

Carbon Fixation in the Chemolithoautotrophic Bacterium Aquifex aeolicus Involves Two Low-Potential Ferredoxins as Partners of the PFOR and OGOR Enzymes

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. A. aeolicus Growth Condition
2.2. Enrichment of PFOR and OGOR from A. aeolicus Soluble Fraction
2.3. Purification of Native Ferredoxins
2.4. Heterologous Expression and Purification of Recombinant Fd7
2.5. Protein Concentration, Sequencing, Spectroscopy, Electrochemistry, and MALDI-TOF Mass Spectrometry
2.6. Gel Electrophoreses
2.7. Enzymatic Activities
2.8. Pull-Down Assay
2.9. Protein Identification and Proteomic Profiles by Mass Spectrometry
3. Results
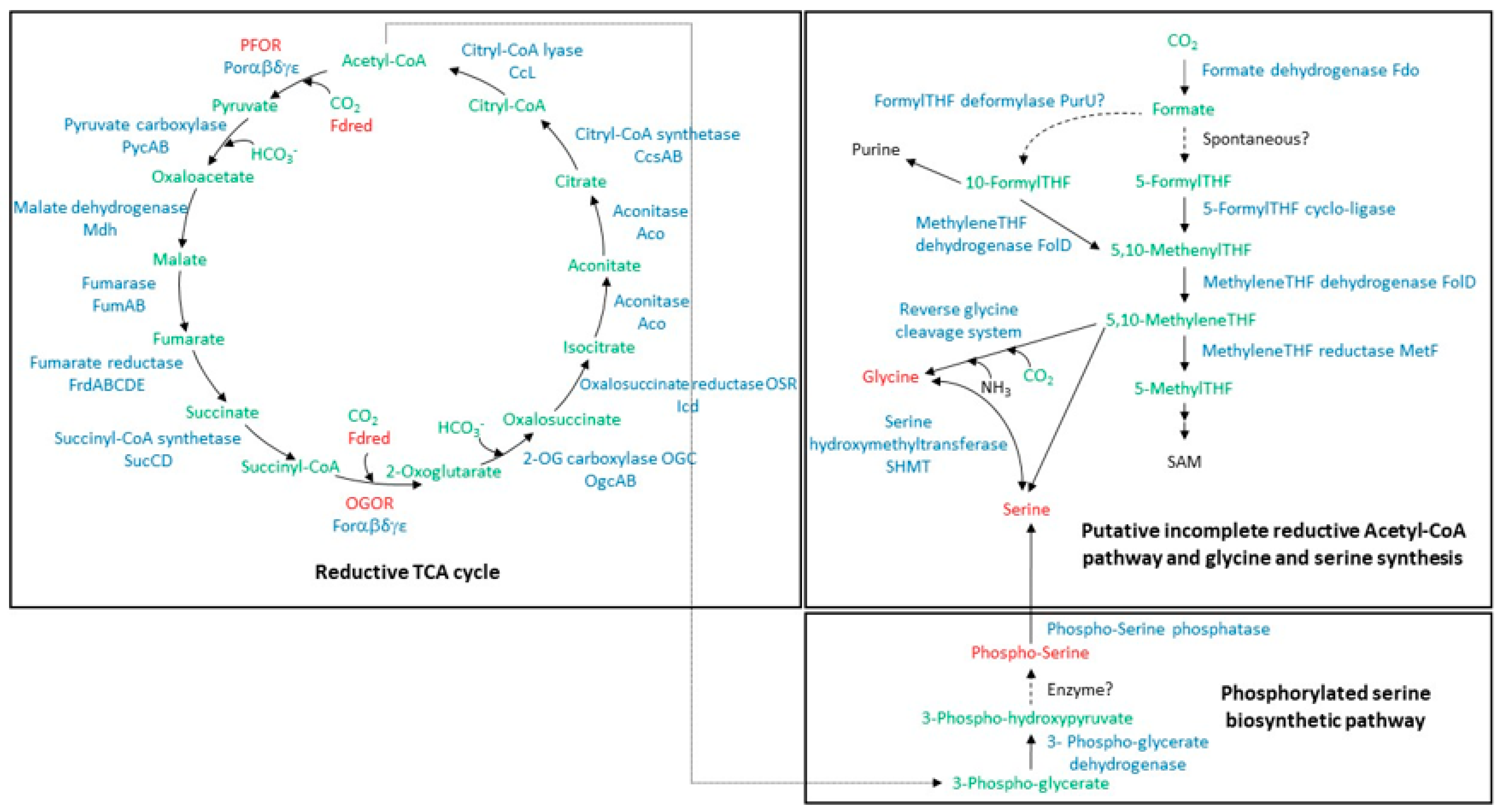
3.1. Carbon Fixation and Serine and Glycine Synthesis Pathways: Proteomic Profile of A. aeolicus Grown with H2 and Thiosulfate
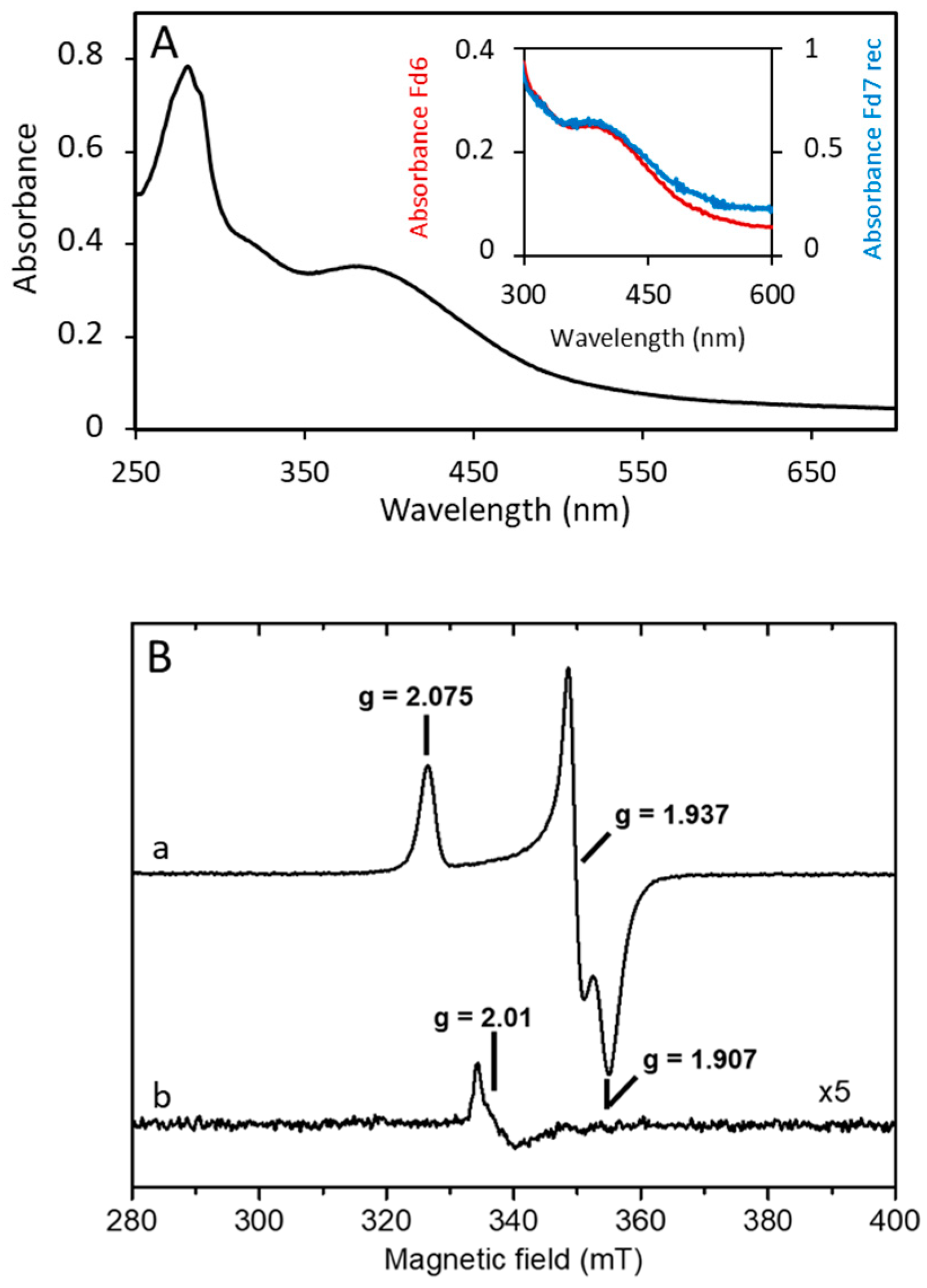
3.2. A. aeolicus Fd6 and Fd7 Are Low-Potential, Oxygen-Stable Ferredoxins
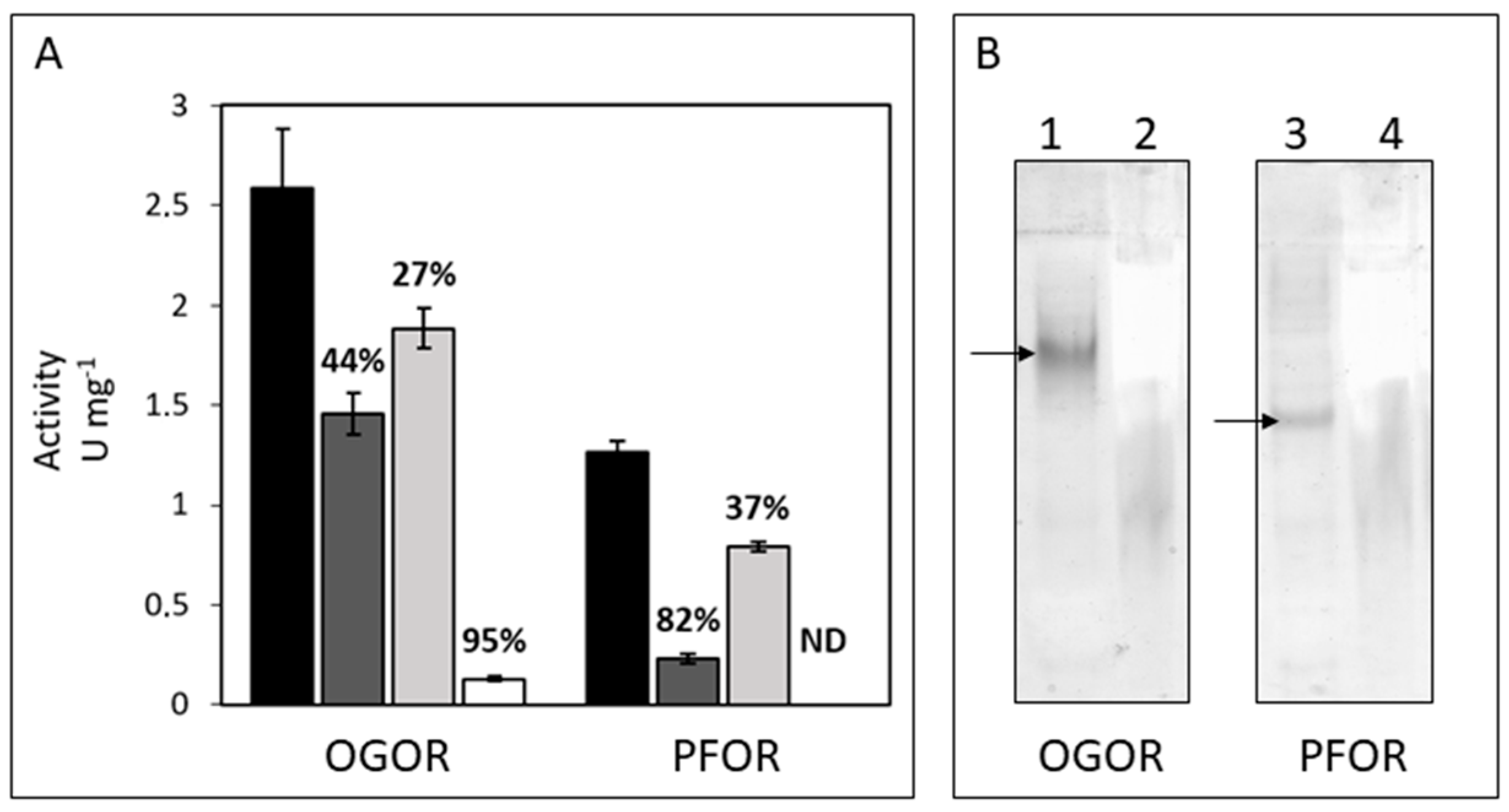
3.3. Identification, Activity, and Oxygen-Sensitivity of A. aeolicus PFOR and OGOR
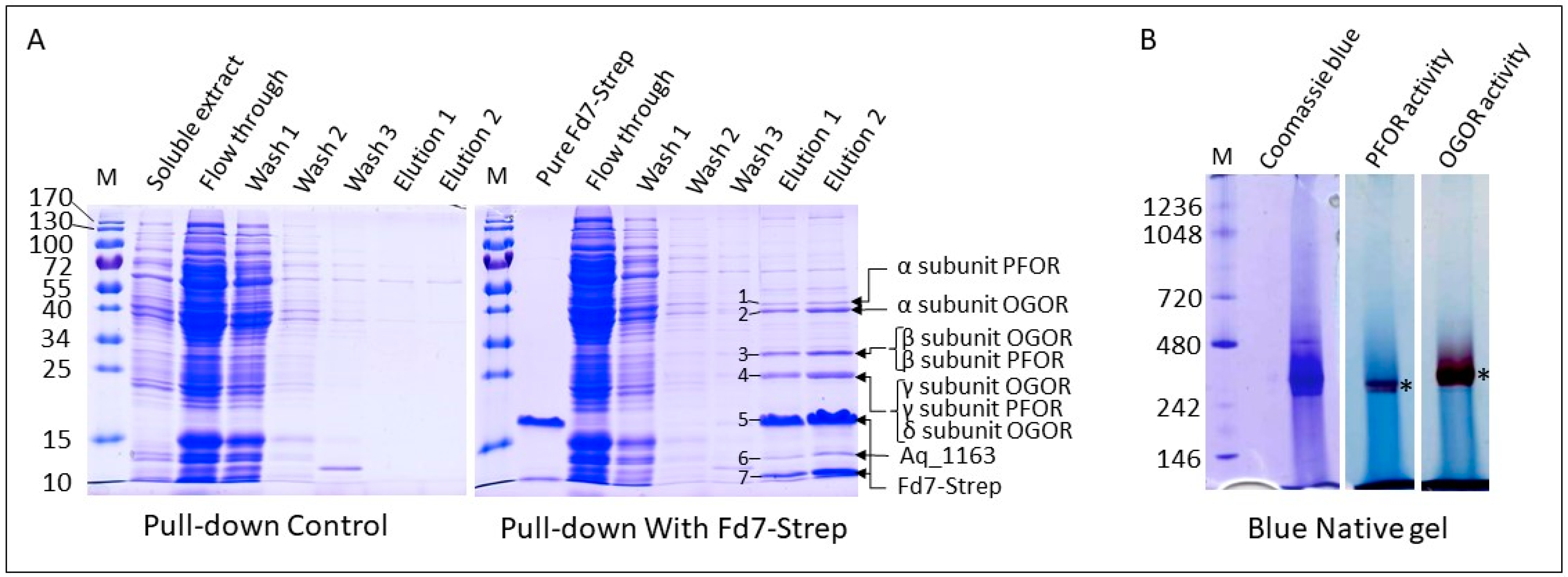
3.4. Fd6 and Fd7 Are Involved in the Reconstructed rTCA Cycle
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braakman, R.; Smith, E. Metabolic Evolution of a Deep-Branching Hyperthermophilic Chemoautotrophic Bacterium. PLoS ONE 2014, 9, e87950. [Google Scholar] [CrossRef]
- Fuchs, G. Alternative Pathways of Carbon Dioxide Fixation: Insights into the Early Evolution of Life? Annu. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar] [CrossRef]
- Gibson, M.I.; Chen, P.Y.-T.; Drennan, C.L. A Structural Phylogeny for Understanding 2-Oxoacid Oxidoreductase Function. Curr. Opin. Struct. Biol. 2016, 41, 54–61. [Google Scholar] [CrossRef]
- Chen, P.Y.-T.; Li, B.; Drennan, C.L.; Elliott, S.J. A Reverse TCA Cycle 2-Oxoacid:Ferredoxin Oxidoreductase That Makes C-C Bonds from CO2. Joule 2019, 3, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Ochiai, T.; Morita, S.; Nishiyama, A.; Yamada, E.; Arai, H.; Ishii, M.; Igarashi, Y. Anabolic Five Subunit-Type Pyruvate:Ferredoxin Oxidoreductase from Hydrogenobacter thermophilus TK-6. Biochem. Biophys. Res. Commun. 2006, 340, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Yamamoto, M.; Arai, H.; Ohmori, D.; Ishii, M.; Igarashi, Y. Enzymatic and Electron Paramagnetic Resonance Studies of Anabolic Pyruvate Synthesis by Pyruvate: Ferredoxin Oxidoreductase from Hydrogenobacter thermophilus. FEBS J. 2010, 277, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Arai, H.; Ishii, M.; Igarashi, Y. Characterization of Two Different 2-Oxoglutarate:Ferredoxin Oxidoreductases from Hydrogenobacter thermophilus TK-6. Biochem. Biophys. Res. Commun. 2003, 312, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ikeda, T.; Arai, H.; Ishii, M.; Igarashi, Y. Carboxylation Reaction Catalyzed by 2-Oxoglutarate:Ferredoxin Oxidoreductases from Hydrogenobacter thermophilus. Extremophiles 2010, 14, 79–85. [Google Scholar] [CrossRef]
- Yamamoto, M.; Arai, H.; Ishii, M.; Igarashi, Y. Role of Two 2-Oxoglutarate:Ferredoxin Oxidoreductases in Hydrogenobacter thermophilus under Aerobic and Anaerobic Conditions. FEMS Microbiol. Lett. 2006, 263, 189–193. [Google Scholar] [CrossRef]
- Yun, N.-R.; Yamamoto, M.; Arai, H.; Ishii, M.; Igarashi, Y. A Novel Five-Subunit-Type 2-Oxoglutalate:Ferredoxin Oxidoreductases from Hydrogenobacter thermophilus TK-6. Biochem. Biophys. Res. Commun. 2002, 292, 280–286. [Google Scholar] [CrossRef]
- Ikeda, T.; Yamamoto, M.; Arai, H.; Ohmori, D.; Ishii, M.; Igarashi, Y. Two Tandemly Arranged Ferredoxin Genes in the Hydrogenobacter thermophilus Genome: Comparative Characterization of the Recombinant [4Fe-4S] Ferredoxins. Biosci. Biotechnol. Biochem. 2005, 69, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Elliott, S.J. The Catalytic Bias of 2-Oxoacid:Ferredoxin Oxidoreductase in CO2: Evolution and Reduction through a Ferredoxin-Mediated Electrocatalytic Assay. Electrochim. Acta 2016, 199, 349–356. [Google Scholar] [CrossRef]
- Deckert, G.; Warren, P.V.; Gaasterland, T.; Young, W.G.; Lenox, A.L.; Graham, D.E.; Overbeek, R.; Snead, M.A.; Keller, M.; Aujay, M.; et al. The Complete Genome of the Hyperthermophilic Bacterium Aquifex aeolicus. Nature 1998, 392, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Hügler, M.; Huber, H.; Molyneaux, S.J.; Vetriani, C.; Sievert, S.M. Autotrophic CO2 Fixation via the Reductive Tricarboxylic Acid Cycle in Different Lineages within the Phylum Aquificae: Evidence for Two Ways of Citrate Cleavage. Environ. Microbiol. 2007, 9, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Braakman, R.; Smith, E. The Emergence and Early Evolution of Biological Carbon-Fixation. PLoS Comput. Biol. 2012, 8, e1002455. [Google Scholar] [CrossRef]
- Giovannelli, D.; Sievert, S.M.; Hügler, M.; Markert, S.; Becher, D.; Schweder, T.; Vetriani, C. Insight into the Evolution of Microbial Metabolism from the Deep-Branching Bacterium, Thermovibrio ammonificans. eLife 2017, 6, e18990. [Google Scholar] [CrossRef]
- Kim, K.; Chiba, Y.; Kobayashi, A.; Arai, H.; Ishii, M. Phosphoserine Phosphatase Is Required for Serine and One-Carbon Unit Synthesis in Hydrogenobacter thermophilus. J. Bacteriol. 2017, 199, e00409-17. [Google Scholar] [CrossRef]
- Guiral, M.; Tron, P.; Aubert, C.; Gloter, A.; Iobbi-Nivol, C.; Giudici-Orticoni, M.-T. A Membrane-Bound Multienzyme, Hydrogen-Oxidizing, and Sulfur-Reducing Complex from the Hyperthermophilic Bacterium Aquifex aeolicus. J. Biol. Chem. 2005, 280, 42004–42015. [Google Scholar] [CrossRef]
- Kpebe, A.; Benvenuti, M.; Guendon, C.; Rebai, A.; Fernandez, V.; Le Laz, S.; Etienne, E.; Guigliarelli, B.; García-Molina, G.; de Lacey, A.L.; et al. A New Mechanistic Model for an O2-Protected Electron-Bifurcating Hydrogenase, Hnd from Desulfovibrio fructosovorans. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1302–1312. [Google Scholar] [CrossRef]
- Gauquelin, C.; Baffert, C.; Richaud, P.; Kamionka, E.; Etienne, E.; Guieysse, D.; Girbal, L.; Fourmond, V.; André, I.; Guigliarelli, B.; et al. Roles of the F-Domain in [FeFe] Hydrogenase. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 69–77. [Google Scholar] [CrossRef]
- Schwartz, C.J.; Giel, J.L.; Patschkowski, T.; Luther, C.; Ruzicka, F.J.; Beinert, H.; Kiley, P.J. IscR, an Fe-S Cluster-Containing Transcription Factor, Represses Expression of Escherichia coli Genes Encoding Fe-S Cluster Assembly Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 14895–14900. [Google Scholar] [CrossRef] [PubMed]
- Dehé, P.-M.; Pamblanco, M.; Luciano, P.; Lebrun, R.; Moinier, D.; Sendra, R.; Verreault, A.; Tordera, V.; Géli, V. Histone H3 Lysine 4 Mono-Methylation Does Not Require Ubiquitination of Histone H2B. J. Mol. Biol. 2005, 353, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lojou, É.; Pieulle, L.; Guerlesquin, F.; Bianco, P. From the Protein–Polypeptide Model System to the Interaction between Physiological Partners Using Electrochemistry. J. Electroanal. Chem. 2002, 523, 150–159. [Google Scholar] [CrossRef]
- Guiral, M.; Prunetti, L.; Lignon, S.; Lebrun, R.; Moinier, D.; Giudici-Orticonit, M.-T. New Insights into the Respiratory Chains of the Chemolithoautotrophic and Hyperthermophilic Bacterium Aquifex aeolicus. J. Proteome Res. 2009, 8, 1717–1730. [Google Scholar] [CrossRef]
- Dementin, S.; Burlat, B.; Lacey, A.L.D.; Pardo, A.; Adryanczyk-Perrier, G.; Guigliarelli, B.; Fernandez, V.M.; Rousset, M. A Glutamate Is the Essential Proton Transfer Gate during the Catalytic Cycle of the [NiFe] Hydrogenase. J. Biol. Chem. 2004, 279, 10508–10513. [Google Scholar] [CrossRef] [PubMed]
- Brugna-Guiral, M.; Tron, P.; Nitschke, W.; Stetter, K.-O.; Burlat, B.; Guigliarelli, B.; Bruschi, M.; Giudici-Orticoni, M.T. [NiFe] Hydrogenases from the Hyperthermophilic Bacterium Aquifex aeolicus: Properties, Function, and Phylogenetics. Extrem. Life Extreme Cond. 2003, 7, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sadygov, R.G.; Yates, J.R. A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef]
- Braakman, R.; Smith, E. The Compositional and Evolutionary Logic of Metabolism. Phys. Biol. 2013, 10, 011001. [Google Scholar] [CrossRef]
- Figueroa, I.A.; Barnum, T.P.; Somasekhar, P.Y.; Carlström, C.I.; Engelbrektson, A.L.; Coates, J.D. Metagenomics-Guided Analysis of Microbial Chemolithoautotrophic Phosphite Oxidation Yields Evidence of a Seventh Natural CO2 Fixation Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E92–E101. [Google Scholar] [CrossRef]
- Sánchez-Andrea, I.; Guedes, I.A.; Hornung, B.; Boeren, S.; Lawson, C.E.; Sousa, D.Z.; Bar-Even, A.; Claassens, N.J.; Stams, A.J.M. The Reductive Glycine Pathway Allows Autotrophic Growth of Desulfovibrio desulfuricans. Nat. Commun. 2020, 11, 5090. [Google Scholar] [CrossRef]
- Desmarais, J.J.; Flamholz, A.I.; Blikstad, C.; Dugan, E.J.; Laughlin, T.G.; Oltrogge, L.M.; Chen, A.W.; Wetmore, K.; Diamond, S.; Wang, J.Y.; et al. DABs Are Inorganic Carbon Pumps Found throughout Prokaryotic Phyla. Nat. Microbiol. 2019, 4, 2204–2215. [Google Scholar] [CrossRef]
- Cao, X.; Hong, Y.; Zhu, L.; Hu, Y.; Cronan, J.E. Development and Retention of a Primordial Moonlighting Pathway of Protein Modification in the Absence of Selection Presents a Puzzle. Proc. Natl. Acad. Sci. USA 2018, 115, 647–655. [Google Scholar] [CrossRef]
- Shirakawa, T.; Takahashi, Y.; Wada, K.; Hirota, J.; Takao, T.; Ohmori, D.; Fukuyama, K. Identification of Variant Molecules of Bacillus thermoproteolyticus Ferredoxin: Crystal Structure Reveals Bound Coenzyme A and an Unexpected [3Fe-4S] Cluster Associated with a Canonical [4Fe-4S] Ligand Motif. Biochemistry 2005, 44, 12402–12410. [Google Scholar] [CrossRef]
- Gorst, C.M.; Zhou, Z.H.; Ma, K.; Teng, Q.; Howard, J.B.; Adams, M.W.; La Mar, G.N. Participation of the Disulfide Bridge in the Redox Cycle of the Ferredoxin from the Hyperthermophile Pyrococcus furiosus: 1H Nuclear Magnetic Resonance Time Resolution of the Four Redox States at Ambient Temperature. Biochemistry 1995, 34, 8788–8795. [Google Scholar] [CrossRef]
- Macedo, A.L.; Moura, I.; Surerus, K.K.; Papaefthymiou, V.; Liu, M.Y.; LeGall, J.; Münck, E.; Moura, J.J. Thiol/Disulfide Formation Associated with the Redox Activity of the [Fe3S4] Cluster of Desulfovibrio gigas Ferredoxin II. 1H NMR and Mössbauer Spectroscopic Study. J. Biol. Chem. 1994, 269, 8052–8058. [Google Scholar] [CrossRef]
- Meyer, J.; Clay, M.D.; Johnson, M.K.; Stubna, A.; Münck, E.; Higgins, C.; Wittung-Stafshede, P. A Hyperthermophilic Plant-Type [2Fe-2S] Ferredoxin from Aquifex Aeolicus Is Stabilized by a Disulfide Bond. Biochemistry 2002, 41, 3096–3108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Adams, M.W. Site-Directed Mutations of the 4Fe-Ferredoxin from the Hyperthermophilic Archaeon Pyrococcus furiosus: Role of the Cluster-Coordinating Aspartate in Physiological Electron Transfer Reactions. Biochemistry 1997, 36, 10892–10900. [Google Scholar] [CrossRef] [PubMed]
- Guigliarelli, B.; Bertrand, P. Application of EPR Spectroscopy to the Structural and Functional Study of Iron-Sulfur Proteins. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 47, pp. 421–497. [Google Scholar]
- Lanciano, P.; Savoyant, A.; Grimaldi, S.; Magalon, A.; Guigliarelli, B.; Bertrand, P. New Method for the Spin Quantitation of [4Fe-4S](+) Clusters with S = (3)/(2). Application to the FS0 Center of the NarGHI Nitrate Reductase from Escherichia coli. J. Phys. Chem. B 2007, 111, 13632–13637. [Google Scholar] [CrossRef]
- Maiocco, S.J.; Arcinas, A.J.; Booker, S.J.; Elliott, S.J. Parsing Redox Potentials of Five Ferredoxins Found within Thermotoga maritima. Protein Sci. Publ. Protein Soc. 2019, 28, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.-E.; Nitschke, W.; Infossi, P.; Giudici-Orticoni, M.-T.; Bill, E.; Lubitz, W. Characterization of a Unique [FeS] Cluster in the Electron Transfer Chain of the Oxygen Tolerant [NiFe] Hydrogenase from Aquifex aeolicus. Proc. Natl. Acad. Sci. USA 2011, 108, 6097–6102. [Google Scholar] [CrossRef] [PubMed]
- Menon, A.L.; Hendrix, H.; Hutchins, A.; Verhagen, M.F.; Adams, M.W. The Delta-Subunit of Pyruvate Ferredoxin Oxidoreductase from Pyrococcus furiosus Is a Redox-Active, Iron-Sulfur Protein: Evidence for an Ancestral Relationship with 8Fe-Type Ferredoxins. Biochemistry 1998, 37, 12838–12846. [Google Scholar] [CrossRef] [PubMed]
- Chabrière, E.; Charon, M.H.; Volbeda, A.; Pieulle, L.; Hatchikian, E.C.; Fontecilla-Camps, J.C. Crystal Structures of the Key Anaerobic Enzyme Pyruvate:Ferredoxin Oxidoreductase, Free and in Complex with Pyruvate. Nat. Struct. Biol. 1999, 6, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.S.; Borrel, G.; Gribaldo, S. Evolutionary History of Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase, One of the Oldest Enzymatic Complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E1166–E1173. [Google Scholar] [CrossRef]
- Burkhart, B.W.; Febvre, H.P.; Santangelo, T.J. Distinct Physiological Roles of the Three Ferredoxins Encoded in the Hyperthermophilic Archaeon Thermococcus kodakarensis. mBio 2019, 10, e02807-18. [Google Scholar] [CrossRef] [PubMed]
- Pieulle, L.; Nouailler, M.; Morelli, X.; Cavazza, C.; Gallice, P.; Blanchet, S.; Bianco, P.; Guerlesquin, F.; Hatchikian, E.C. Multiple Orientations in a Physiological Complex: The Pyruvate-Ferredoxin Oxidoreductase-Ferredoxin System. Biochemistry 2004, 43, 15480–15493. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.S.; Ishii, M.; Kodama, T.; Igarashi, Y. Purification and Characterization of Pyruvate:Ferredoxin Oxidoreductase from Hydrogenobacter thermophilus TK-6. Arch. Microbiol. 1997, 167, 275–279. [Google Scholar] [CrossRef]
- Bayer, B.; Saito, M.A.; McIlvin, M.R.; Lücker, S.; Moran, D.M.; Lankiewicz, T.S.; Dupont, C.L.; Santoro, A.E. Metabolic Versatility of the Nitrite-Oxidizing Bacterium Nitrospira marina and Its Proteomic Response to Oxygen-Limited Conditions. ISME J. 2021, 15, 1025–1039. [Google Scholar] [CrossRef]
- Levicán, G.; Ugalde, J.A.; Ehrenfeld, N.; Maass, A.; Parada, P. Comparative Genomic Analysis of Carbon and Nitrogen Assimilation Mechanisms in Three Indigenous Bioleaching Bacteria: Predictions and Validations. BMC Genom. 2008, 9, 581. [Google Scholar] [CrossRef]
- Lücker, S.; Nowka, B.; Rattei, T.; Spieck, E.; Daims, H. The Genome of Nitrospina gracilis Illuminates the Metabolism and Evolution of the Major Marine Nitrite Oxidizer. Front. Microbiol. 2013, 4, 27. [Google Scholar] [CrossRef]
- Yu, H.; Leadbetter, J.R. Bacterial Chemolithoautotrophy via Manganese Oxidation. Nature 2020, 583, 453–458. [Google Scholar] [CrossRef]
- Berg, I.A. Ecological Aspects of the Distribution of Different Autotrophic CO2 Fixation Pathways. Appl. Environ. Microbiol. 2011, 77, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Bar-Even, A.; Noor, E.; Milo, R. A Survey of Carbon Fixation Pathways through a Quantitative Lens. J. Exp. Bot. 2012, 63, 2325–2342. [Google Scholar] [CrossRef] [PubMed]
- Guiral, M.; Prunetti, L.; Aussignargues, C.; Ciaccafava, A.; Infossi, P.; Ilbert, M.; Lojou, E.; Giudici-Orticoni, M.-T. The Hyperthermophilic Bacterium Aquifex aeolicus: From Respiratory Pathways to Extremely Resistant Enzymes and Biotechnological Applications. Adv. Microb. Physiol. 2012, 61, 125–194. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Thauer, R.K. Flavin-Based Electron Bifurcation, Ferredoxin, Flavodoxin, and Anaerobic Respiration with Protons (Ech) or NAD+ (Rnf) as Electron Acceptors: A Historical Review. Front. Microbiol. 2018, 9, 401. [Google Scholar] [CrossRef]
- Poudel, S.; Dunham, E.C.; Lindsay, M.R.; Amenabar, M.J.; Fones, E.M.; Colman, D.R.; Boyd, E.S. Origin and Evolution of Flavin-Based Electron Bifurcating Enzymes. Front. Microbiol. 2018, 9, 1762. [Google Scholar] [CrossRef]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A Web Tool for Hydrogenase Classification and Analysis. Sci. Rep. 2016, 6, 34212. [Google Scholar] [CrossRef]
- Rousset, M.; Montet, Y.; Guigliarelli, B.; Forget, N.; Asso, M.; Bertrand, P.; Fontecilla-Camps, J.C.; Hatchikian, E.C. [3Fe-4S] to [4Fe-4S] Cluster Conversion in Desulfovibrio fructosovorans [NiFe] Hydrogenase by Site-Directed Mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11625–11630. [Google Scholar] [CrossRef]
- Ueda, Y.; Yamamoto, M.; Urasaki, T.; Arai, H.; Ishii, M.; Igarashi, Y. Sequencing and Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis of Four Hydrogenase Gene Clusters from an Obligately Autotrophic Hydrogen-Oxidizing Bacterium, Hydrogenobacter thermophilus TK-6. J. Biosci. Bioeng. 2007, 104, 470–475. [Google Scholar] [CrossRef]
- Boughanemi, S.; Lyonnet, J.; Infossi, P.; Bauzan, M.; Kosta, A.; Lignon, S.; Giudici-Orticoni, M.-T.; Guiral, M. Microbial Oxidative Sulfur Metabolism: Biochemical Evidence of the Membrane-Bound Heterodisulfide Reductase-like Complex of the Bacterium Aquifex aeolicus. FEMS Microbiol. Lett. 2016, 363, fnw156. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Kayastha, K.; Koch, T.; Venceslau, S.S.; Pereira, I.A.C.; Demmer, U.; Ermler, U.; Dahl, C. Structural and Spectroscopic Characterization of a HdrA-like Subunit from Hyphomicrobium denitrificans. FEBS J. 2021, 288, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G.; Moura, I. Molybdenum and Tungsten-Dependent Formate Dehydrogenases. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 2015, 20, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Niks, D.; Hille, R. Reductive Activation of CO2 by Formate Dehydrogenases. Methods Enzymol. 2018, 613, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Modularity of Methylotrophy, Revisited. Environ. Microbiol. 2011, 13, 2603–2622. [Google Scholar] [CrossRef]
- Rubin-Blum, M.; Dubilier, N.; Kleiner, M. Genetic Evidence for Two Carbon Fixation Pathways (the Calvin-Benson-Bassham Cycle and the Reverse Tricarboxylic Acid Cycle) in Symbiotic and Free-Living Bacteria. mSphere 2019, 4, e00394-18. [Google Scholar] [CrossRef]
- Cao, X.; Koch, T.; Steffens, L.; Finkensieper, J.; Zigann, R.; Cronan, J.E.; Dahl, C. Lipoate-binding proteins and specific lipoate-protein ligases in microbial sulfur oxidation reveal an atpyical role for an old cofactor. Elife 2018, 7, e37439. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.J.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange Consortium at 10 Years: 2023 Update. Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Substrate → Product | Subunit/ Protein | Accession 1 | Locus Tag | Gene | Mw 2 | PSM 3 | Cov 4 | Pep 5 | |
|---|---|---|---|---|---|---|---|---|---|---|
| REVERSE TCA CYCLE | ||||||||||
| Pyruvate: ferredoxin oxidoreductase (PFOR) | Acetyl-CoA → Pyruvate | Porα | O67254 | aq_1195 | forA1 | 45.1 | 178 | 63 | 20 | |
| Porβ | O67255 | aq_1196 | forB1 | 32.2 | 44 | 55 | 14 | |||
| Porγ | O67256 | aq_1200 | forG1 | 26.7 | 84 | 72 | 14 | |||
| Porε (Fdx3) | O67251 | aq_1192A | forD1 | 9 | 11 | 85 | 5 | |||
| Porδ | O67252 | aq_1192 | aq_1192 | 21.3 | 43 | 66 | 10 | |||
| PEP synthase | Pyruvate → Phosphoenolpyruvate | PpsA | O67899 | aq_2142 | ppsA | 96.4 | 220 | 64 | 59 | |
| Pyruvate carboxylase (PYC) | Pyruvate → Oxaloacetate | PycA | O67544 | aq_1614 | oadA | 70.4 | 108 | 55 | 35 | |
| PycB | O67449 | aq_1470 | accC2 | 53.7 | 64 | 55 | 29 | |||
| Malate dehydrogenase (MDH) | Oxaloacetate → Malate | Mdh1 | O67655 | aq_1782 | mdh1 | 36.7 | 85 | 71 | 19 | |
| Mdh2 | O67581 | aq_1665 | mdh2 | 36.7 | 9 | 27 | 7 | |||
| Fumarase (FUM) | Malate → Fumarate | FumA | O67654 | aq_1780 | fumB | 30.9 | 25 | 38 | 12 | |
| FumB | O67590 | aq_1679 | fumX | 20.4 | 21 | 61 | 11 | |||
| Fumarate reductase (FRD) | Fumarate → Succinate | FrdA | O66855 | aq_594 | frdA | 63.9 | 105 | 69 | 38 | |
| FrdB | O66828 | aq_553 | frdB1 | 27 | 4 | 16 | 3 | |||
| FrdC | O66518 | aq_116 | aq_116 | 59.5 | 21 | 28 | 12 | |||
| FrdD | O67007 | aq_835 | nox | 53.3 | 26 | 59 | 18 | |||
| FrdE | O66481 | aq_067 | dmsB2 | 19.4 | 5 | 18 | 3 | |||
| Succinyl-CoA synthetase (SUC) | Succinate → Succinyl-CoA | SucC | O67546 | aq_1620 | sucC | 42.2 | 97 | 69 | 30 | |
| SucD | O67547 | aq_1622 | sucD2 | 32.2 | 105 | 58 | 12 | |||
| 2-Oxoglutarate: ferredoxin oxidoreductase (OGOR) | Succinyl-CoA → 2-Oxoglutarate | Forα | O67229 | aq_1167 | forA2 | 44.7 | 253 | 72 | 26 | |
| Forβ | O67230 | aq_1168 | forB2 | 32.6 | 76 | 69 | 20 | |||
| Forγ | O67231 | aq_1169 | forG2 | 25.5 | 89 | 92 | 18 | |||
| Forε (Fdx2) | O67232 | aq_1171 | forD2 | 9 | 30 | 91 | 7 | |||
| Forδ | O67228 | aq_1166 | aq-1166 | 27.6 | 149 | 84 | 21 | |||
| 2-Oxoglutarate carboxylase (OGC) | 2-Oxoglutarate → Oxalosuccinate | OgcA (CfiA) | O67484 | aq_1520 | pycA | 73.6 | 472 | 74 | 56 | |
| OgcB (CfiB) | O67483 | aq_1517 | pycB | 52.8 | 244 | 74 | 33 | |||
| Oxalosuccinate reductase (OSR) | Oxalosuccinate → Isocitrate | Icd | O67480 | aq_1512 | icd | 46.9 | 54 | 48 | 23 | |
| Aconitase | Isocitrate → Aconitate → Citrate | Aco | O67656 | aq_1784 | aco | 72.5 | 133 | 67 | 38 | |
| Citryl-CoA synthetase (CCS) | Citrate → Citryl-CoA | CcsA | O67330 | aq_1306 | sucC1 | 48.3 | 231 | 77 | 35 | |
| CcsB | O67729 | aq_1888 | sucD1 | 39.8 | 164 | 78 | 30 | |||
| Citryl-CoA lyase (CCL) | Citryl-CoA → Acetyl-CoA + Oxaloacetate | Ccl | O66541 | aq_150 | gltA | 29.1 | 95 | 80 | 21 | |
| PUTATIVE INCOMPLETE REDUCTIVE ACETYL-CoA PATHWAY and GLYCINE AND SERINE SYNTHESIS | ||||||||||
| Formate dehydrogenase | CO2 → Formate | FdoI FdoH FdoG | O67148 O67147 O67146 | aq_1049 aq_1046 aq_1039 | fdoI fdoH fdoG | 24.3 33.4 114.5 | - 10 17 | - 24 14 | - 6 12 | |
| Formyltetrahydrofolate deformylase | Formate → 10-formylTHF | PurU | O67681 | aq_1818 | purU | 32.8 | 8 | 41 | 8 | |
| 5-Formyltetrahydrofolate cyclo-ligase | 5-formylTHF → 5,10-methenylTHF | MTHFS | O67621 | aq_1731 | aq_1731 | 21.2 | - | - | - | |
| Methylenetetrahydrofolate dehydrogenase | 10-formylTHF → 5,10-methenylTHF → 5,10-methyleneTHF | FolD | O67736 | aq_1898 | folD | 31.9 | 18 | 37 | 9 | |
| 5,10-methylenetetrahydrofolate reductase | 5,10-methyleneTHF → 5-methylTHF | MetF | O67422 | aq_1429 | metF | 33.9 | 19 | 49 | 13 | |
| Dihydrolipoyl dehydrogenase |  | GcvL/Lpd | O66945 | aq_736 | lpdA | 51.6 | 54 | 72 | 25 | |
| Aminomethyl transferase | GcvT | O67441 | aq_1458 | gcvT | 40.3 | 27 | 46 | 17 | ||
| Glycine dehydrogenase (decarboxylating) subunit 1 | Reversible glycine cleavage system | GcsP2 | O67193 | aq_1109 | gcvPA | 49.7 | 32 | 40 | 19 | |
| Glycine dehydrogenase (decarboxylating) subunit 2 | GcsP1 | O67740 | aq_1903 | gcvPB | 55 | 30 | 45 | 18 | ||
| Glycine cleavage system H protein 1 | CO2 +NH3 + 5,10-methyleneTHF → Glycine + THF | GcsH | O67151 | aq_1052 | gcvH1 | 16.2 | - | - | - | |
| Glycine cleavage system H protein 2 | GcsH | O67573 | aq_1657 | gcvH2 | 18.2 | 3 | 29 | 3 | ||
| Glycine cleavage system H protein 3 | GcsH | O67080 | aq_944 | gcvH3 | 18.1 | - | - | - | ||
| Glycine cleavage system H protein 4 | GcsH | O67192 | aq_1108 | gcvH4 | 19.6 | - | - | - | ||
| Serine hydroxymethyltransferase | Glycine + 5,10-methyleneTHF → Serine + THF | SHMT | O66776 | aq_479 | glyA | 47.4 | 63 | 46 | 21 | |
| SERINE AND GLYCINE SYNTHESIS via phosphorylated serine | ||||||||||
| Phospho-serine phosphatase | Phospho-serine → Serine | PspA | O67797 | aq_1990 | pgmA | 24.4 | 17 | 56 | 10 | |
| Serine hydroxymethyltransferase | Serine + THF → Glycine + 5,10-methyleneTHF | SHMT | O66776 | aq_479 | glyA | 47.4 | 63 | 46 | 21 | |
| Description | Subunit | Accession 1 | Locus Tag | Gene | Mw 2 | PSM 3 | Cov 4 | Pep 5 | |
|---|---|---|---|---|---|---|---|---|---|
| OGOR in-gel activity staining | Alpha subunit OGOR | Forα | O67229 | aq_1167 | forA2 | 42.7 | 62 | 58 | 17 |
| Beta subunit OGOR | Forβ | O67230 | aq_1168 | forB2 | 32.6 | 30 | 26 | 8 | |
| Gamma subunit OGOR | Forγ | O67231 | aq_1169 | forG2 | 25.5 | 25 | 84 | 12 | |
| Epsilon subunit OGOR | Forε | O67232 | aq_1171 | forD2 (fdx2) | 9 | 4 | 43 | 3 | |
| Delta subunit OGOR | Forδ | O67228 | aq_1166 | aq_1166 | 27.6 | 23 | 57 | 10 | |
| PFOR in-gel activity staining | Alpha subunit PFOR | Forα | O67254 | aq_1195 | forA1 | 45.1 | 113 | 66 | 19 |
| Beta subunit PFOR | Porβ | O67255 | aq_1196 | forB1 | 32.2 | 28 | 47 | 10 | |
| Gamma subunit PFOR | Porγ | O67256 | aq_1200 | forG1 | 26.6 | 20 | 63 | 10 | |
| Epsilon subunit PFOR | Porε | O67251 | aq_1192a | forD1 (fdx3) | 9.1 | 7 | 26 | 3 | |
| Delta subunit PFOR | Porδ | O67252 | aq_1192 | aq_1192 | 21.3 | 17 | 61 | 7 | |
| Uma2 domain-containing protein | O67253 | aq_1194 | aq_1194 | 20.9 | - | - | - |
| Electron Acceptor 2 | Activity (U mg−1) | |
|---|---|---|
| OGOR 1 | MeV | 1.71 ± 0.10 |
| MTZ | ND | |
| MTZ + Fd6 | 0.30 ± 0.01 | |
| MTZ + Fd7 | 0.26 ± 0.02 | |
| PFOR 1 | MeV | 0.33 ± 0.02 |
| MTZ | ND | |
| MTZ + Fd6 | ND | |
| MTZ + Fd7 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prioretti, L.; D’Ermo, G.; Infossi, P.; Kpebe, A.; Lebrun, R.; Bauzan, M.; Lojou, E.; Guigliarelli, B.; Giudici-Orticoni, M.-T.; Guiral, M. Carbon Fixation in the Chemolithoautotrophic Bacterium Aquifex aeolicus Involves Two Low-Potential Ferredoxins as Partners of the PFOR and OGOR Enzymes. Life 2023, 13, 627. https://doi.org/10.3390/life13030627
Prioretti L, D’Ermo G, Infossi P, Kpebe A, Lebrun R, Bauzan M, Lojou E, Guigliarelli B, Giudici-Orticoni M-T, Guiral M. Carbon Fixation in the Chemolithoautotrophic Bacterium Aquifex aeolicus Involves Two Low-Potential Ferredoxins as Partners of the PFOR and OGOR Enzymes. Life. 2023; 13(3):627. https://doi.org/10.3390/life13030627
Chicago/Turabian StylePrioretti, Laura, Giulia D’Ermo, Pascale Infossi, Arlette Kpebe, Régine Lebrun, Marielle Bauzan, Elisabeth Lojou, Bruno Guigliarelli, Marie-Thérèse Giudici-Orticoni, and Marianne Guiral. 2023. "Carbon Fixation in the Chemolithoautotrophic Bacterium Aquifex aeolicus Involves Two Low-Potential Ferredoxins as Partners of the PFOR and OGOR Enzymes" Life 13, no. 3: 627. https://doi.org/10.3390/life13030627
APA StylePrioretti, L., D’Ermo, G., Infossi, P., Kpebe, A., Lebrun, R., Bauzan, M., Lojou, E., Guigliarelli, B., Giudici-Orticoni, M.-T., & Guiral, M. (2023). Carbon Fixation in the Chemolithoautotrophic Bacterium Aquifex aeolicus Involves Two Low-Potential Ferredoxins as Partners of the PFOR and OGOR Enzymes. Life, 13(3), 627. https://doi.org/10.3390/life13030627