
Embryonic Hyperglycemia Disrupts Myocardial Growth, Morphological Development, and Cellular Organization: An In Vivo Experimental Study

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Embryos
2.2. Weight and Body Development
2.3. Histology
2.4. Scanning Electron Microscopy
2.5. Immunostaining and Detection by Confocal Microscopy
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
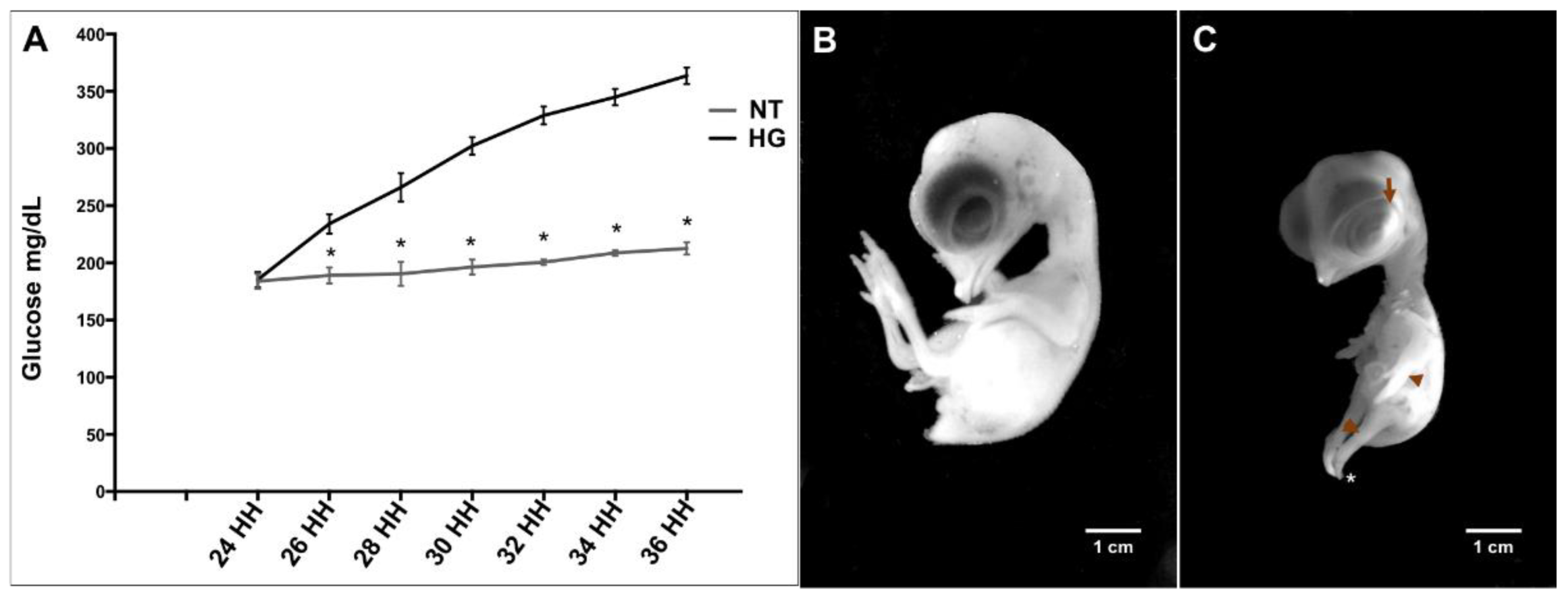
3.1. Induction of Hyperglycemia
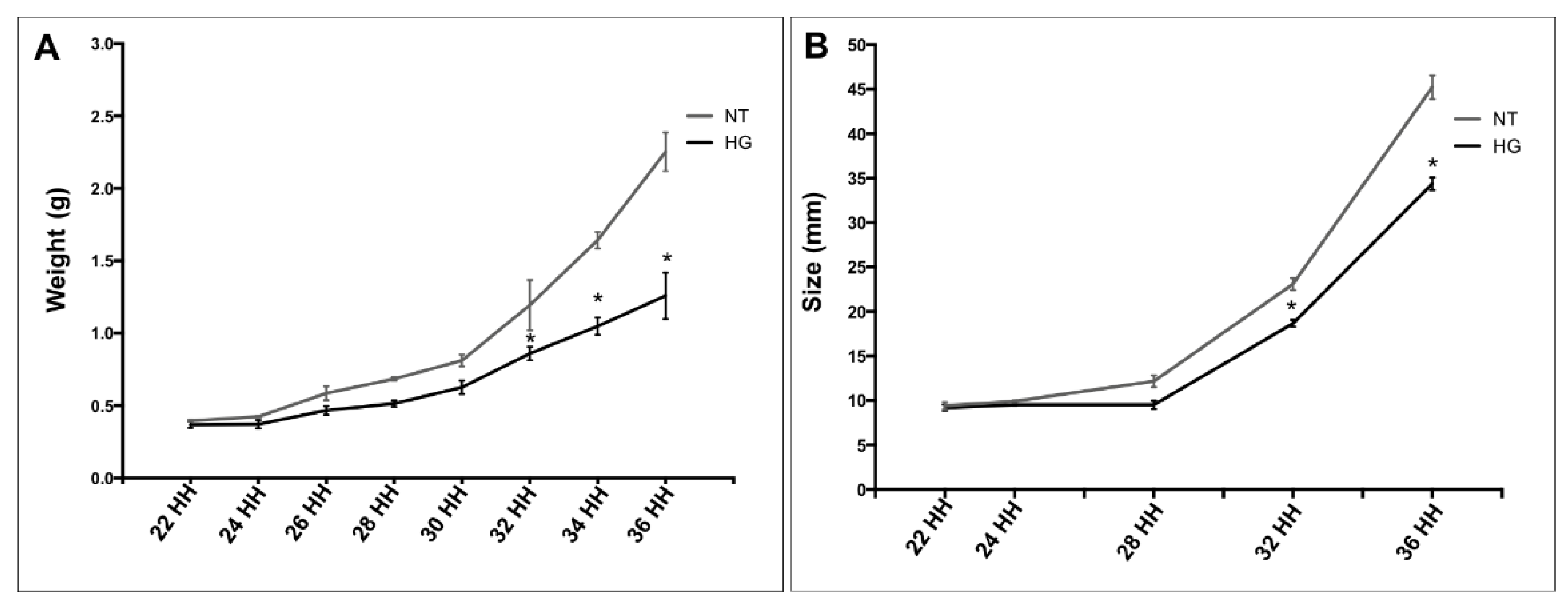
3.2. Weight and Body Development
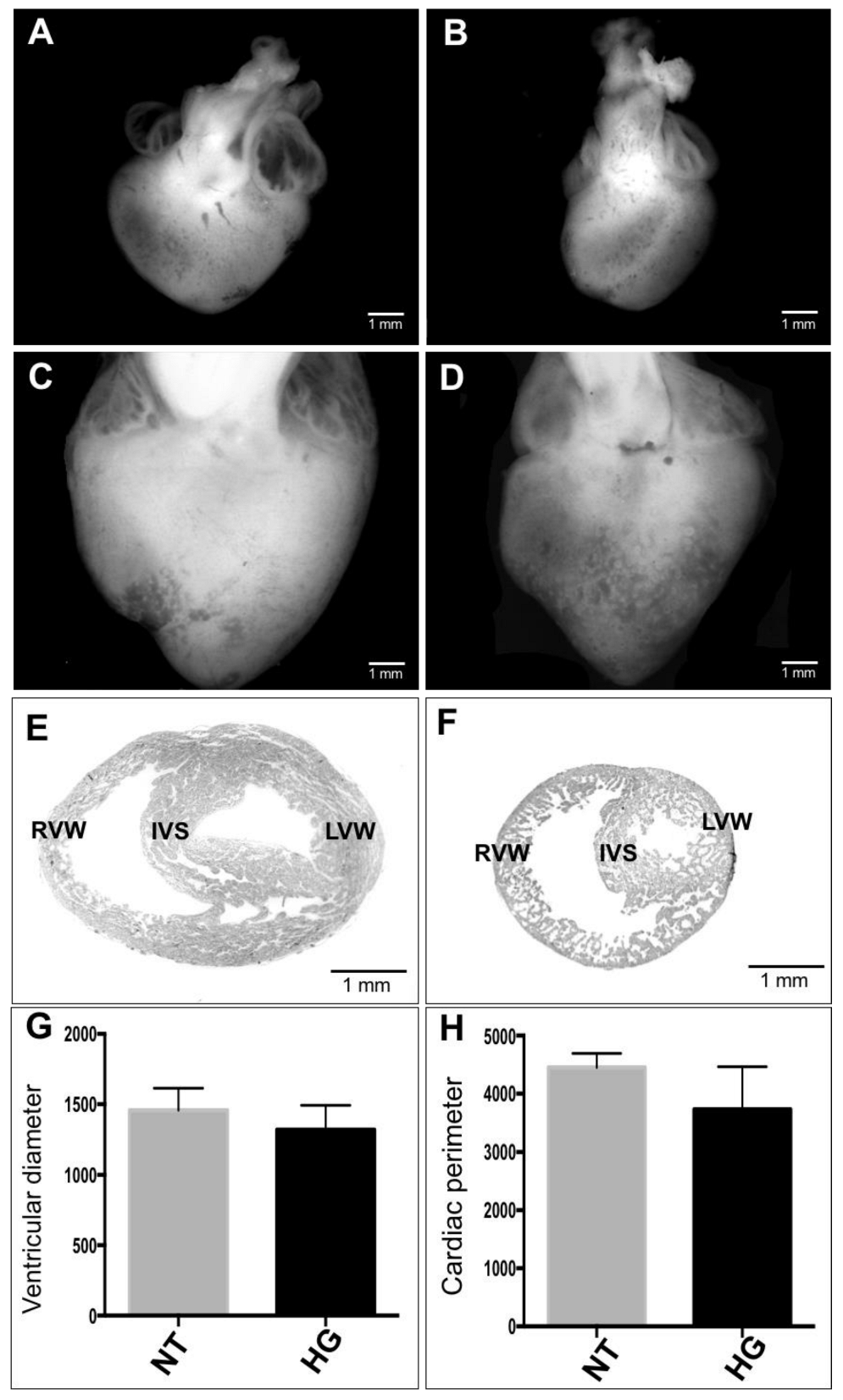
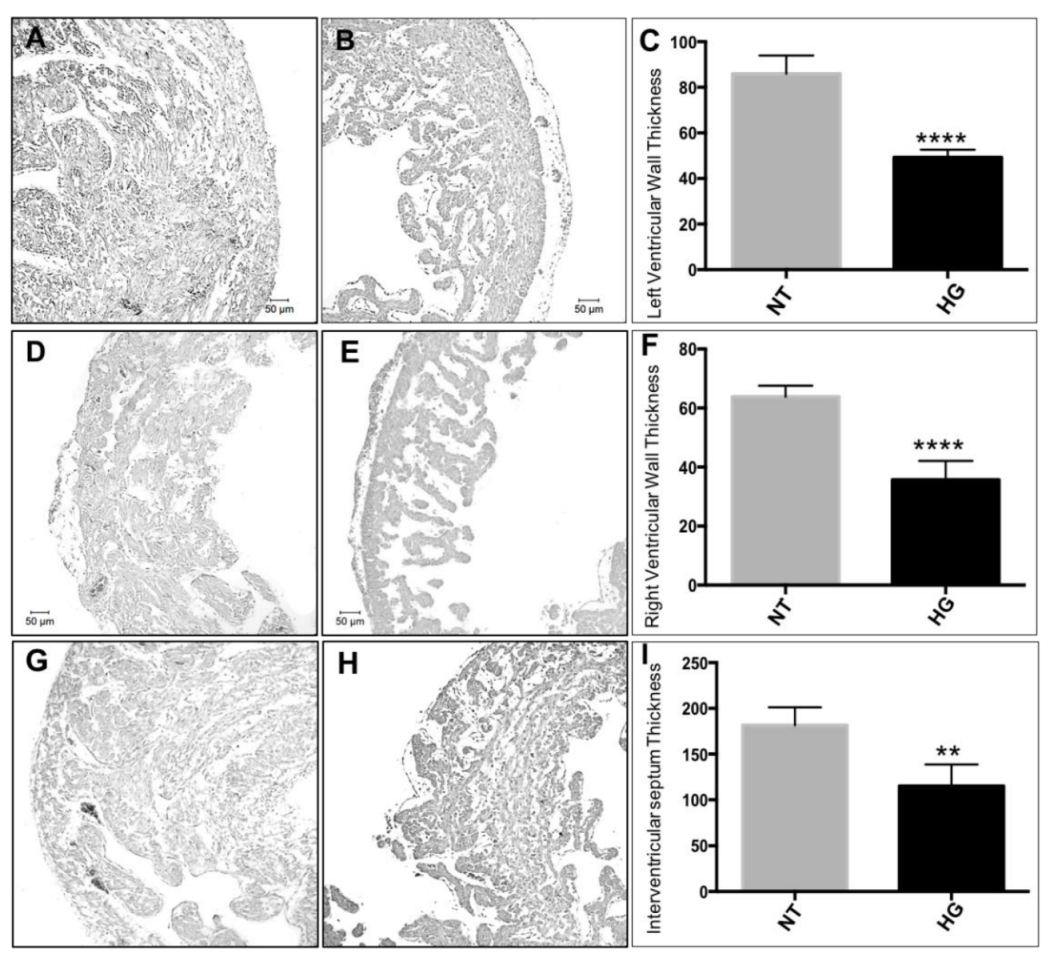
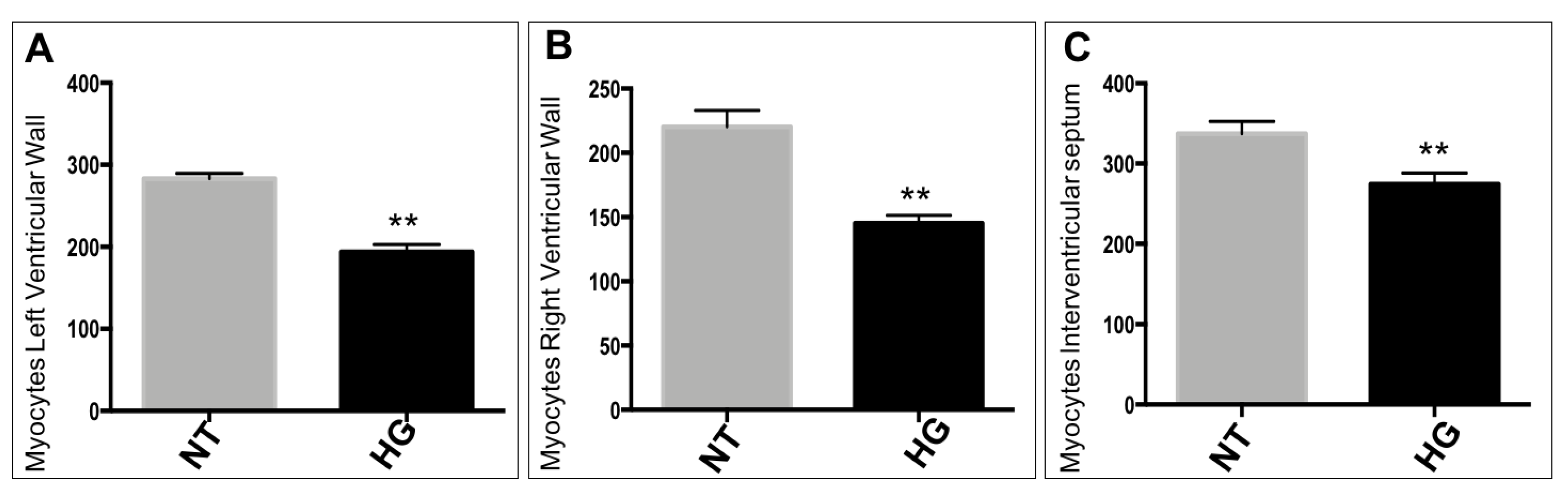
3.3. Morphometric and Histological Analyses of Embryonic Heart
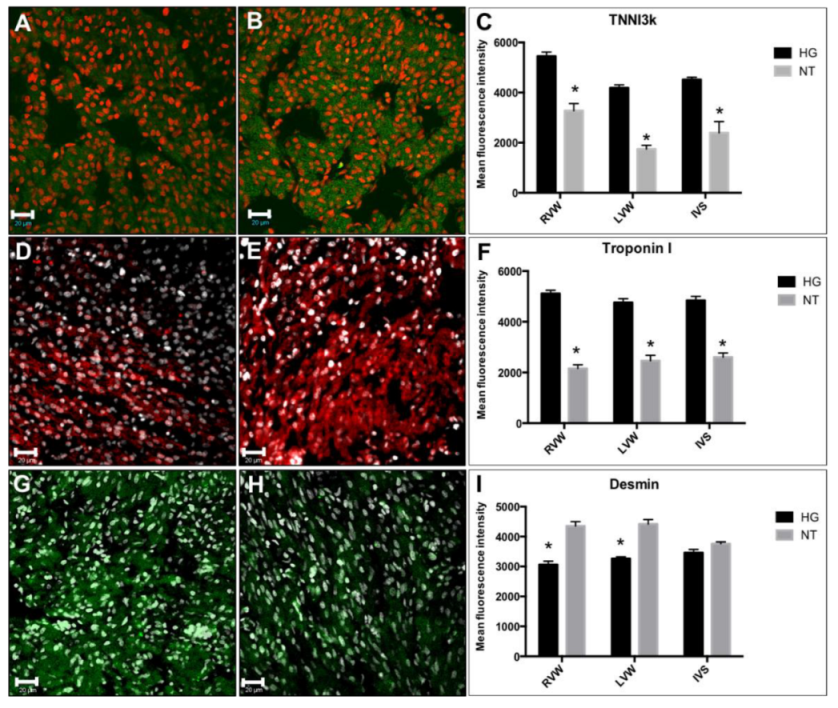
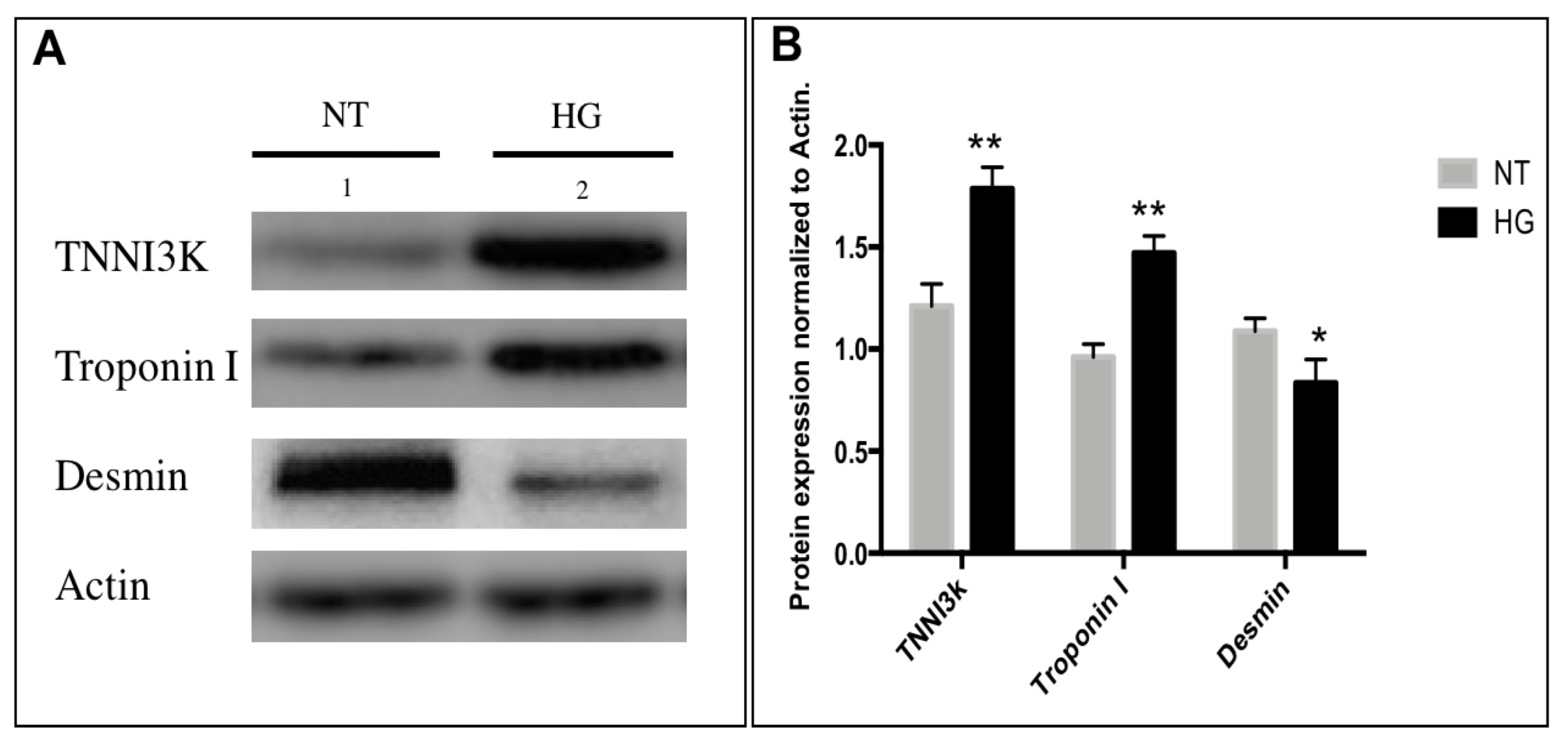
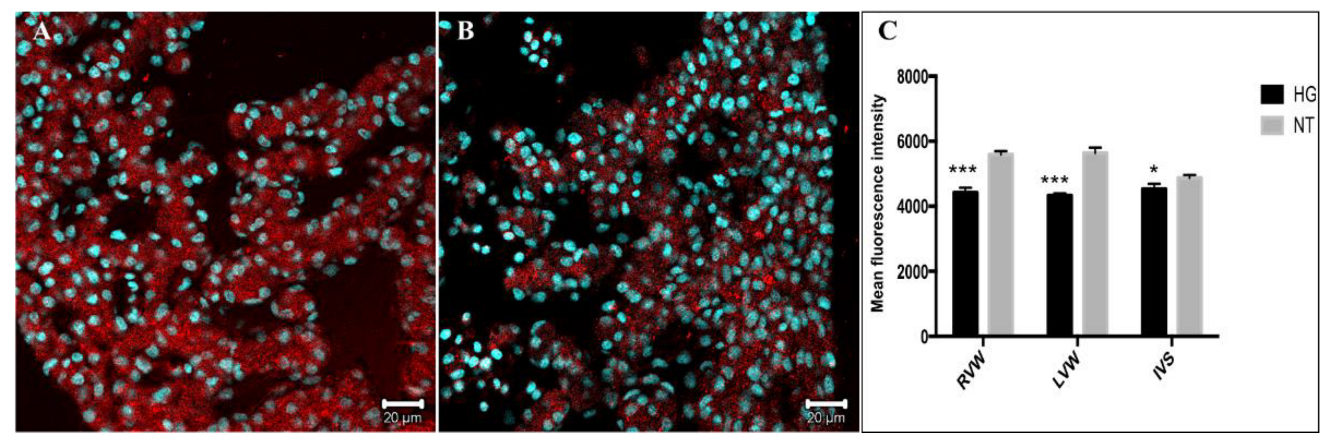
3.4. Immunodetection of Myocardial Damage Biomarkers
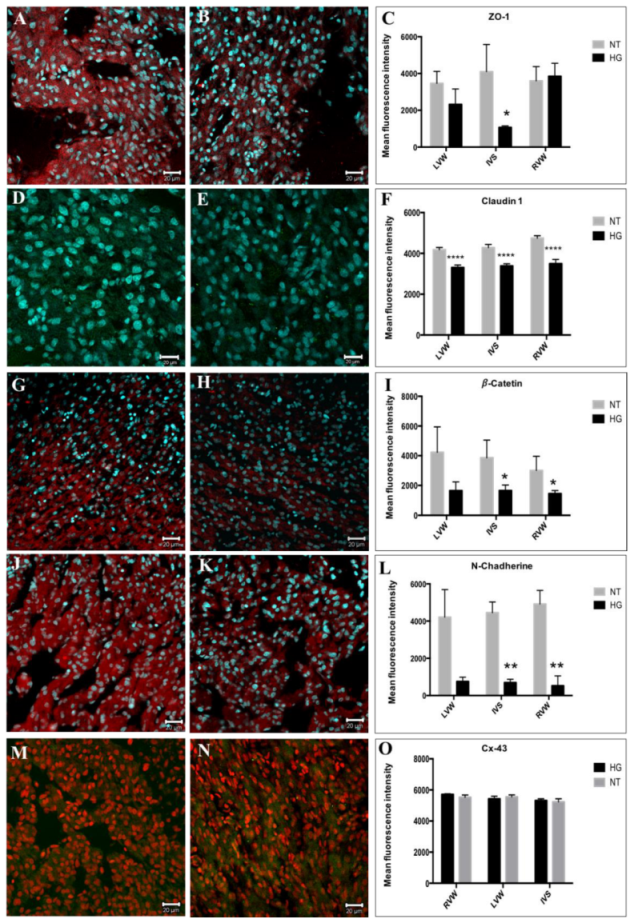
3.5. Evaluation of Cell Junction Proteins
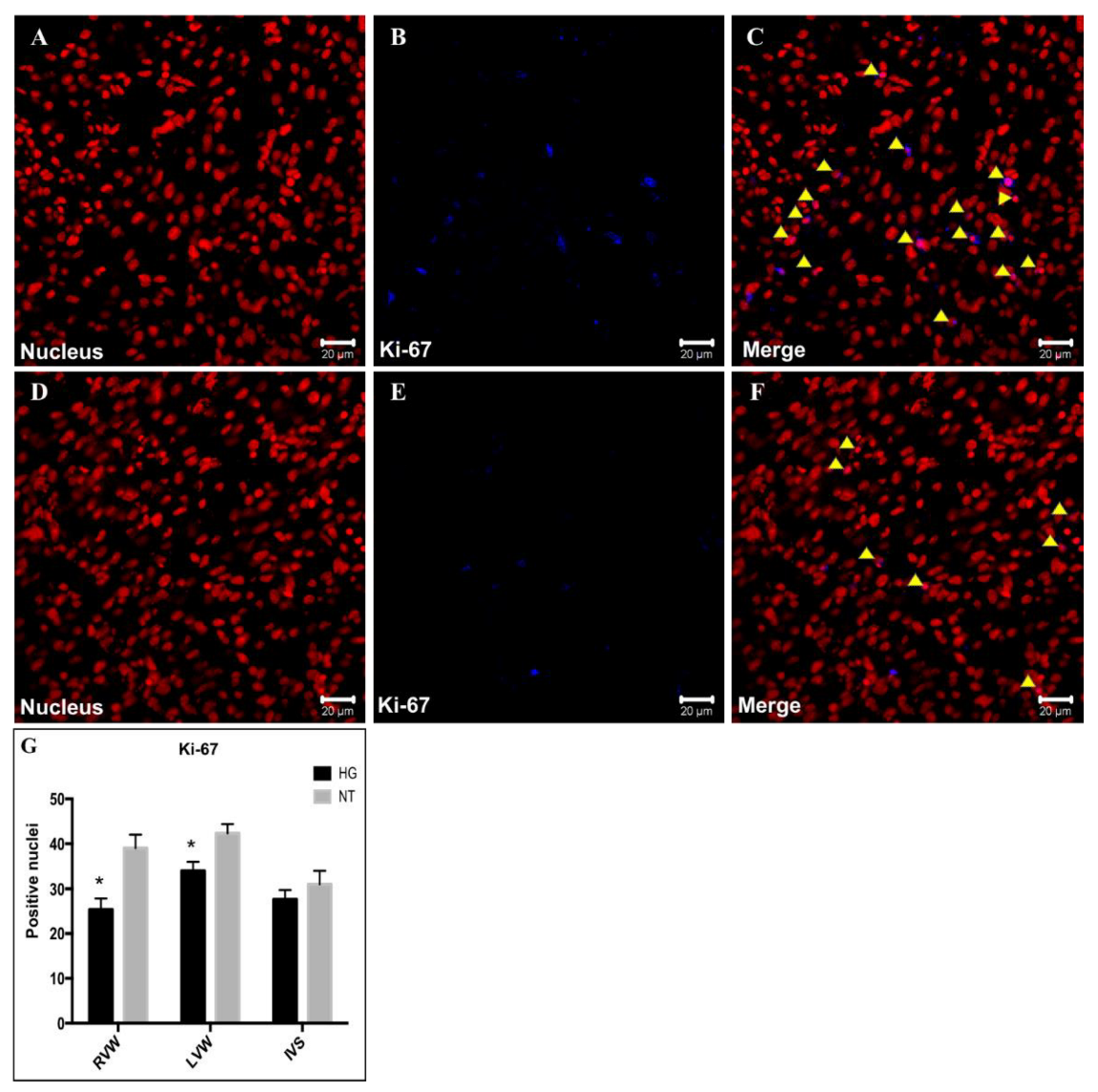
3.6. Evaluation of Cell Proliferation
4. Discussion
4.1. Importance of the Chicken Embryo Hyperglycemia Model
4.2. Effects of Hyperglycemia on Cardiac Morphology and Histology
4.3. Altered Expression Patterns of Sarcomeric Proteins Associated with Myocardial Damage
4.4. Effects of Embryonic Hyperglycemia on Glucose Transport in the Heart
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Z.; Reece, A. New concepts in diabetic embryopathy. Lab. Med. Clin. 2013, 33, 207–223. [Google Scholar] [CrossRef] [Green Version]
- Ogurtsova, K.; da Rocha Fernandes, J.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Res. Clin. Pract. 2019, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, M.C.; Bakris, G.; Blonde, L.; Boulton, A.J.M.; D’alessio, D.; DiMeglio, L.A. Standards of Medical Care in Diabetes. American Diabetes Association. Diabetes Care 2019, 42, S34–S60. [Google Scholar]
- World Health Organization. Informe Mundial Sobre la Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- DeSisto, C.; Kim, S.; Sharma, A. Prevalence Estimates of Gestational Diabetes Mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007–2010. Prev. Chronic. Dis. 2014, 11, E104. [Google Scholar] [CrossRef] [Green Version]
- Tkáč, I. Cardiovascular Importance of Hyperglycemia and Hypoglycemia. Diabetes Care 2013, 36, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Sánchez-Gómez, C.; Contreras, A.; Carrillo, B.; Revilla, C. Los segmentos cardiacos primitivos su implicacion en la cardiogenesis normal aplicada a la cardiologia pediatrica. Arch. Mex. Cardiol. 2006, 76, 46–51. [Google Scholar]
- Villavicencio, G.; Salazar Marcela Jaime, C.; Sánchez Gómez, C. Incorporation of the first and second heart fields and prospective fate of the straight heart tube via in vivo labeling of chicken embryos. PLoS ONE 2020, 15, e0234069. [Google Scholar] [CrossRef]
- Villavicencio-Guzmán, L.; Sánchez-Gómez, C.; Jaime-Cruz, R.; Ramírez-Fuentes, T.C.; Patiño-Morales, C.C.; Salazar-García, M. Human Heart Morphogenesis: A New Vision Based on In Vivo Labeling and Cell Tracking. Life 2023, 13, 165. [Google Scholar] [CrossRef]
- Sedmera, D.; Thompson, R. Myocyte Proliferation in the Developing Heart. Dev. Dyn. 2011, 240, 1322–1334. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.E. (Ed.) Guyton y Hall, Tratado de fisiología médica. In Tratado de Fisiología Médica; Elsevier Health Sciences: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Kraker, J.; Viswanathan, S.K.; Knöll, R.; Sadayappan, O. Recent Advances in the Molecular Genetics of Familial Hypertrophic Cardiomyopathy. Front. Physiol. 2016, 7, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Huidobro, M.; Andrea, F.; Carrasco, E. Incidencia de diabetes gestacional y su relación con obesidad en embarazadas. Rev. Médica Chile 2004, 132, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Davey, M.; Tickle, C. The chicken as a model for embryonic development. Cytogenet. Genome Res. 2007, 117, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, R.; Alcántar, O.; Jaime, R.; García, E.G.; Gómez, L.E. Las nanopartículas de oro de 20 nm inhiben la proliferación e invasión de células de carcinoma mamario humano MCF7, in vivo. Mundo Nano 2015, 8, 96–107. [Google Scholar] [CrossRef]
- Itani, N.; Skeffington, K.L.; Beck, C.; Giussani, D. Sildenafil therapy for fetal cardiovascular dysfunction during hypoxic development: Studies in the chick embryo. J. Physiol. 2016, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Suárez, J.; Bravo, A. Conexinas y sistema cardiovascular. Rev. Argent. Cardiol. 2006, 74, 149–156. [Google Scholar]
- Bullwinkel, J.; Baron-Lühr, B.; Lüdemann, A.; Wohlenberg, C.; Gerdes, J.; Scholzen, T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J. Cell Physiol. 2006, 206, 624–635. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Zühlke, L.; Black, G.C.; Choy, M.K.; Li, N.; Keavney, B.D. Global birth prevalence of congenital heart defects 1970–2017: Updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 2019, 48, 455–463. [Google Scholar] [CrossRef]
- Helle, E.; Priest, J.R. Maternal Obesity and Diabetes Mellitus as Risk Factors for Congenital Heart Disease in the Offspring. J. Am. Heart Assoc. 2020, 9, e011541. [Google Scholar] [CrossRef]
- Dervisoglu, P.; Kosecik, M.; Kumbasar, S. Effects of gestational and pregestational diabetes mellitus on the foetal heart: A cross-sectional study. J. Obstet Gynaecol. 2018, 38, 408–412. [Google Scholar] [CrossRef]
- Simeone, R.M.; Devine, O.J.; Marcinkevage, J.A.; Gilboa, S.M.; Razzaghi, H.; Bardenheier, B.H.; Sharma, A.J.; Honein, M.A. Diabetes and congenital heart defects: A systematic review, meta-analysis, and modeling project. Am. J. Prev. Med. 2015, 48, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Elizabeth, K.E.; Ashwin, D.A.; Sobhakumar, S.; Sujatha, T.L. Outcome of large- and small-for-gestational-age babies born to mothers with pre-pregnancy and gestational diabetes mellitus versus without diabetes mellitus. Indian J. Child Health 2018, 5, 592–596. [Google Scholar] [CrossRef]
- Jean-Baptiste, A.; Simeoni, U. Offspring of mothers with hyperglycemia in pregnancy: Short-term consequences for newborns and infants. In Gestational Diabetes; Lapolla, A., Metzger, B.E., Eds.; Karger: Basel, Switzerland, 2020; pp. 194–200. [Google Scholar] [CrossRef]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In Utero Exposure to Maternal Hyperglycemia Increases Childhood Cardiometabolic Risk in Offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Zhou, J.; Hong, K.; Cheng, X.S.; Li, Y.G. MicroRNA-223 displays a protective role against cardiomyocyte hypertrophy by targeting cardiac troponin I-interacting kinase. Cell. Physiol. Biochem. 2015, 35, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Sáenz, S.; Torrejón, M.J.; Bordiú, E.; del Valle, L.; Galindo, M.; Perez, N.; Herraiz, M.A.; Izquierdo, N.; Rubio, M.A.; et al. Introduction of IADPSG Criteria for the Screening and Diagnosis of Gestational Diabetes Mellitus Results in Improved Pregnancy Outcomes at a Lower Cost in a Large Cohort of Pregnant Women: The St. Carlos Gestational Diabetes Study. Diabetes Care 2014, 37, 2442–2450. [Google Scholar] [CrossRef] [Green Version]
- Basu, M.; Garg, V. Maternal hyperglycemia and fetal cardiac development: Clinical impact and underlying mechanisms. Birth Defects Res. 2018, 110, 1504–1516. [Google Scholar] [CrossRef]
- García, M.S.; Maldonado, E.R.; Monsalve, M.C.R.; Guzmán, L.V.; López, A.R.; Sánchez-Gómez, C. Importance of Maternal Diabetes on the Chronological Deregulation of the Intrauterine Development: An Experimental Study in Rat. J. Diabetes Res. 2015, 2015, 54265. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Xiao, K.; Mao, L.; Rockman, H.A.; Marchuk, D.A. Overexpression of TNNI3K, a cardiac-specific MAPKKK, promotes cardiac dysfunction. J. Mol. Cell. Cardiol. 2013, 54, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Icardo, J.M.; Fernandez-Terán, A. Morphologic Study of Ventricular Trabeculation in the Embryonic Chick Heart. Acta Anat 1987, 130, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.; Corrigan, N.; Horgan, S.; Watson, C.J.; Baugh, J.; Downey, P.; McAuliffe, F.M. Cardiomyopathy in Offspring of Pregestational Diabetic Mouse Pregnancy. J. Diabetes Res. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Song, L.; Zhang, Y.J.; Xu, R.; Meng, X. TNNI3K is a novel mediator of myofilament function and phosphorylates cardiac troponin I. Braz. J. Med. Biol. Res. 2013, 46, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, J.; Su, M.; Wang, C.; Chen, J.; Wang, H.; Song, L.; Zou, Y.; Zhang, L.; Zhang, Y.; et al. TNNI3K, a Cardiac-Specific Kinase, Promotes Physiological Cardiac Hypertrophy in Transgenic Mice. PLoS ONE 2013, 8, e58570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagnozzi, R.J.; Gatto, G.J.; Kallander, L.S.; Hoffman, N.E.; Mallilankaraman, K.; Ballard, V.L.T.; Lawhorn, B.G.; Stoy, P.; Philp, J.; Graves, A.P.; et al. Inhibition of the Cardiomyocyte-Specific Kinase TNNI3K Limits Oxidative Stress, Injury, and Adverse Remodeling in the Ischemic Heart. Sci. Transl. Med. 2013, 5, 207ra141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landstrom, A.P.; Ackerman, M.J. Beyond the Cardiac Myofilament: Hypertrophic Cardiomyopathy- Associated Mutations in Genes that Encode Calcium-Handling Proteins. Curr. Mol. Med. 2012, 12, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Sundström, J.; Ingelsson, E.; Berglund, L.; Zethelius, B.; Lind, L.; Venge, P.; Ärnlöv, J. Cardiac troponin-I and risk of heart failure: A community-based cohort study. Eur. Heart J. 2009, 30, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.; Muñoz-Martín, N.; Lodder, E. The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment. Int. J. Mol. Sci. 2021, 22, 6422. [Google Scholar] [CrossRef]
- Shah, A.S.V.; Ferry, A.V.; Mills, N.L. Cardiac Biomarkers and the Diagnosis of Myocardial Infarction in Women. Curr. Cardiol. Rep. 2017, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, L.G.; Dalakas, M.C. Tragedy in a heartbeat: Malfunctioning desmin causes skeletal and cardiac muscle disease. J. Clin. Investig. 2009, 119, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p. A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein–Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molica, F.; Figueroa, X.F.; Kwak, B.R.; Isakson, B.E.; Gibbins, J.M. Connexins and Pannexins in Vascular Function and Disease. Int. J. Mol. Sci. 2018, 19, 1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisewski, U.; Shi, Y.; Wrackmeyer, U.; Fischer, R.; Chen, C.; Schirdewan, A.; Jüttner, R.; Rathjen, F.; Poller, W.; Radke, M.H.; et al. The tight junction protein CAR regulates cardiac conduction and cell–cell communication. J. Exp. Med. 2008, 205, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Vincent, K.; Peter, A.K.; Klos, M.; Cheng, H.; Huang, S.M.; Towne, J.; Ferng, D.; Gu, Y.; Dalton, N.D.; et al. Cardiomyocyte Expression of ZO-1 Is Essential for Normal Atrioventricular Conduction but Does Not Alter Ventricular Function. Circ. Res. 2020, 127, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Nadadur, R.D.; Brennan, J.A.; Smith, H.L.; Shen, K.M.; Gadek, M.; Laforest, B.; Wang, M.; Gemel, J.; Li, Y.; et al. ZO-1 Regulates Intercalated Disc Composition and Atrioventricular Node Conduction. Circ. Res. 2020, 127, e28–e43. [Google Scholar] [CrossRef]
- Simard, A.; Di Pietro, E.; Young, C.R.; Plaza, S.; Ryan, A.K. Alterations in heart looping induced by overexpression of the tight junction protein Claudin-1 are dependent on its C-terminal cytoplasmic tail. Mech. Dev. 2006, 123, 210–227. [Google Scholar] [CrossRef]
- Meng, W.; Takeichi, M. Adherens Junction: Molecular Architecture and Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Goossens, S.; van Hengel, J.; Gao, E.; Cheng, L.; Tyberghein, K.; Shang, X.; De Rycke, R.; van Roy, F.; Radice, G.L. Loss of αT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J. Cell Sci. 2012, 125, 1058–1067. [Google Scholar] [CrossRef] [Green Version]
- Gutstein, D.E.; Liu, F.-Y.; Meyers, M.B.; Choo, A.; Fishman, G. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J. Cell Sci. 2003, 116, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.A.; McBride, M.W.; Smith, G.; George, S.J.; Baker, A. Dysregulation of cadherins in the intercalated disc of the spontaneously hypertensive stroke-prone rat. J. Mol. Cell. Cardiol. 2010, 48, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced Deletion of the N-Cadherin Gene in the Heart Leads to Dissolution of the Intercalated Disc Structure. Circ. Res. 2005, 96, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N. Gap Junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [CrossRef] [Green Version]
- Solano, J.N.; Fornieri, M.V. Cardiomiopatía diabética: Entidad poco conocida y el impacto terapéutico de los inhibidores del cotransporrtador sodio-glucosa tipo 2 en el miocardio diabetico. Rev. Clínica Esc. Med. Univ. Costa Rica 2019, 9, 11–27. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2011, 6, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hess, C.; Hiatt, W.; Goldfine, A. Clinical Update: Cardiovascular Disease in Diabetes Mellitus Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus—Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Joyner, N.T.; Smoak, I.W. In vivo hyperglycemia and its effect on Glut-1 expression in the embryonic heart. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 438–448. [Google Scholar] [CrossRef]
- Wheeler, F.C.; Tang, H.; Marks, O.A.; Hadnott, T.N.; Chu, P.-L.; Mao, L.; Rockman, H.A.; Marchuk, D.A. Tnni3k Modifies Disease Progression in Murine Models of Cardiomyopathy. PLoS Genet. 2009, 5, e1000647. [Google Scholar] [CrossRef] [Green Version]
- Hufnagel, A.; Dearden, L.; Fernandez-Twinn, D.S.; E Ozanne, S. Programming of cardiometabolic health: The role of maternal and fetal hyperinsulinaemia. J. Endocrinol. 2022, 253, R47–R63. [Google Scholar] [CrossRef]
- Sánchez-Soriano, C.; Pearson, E.R.; Reynolds, R.M. The role of genetics in fetal programming of adult cardiometabolic disease. J. Dev. Orig. Health Dis. 2022, 13, 292–299. [Google Scholar] [CrossRef]
- Dong, M.; Zheng, Q.; Ford, S.P.; Nathanielsz, P.W.; Ren, J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J. Mol. Cell. Cardiol. 2013, 55, 111–116. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaime-Cruz, R.; Sánchez-Gómez, C.; Villavicencio-Guzmán, L.; Lazzarini-Lechuga, R.; Patiño-Morales, C.C.; García-Lorenzana, M.; Ramírez-Fuentes, T.C.; Salazar-García, M. Embryonic Hyperglycemia Disrupts Myocardial Growth, Morphological Development, and Cellular Organization: An In Vivo Experimental Study. Life 2023, 13, 768. https://doi.org/10.3390/life13030768
Jaime-Cruz R, Sánchez-Gómez C, Villavicencio-Guzmán L, Lazzarini-Lechuga R, Patiño-Morales CC, García-Lorenzana M, Ramírez-Fuentes TC, Salazar-García M. Embryonic Hyperglycemia Disrupts Myocardial Growth, Morphological Development, and Cellular Organization: An In Vivo Experimental Study. Life. 2023; 13(3):768. https://doi.org/10.3390/life13030768
Chicago/Turabian StyleJaime-Cruz, Ricardo, Concepción Sánchez-Gómez, Laura Villavicencio-Guzmán, Roberto Lazzarini-Lechuga, Carlos César Patiño-Morales, Mario García-Lorenzana, Tania Cristina Ramírez-Fuentes, and Marcela Salazar-García. 2023. "Embryonic Hyperglycemia Disrupts Myocardial Growth, Morphological Development, and Cellular Organization: An In Vivo Experimental Study" Life 13, no. 3: 768. https://doi.org/10.3390/life13030768