveness against onward transmission of SARS-CoV2-infection by variant of concern and time since vaccination, Belgian contact tracing, 2021. Vaccine 2022, 40, 3027–3037. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2021, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- Mukherjee, A.G.; Wanjari, U.R.; Murali, R.; Chaudhary, U.; Renu, K.; Madhyastha, H.; Iyer, M.; Vellingiri, B.; Gopalakrishnan, A.V. Omicron variant infection and the associated immunological scenario. Immunobiology 2022, 227, 152222. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 Variants and Impact in Global Vaccination Programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.; Chatterjee, S.; Sharma, A.R.; Agoramoorthy, G.; Chakraborty, C. D614G mutation and SARS-CoV-2: Impact on S-protein structure, function, infectivity, and immunity. Appl. Microbiol. Biotechnol. 2021, 105, 9035–9045. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv 2021. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Q.; Wei, P.; Chen, Z.; Aviszus, K.; Yang, J.; Downing, W.; Jiang, C.; Liang, B.; Reynoso, L.; et al. The basis of a more contagious 501Y.V1 variant of SARS-CoV-2. Cell Res. 2021, 31, 720–722. [Google Scholar] [CrossRef]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Fabiani, M.; Puopolo, M.; Morciano, C.; Spuri, M.; Spila Alegiani, S.; Filia, A.; D’Ancona, F.; Del Manso, M.; Riccardo, F.; Tallon, M.; et al. Italian Integrated Surveillance of covid-19 study group and Italian covid-19 Vaccines Registry group. Effectiveness of mRNA vaccines and waning of protection against SARS-CoV-2 infection and severe covid-19 during predominant circulation of the delta variant in Italy: Retrospective cohort study. BMJ 2022, 376, e069052. [Google Scholar] [CrossRef]
- Ding, K.; Jiang, W.; Xiong, C.; Lei, M. Turning point: A new global COVID-19 wave or a signal of the beginning of the end of the global COVID-19 pandemic? Immun. Inflamm. Dis. 2022, 10, e606. [Google Scholar] [CrossRef] [PubMed]
- Rajpal, V.R.; Sharma, S.; Kumar, A.; Chand, S.; Joshi, L.; Chandra, A.; Babbar, S.; Goel, S.; Raina, S.N.; Shiran, B. “Is Omicron mild”? Testing this narrative with the mutational landscape of its three lineages and response to existing vaccines and therapeutic antibodies. J. Med. Virol. 2022, 94, 3521–3539. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef]

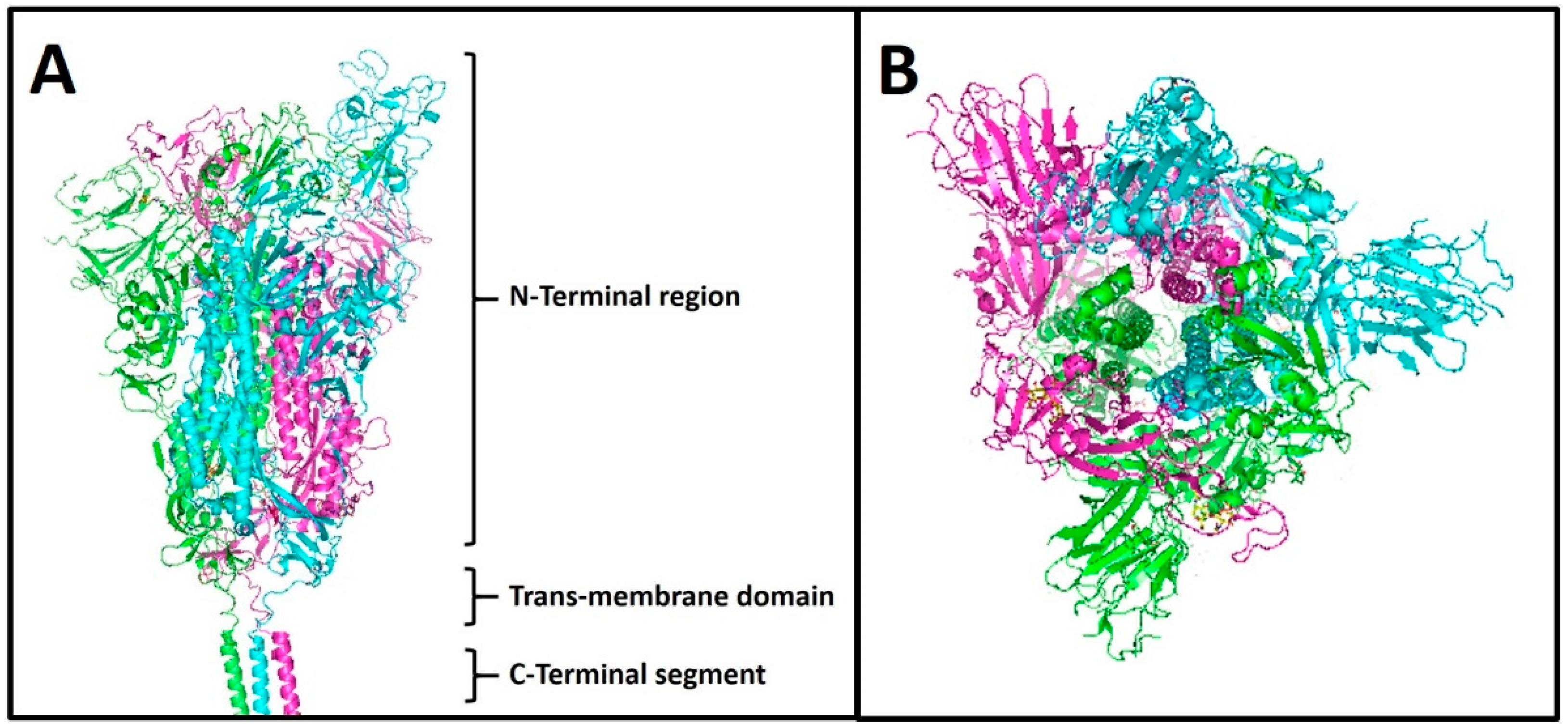
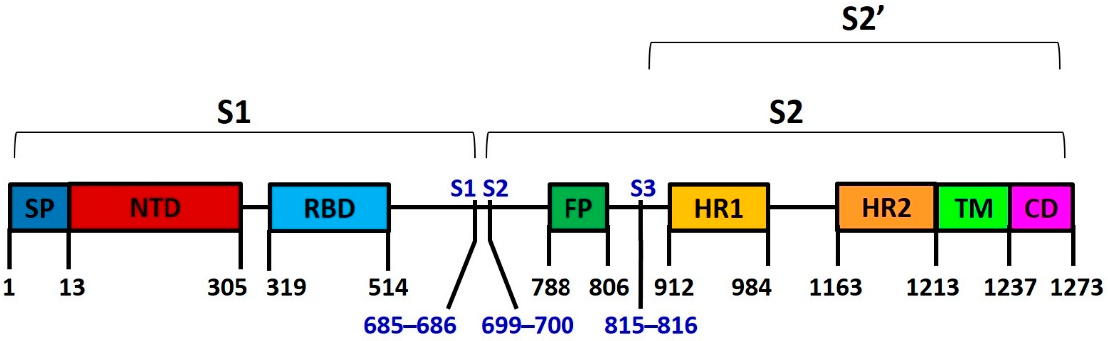

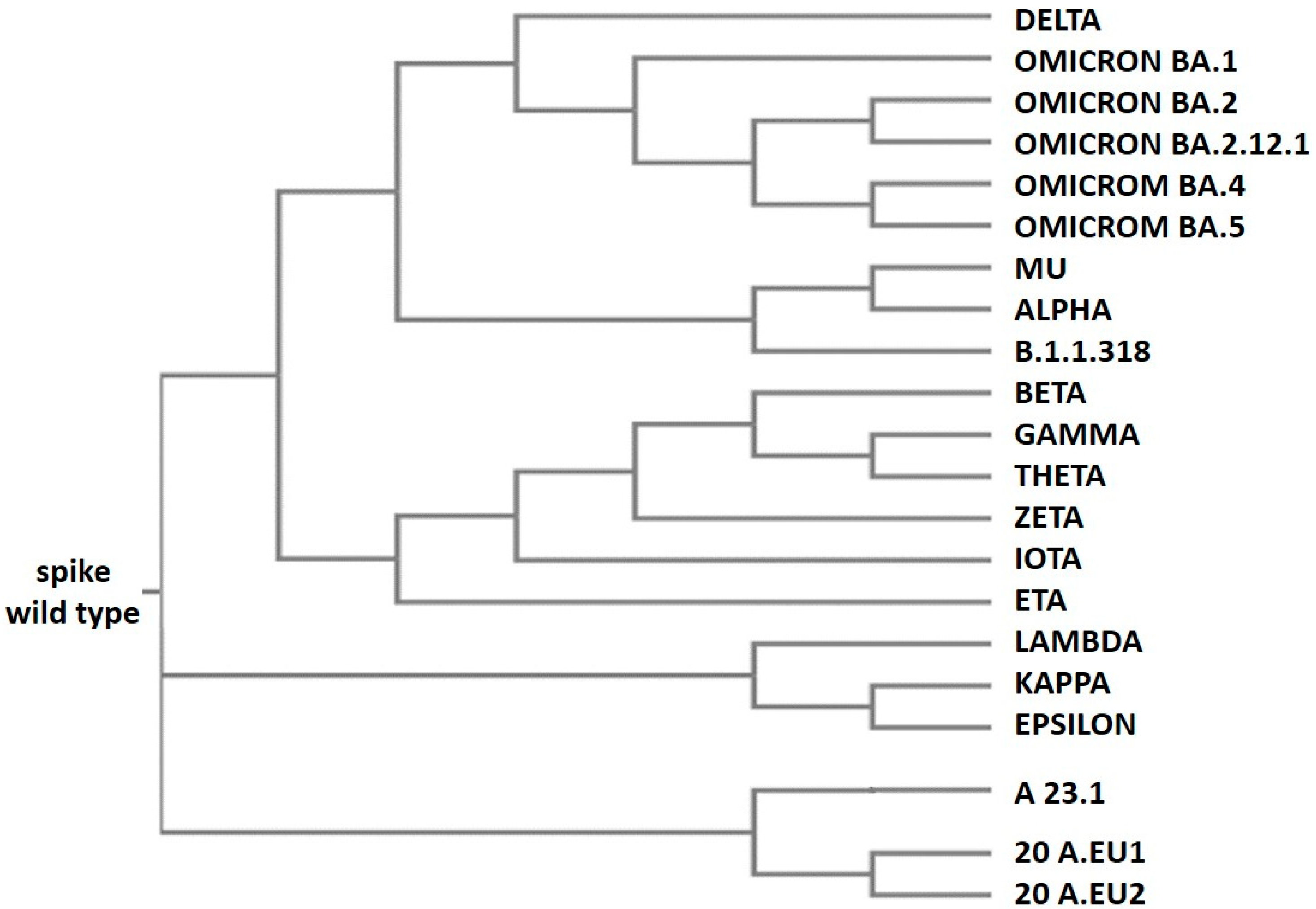

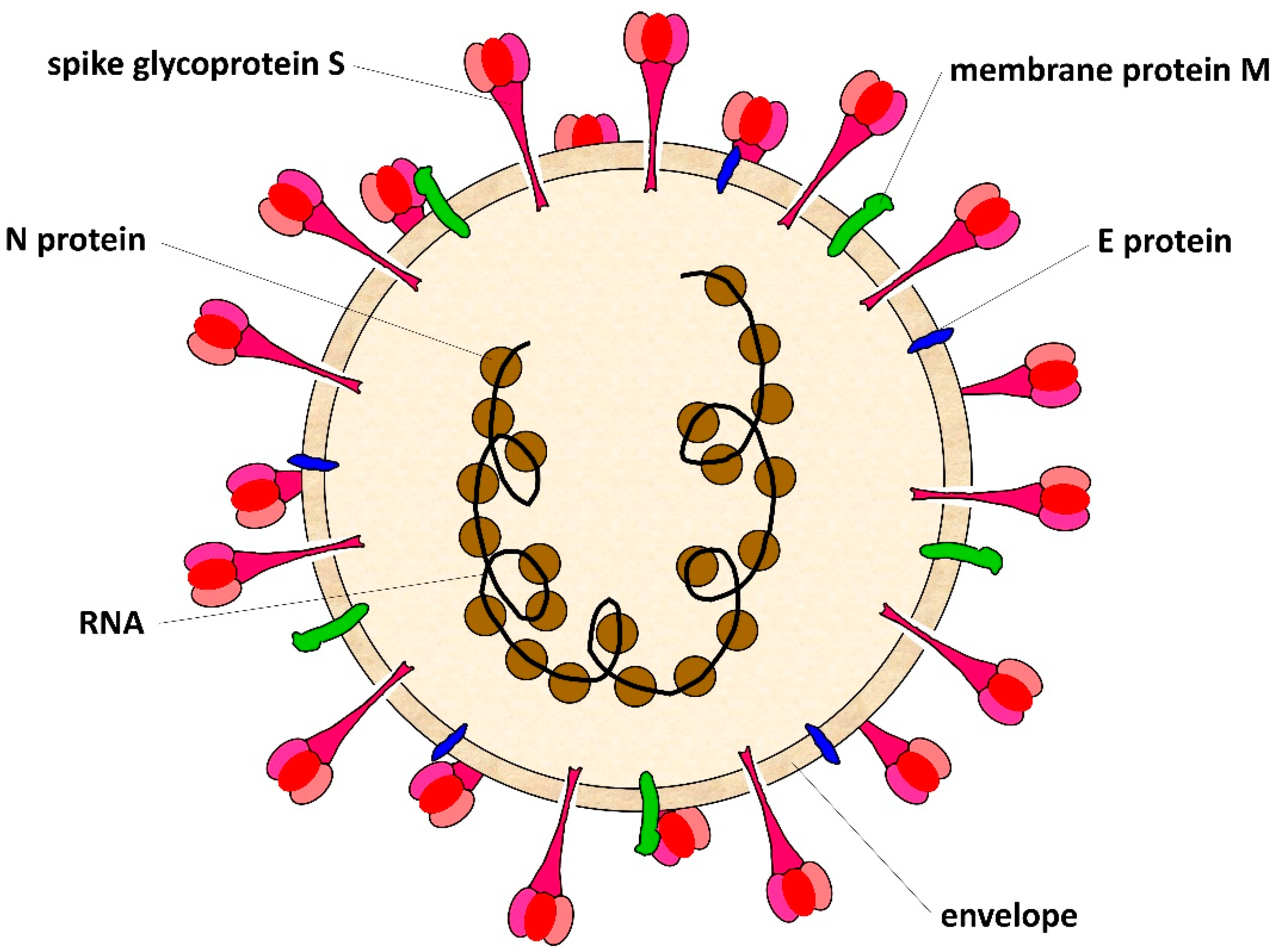{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spike Variants | Isolation Country | Isolation Date |
|---|---|---|
| wild type | China | December 2019 |
| EPSILON | USA | March 2020 |
| ZETA | Brazil | April 2020 |
| BETA | South Africa | May 2020 |
| 20 A.EU2 | Portugal | June2020 |
| 20 A.EU1 | Spain | July 2020 |
| ALPHA | England | September 2020 |
| DELTA | India | October 2020 |
| KAPPA | India | October 2020 |
| A 23.1 | Uganda | October 2020 |
| GAMMA | Brazil | November 2020 |
| IOTA | USA | November 2020 |
| ETA | Multiple countries | November 2020 |
| LAMBDA | Peru | December 2020 |
| THETA | Philippines | January 2021 |
| B.1.1.318 | Multiple countries | January 2021 |
| MU | Columbia | January 2021 |
| OMICRON BA.1 | South Africa | November 2021 |
| OMICRON BA.2 | South Africa | December 2021 |
| OMICRON BA.2.12.1 | North America | December 2021 |
| OMICRON BA.4 | South Africa | January 2022 |
| OMICRON BA.5 | South Africa | January 2022 |
| Spike Variants | Mutations |
|---|---|
| ALPHA | A570D T716I S982A D1118H |
| BETA | D80A D215G Del241-243 K417N A701V |
| GAMMA | L18F T20N D138Y R190S T1027I |
| DELTA | Del156-157 R158G |
| EPSILON | S13I W152C |
| ETA | Q52R A67V Q677H F888L |
| THETA | E1092K H1101Y |
| IOTA | L5F D253G |
| KAPPA | E154K Q1071H |
| LAMBDA | T76I R246N Del247-252 F490S T859N |
| MU | Ins147N Y147N R346K |
| 20 A.EU1 | A222V |
| A 23.1 | R102I F157L V367F |
| B.1.1.318 | T95I |
| OMICRON BA.1 | Del143-144 N211I L212V Ins213-214 V215P G446S G449S T547K N856K L981F |
| OMICRON BA.2.12.1 | S704L |
| OMICRON BA.4 | V3G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputo, E.; Mandrich, L. Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life 2022, 12, 1245. https://doi.org/10.3390/life12081245
Caputo E, Mandrich L. Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life. 2022; 12(8):1245. https://doi.org/10.3390/life12081245
Chicago/Turabian StyleCaputo, Emilia, and Luigi Mandrich. 2022. "Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants" Life 12, no. 8: 1245. https://doi.org/10.3390/life12081245
APA StyleCaputo, E., & Mandrich, L. (2022). Structural and Phylogenetic Analysis of SARS-CoV-2 Spike Glycoprotein from the Most Widespread Variants. Life, 12(8), 1245. https://doi.org/10.3390/life12081245